
ABSTRACT
We characterized the complete mitogenome of Pipistrellus coromandra (Indian pipistrelle) for comparative analysis of mitogenomes and for resolving the phylogenetic relationship of four tribes in the subfamily Vespertilioninae. The mitogenome size of P. coromandra was 17,153 bp, with a control region and a typical set of 37 mitochondrial genes. The nucleotide composition of the P. coromandra mitogenome showed an AT bias with a nucleotide composition of 33.5% A, 30.7% T, 13.3% G, and 22.5% C. The mitochondrial protein-coding genes in P. coromandra use the standard start codon (ATN), two stop codons (TAA and AGA), and two incomplete stop codons (TA- and T--). The intertribal relationship of four tribes was highly resolved from the phylogenetic analysis of mitogenome sequences.
1. Introduction
Animal mitogenomes are generally 16–18-kb double-stranded circular molecules (Boore Citation1999; Jeon & Park Citation2015) that consist of 13 protein-coding genes (PCGs), two ribosomal RNA genes (12S rRNA and 16S rRNA), 22 transfer RNA genes (tRNAs), and a non-coding control region (CR) that contains signals required for replication and transcription (Wolstenholme Citation1992; Ruokonen & Kvist Citation2002; Gissi et al. Citation2008). Mitogenomes have been used as reliable tools in various phylogenetic and molecular evolution studies because of their special characteristics, such as uniparental inheritance, a unique genetic code, high mutation rates, and low number of recombination events. A few rapidly evolving regions (e.g. the CR) within the mitogenome have been used in phylogenetic analyses to investigate intraspecific relationships and relationships between closely related species (Kim et al. Citation2010), while genome-level analyses, which include base compositions, codon usages, gene order arrangements, and secondary structures of tRNA and rRNA genes, have been used to identify higher level phylogeny among taxa (Boore Citation1999; Lei et al. Citation2010). Thus, the generation and identification of characteristics of complete mitogenome sequences are useful when investigating phylogenetic relationships among taxa at higher levels.
The Chiroptera is the second largest order in mammals and includes about 1240 bat species (Schnitzler & Kalko Citation2001). Among bats, the family Vespertilionidae has over 300 species distributed worldwide and is the largest and most well-known family. Approximately, 240 species of this family have been placed in the subfamily Vespertilioninae (Simmons Citation2005). Although various studies have assessed the phylogenetic relationships within Vespertilioninae (Roehrs et al. Citation2010; Koubínová et al. Citation2013), intertribal relationships based on complete mitogenome sequences have not yet been determined.
In the present study, the complete mitogenome of the Indian pipistrelle, Pipistrellus coromandra (Vespertilioninae), was characterized, and compared with previously published Vespertilioninae mitogenomes. Based on the mitogenome DNA sequences, we investigated the intertribal relationships in the subfamily Vespertilioninae, including the tribes Pipistrellini, Vespertilionini, Plecotini, and Lasiurini.
2. Materials and methods
2.1. Sample collection and DNA extraction
P. coromandra is distributed throughout most of South Asia, parts of southern China, and much of mainland Southeast Asia. A wing membrane tissue sample was collected from a P. coromandra individual after it was caught in the academic research forests of the University Putra Malaysia (Malaysia) using a mist-net (Avinet, Dryden, NY, USA). The tissue sample was stored at −40°C until total genomic DNA extraction. Total genomic DNA was extracted from the wing membrane tissue sample using the DNeasy® Blood & Tissue Kit (Qiagen, Valencia, CA, USA), according to the manufacturer's protocols.
2.2. Primer design, PCR amplification, and DNA sequencing
For PCR amplification of the complete mitogenome of P. coromandra, we used 10 newly designed primer sets (Supplementary Table 1) from sequence-conserved regions on the multiple alignments of the complete mitogenomes of Vespertilio sinensis (NC024558) and Pipistrellus abramus (NC005436), which are available from GenBank (Nikaido et al. Citation2001; Yoon et al. Citation2014).
PCR amplification was performed in a final reaction volume of 25 μL, which contained 10 mM Tris-HCl (pH 8.4), 50 mM KCl, 4 mM MgCl2, 200 mM of each dNTP, 50 pmol of each primer, 2 U ExTaq polymerase, and 1 μL of DNA sample, under the following conditions: 94°C for 5 min (initial denaturation); then 94°C for 1 min (denaturation), 48–56°C for 30 s (annealing), and 72°C for 1 min (extension) for 35 cycles; with a final extension at 72°C for 10 min. The PCR products were resolved by electrophoresis in 1.0% agarose gel, and were extracted using a DNA Gel Extraction Kit (Qiagen, Valencia, CA, USA). The extracts were then sent to Biomedic Co. Ltd. (Bucheon, South Korea) for sequencing from both directions using a primer-walking strategy.
2.3. Genome annotation
The complete mitogenome of P. coromandra was annotated and characterized based on the V. sinensis and P. abramus mitogenomes, and then aligned with the other six Vespertilioninae species using the Clustal W program found in Geneious Pro 5.5.9 (Biomatters, Auckland, New Zealand). The 13 mitochondrial PCG sequences were translated into amino acid sequences using the vertebrate mitogenome genetic code. The tRNA scan-SE search server and the ARWEN web server with default parameters were used to identify the tRNA genes and potential stem-loop secondary structures within these tRNA genes (Lowe & Eddy Citation1997; Bernt et al. Citation2013). Nucleotide skewness was calculated according to the formulae: AT skew = [A − T]/[A + T] and GC skew = [G − C]/[G + C] (Lobry Citation1996). The tandem repeats were searched in the CR using the Tandem Repeats Finder program (Benson Citation1999).
2.4. Phylogenetic analysis
Phylogenetic relationships among the four tribes (Pipistrellini, Vespertilionini, Lasiurini, and Plecotini) were inferred using a concatenated nucleotide dataset (11,785 bp) of the mitochondrial 13 PCGs, two RNA genes (12S rRNA and 16S rRNA), and 22 tRNA genes except for the CR and the 3rd codon positions and stop codons of 13 PCGs, which are highly variable. Among the complete mitogenome sequences available from GenBank, we selected two mitogenome sequences from closely (Phyllostomidae: Artibeus lituratus) (NC016871) and more distantly (Rhinolophidae: Rhinolophus pumilus) (NC005434) related families to the family Vespertilionidae as outgroup (Agnarsson et al. Citation2011).
The phylogenetic relationships were inferred using Bayesian inference (BI) procedures in MrBayes version 3.2.2 program (Ronquist et al. Citation2012) and the maximum likelihood (ML) method in MEGA v.6.06 (Adachi & Hasegawa Citation1996), respectively. For the BI analysis, general time reversible model (GTR) + I + G was selected as the best substitution model under Akaike information criterion by Modetest 3.7 (Posada & Crandall Citation1998). The concatenated multi-gene dataset was partitioned as three parts of 13 PCGs, two rRNA genes (12S rRNA and 16S rRNA), and 22 tRNA genes. The BI analysis was performed with two simultaneous runs of two million generations and one out of every 500 generations was sampled. The first 25% of trees were discarded and the resulting trees were used to generate a majority consensus tree based on posterior probabilities. For the ML analysis, the GTR with gamma distributed invariant sites (G + I) was selected as the best model of evolution by the MEGA v 6.06. The number of discrete gamma categories was specified by the default. The ML heuristic method for tree search was the Nearest Neighbor Interchange starting with the initial tree selected automatically under the default NJ/BioNJ. The reliability of the phylogenetic tree was supported with 1000 bootstrap replications.
3. Results and discussion
3.1. Genome organization
The complete mitogenome (KP688404) of P. coromandra is a circular molecule that consists of a CR and a typical set of 37 vertebrate mitochondrial genes containing 13 PCGs, 22 tRNA genes, and 2 rRNA genes (12s rRNA and 16s rRNA) (). One Nd6 and eight tRNAs were transcribed from the light strand, while the other 12 PCGs, 14 tRNAs, and 2 rRNAs were located on the heavy strand. The gene order and orientation of the P. coromandra mitogenome were identical to those of all the other Vespertilioninae species ().
Figure 1. Map of the P. coromandra mitogenome. Gray color indicates the PCG regions. Genes for tRNAs are designated by a single letter code for the corresponding amino acid. The heavy strand in the outer circle encodes 28 genes, whereas 9 genes are encoded in the light strand in the inner circle.
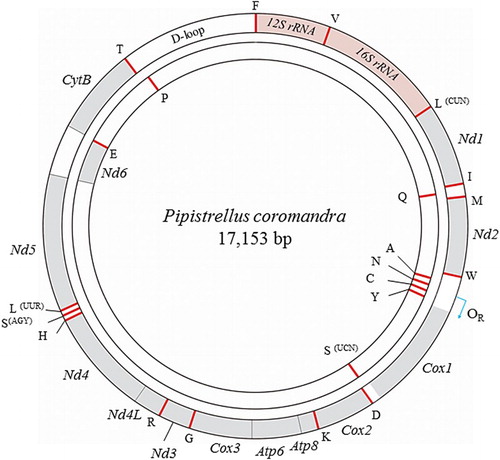
Table 1. Gene organization and characterization of P. coromandra mitogenome.
The total P. coromandra mitogenome was 17,153 bp, which is similar in size to the other completely sequenced mitogenomes of the subfamily Vespertilioninae (). A greater size variation was observed in the CRs when compared with variations in the other mitochondrial gene regions. The variations in the CRs are mainly due to differences in the number of repeated motifs at the 3′-end of the CR.
Table 2. Size and nucleotide composition of the subfamily Vespertilioninae mitogenomes.
3.2. Nucleotide composition
The mitogenome of P. coromandra showed AT bias with a nucleotide composition of 33.5% A, 30.7% T, 13.3% G, and 22.5% C, which is similar to that in the other Vespertilioninae mitogenomes (). As observed in the mitogenomes of other mammals (Reyes et al. Citation1998; Gibson et al. Citation2005), the Vespertilioninae mitogenomes, including that of P. coromandra, have a nucleotide composition order of A > T > C > G, which indicates that the AT content is higher than the GC content (). The AT skew of all the Vespertilioninae mitogenomes was positive and their GC skew was negative, respectively. The GC skew value was −0.26 for P. coromandra, Plecotus auritus, and V. sinensis mitogenomes, and their mitochondrial GC skew value was less negative than that (−0.28) for Chalinolobus tuberculatus and Plecotus rafinesquii. The asymmetrical base composition seen in the Vespertilioninae mitogenomes was probably due to the selection pressures such as mutational pressure (Sueoka Citation1962, 1994; Reyes et al. Citation1998; Hassanin et al. Citation2005; Castellana et al. Citation2011), purifying (Ruiz-Pesini et al. Citation2004; Castellana et al. Citation2011; Bi & Sokouri Citation2015), and adaptive selection (Ruiz-Pesini et al. Citation2004; da Fonseca et al. Citation2008; Castellana et al. Citation2011).
3.3. Codon usage and the sequence features of PCGs
The total length of the 13 mitochondrial PCGs in P. coromandra was 11,408 bp, which could be translated into 3793 amino acids (). It has been reported that mammalian mitogenomes contain some compact and overlapping regions between PCGs (Fernandez-Silva et al. Citation2003). We observed a 43 bp overlap between Atp8 and Atp6, a 17 bp overlap between Nd5 and Nd6, a 7 bp overlap between Nd4L and Nd4, and a single bp overlap between Atp6 and Cox3 (). Similar gene overlapping has been observed in other bat mitogenomes (Meganathan et al. Citation2012).
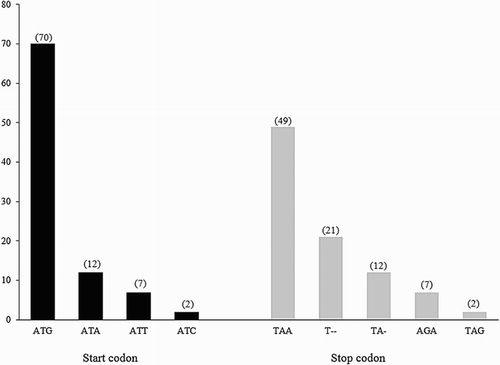
The 13 mitochondrial PCGs in P. coromandra use the standard start codon (ATN), two stop codons (TAA and AGA), and two incomplete stop codons (TA- and T--) for translation initiation and termination (). Ten PCGs used ATG as a start codon; the exceptions were Nd2 and Nd5 (ATA), and Nd3 (ATT). TAA was used as a stop codon in seven PCGs (Cox1, Cox2, Atp8, Atp6, Nd4L, Nd5, and Nd6). The stop codon AGA was used in Cytb and Nd2, and the incomplete stop codon (T--) was used in Nd4, which may be completed by poly-adenylation of the 3′-end of the mRNA after transcription (Anderson et al. Citation1981; Boore Citation1999; Yoon et al. Citation2014).
Loss of unnecessary genes, gene transfer to the nucleus, and reduction of genome size required for rapid organelle replication could account for the compactness of mitochondrial genomes (Selosse et al. Citation2001). Thus, incomplete stop codons or overlaps between genes may be a product of the selective pressure to reduce genome size in mitochondria (Rand Citation1993; Selosse et al. Citation2001).
Codon usage bias for the start and stop codons in the seven Vespertilioninae mitogenomes is shown in and Supplementary Table 2. The four different start codons (ATG, ATA, ATT, and ATC) were detected in all 13 Vespertilioninae mitogenome PCGs. ATG was the most common start codon, accounting for 77.78% of the overall 91 start codons and appearing in 10 PCGs (excluding Nd2, Nd3, and Nd5), whereas the rare start codon ATC was only used in the Nd2 of P. abramus and P. auritus. The three stop codons (TAA, AGA, and TAG) and the two incomplete stop codons (TA and T--) were detected in 13 Vespertilioninae mitogenome PCGs. The most common stop codon was TAA, which accounted for 53.85% of the 91 stop codons and appeared in Cox1, Cox2, Atp8, Atp6, Nd4L, Nd5, and Nd6. The rare stop codon AGA was only detected in Cytb. The codon usage pattern of the P. coromandra mitogenome, apart from the absence of the start codon ATC, was similar to the other Vespertilioninae mitogenomes.
Figure 2. Usage bias of the start and stop codons in the 13 mitochondrial PCGs of the seven Vespertilioninae species. The number in the parenthesis on each codon indicates the number of times the codon appears in the 13 mitochondrial PCGs of the seven Vespertilioninae species.
3.4. Ribosomal RNA and transfer RNA genes
The rRNA genes in the P. coromandra mitogenome were located between tRNAPhe and tRNALeu (CUN) and separated by tRNAVal (). The combined size of the two P. coromandra rRNA genes was 2533 bp, which was longer than the other Vespertilioninae mitogenomes, with the exception of the Lasiurus borealis mitogenome ().
The 22 tRNA genes of the P. coromandra mitogenome were interspersed in the mitogenome, and the combined size of the 22 tRNA genes was 1524 bp, ranging from 60 bp in tRNASer (AGY) to 76 bp in tRNALeu (CUR) (). Its size is a little longer than that of the other Vespertilioninae mitogenomes (). Most of the tRNA genes of P. coromandra mitogenome could be folded into the canonical cloverleaf secondary structure. The DHU arm had been deleted in the secondary structure of the tRNASer (AGY), which is thought to be common in the metazoan mitogenome (Lavrov et al. Citation2000; Kilpert & Podsiadlowski Citation2006).
3.5. Non-coding regions
The metazoan mitogenome contains some non-coding regions, such as the origin of replication (OR), the CR, and some intergenic spacers. These non-coding regions are important during the replication and maintenance of the mitogenome (Annex & Williams Citation1990; Fernandez-Silva et al. Citation2003).
The mitochondrial OR of P. coromandra is located between tRNAAsn and tRNACys in the WANCY region, which consists of a cluster of five tRNA genes (tRNATrp, tRNAAla, tRNAAsn, tRNACys, and tRNATyr) (), as in other vertebrates (Seutin et al. Citation1994). The size mitochondrial OR in the Vespertilioninae, including P. coromandra, is generally 35 bp except for two species P. rafinesquii (31 bp) and L. borealis (32 bp), respectively ().
The CR of P. coromandra is located between tRNAPro and tRNAPhe, and 1698 bp long (). It is longer than that of the other Vespertilioninae bats, ranging from 1013 bp long in P. rafinesquii to 1591 bp long in L. borealis. In comparison with the other genes, the CR showed the higher variation in size, which is caused of repeated sequences.
Previous studies show the presence of termination-associated sequences, many complex repeat regions, and some conserved regions within the CR of bat mitogenomes (Wilkinson et al. Citation1997; Pumo et al. Citation1998). These features have been observed in the mitogenomes of all Vespertilioninae species.
Intergenic spacers were present at 10 sites in the P. coromandra mitogenome, and ranged from a single bp spacer (between tRNATyr and Cox1, tRNALys and Atp8, tRNAArg and Nd3, and tRNAArg and Nd4L) to a 7-bp spacer (between tRNAtrp and tRNAAla) (). In Vespertilioninae, the combined sizes of the mitochondrial intergenic spacers ranged from 30 bp in V. sinensis to 43 bp in C. tuberculatus. Such intergenic spacers have generally been observed in various metazoan mitogenomes, including mammalian mitogenomes (Fernandez-Silva et al. Citation2003).
3.6. Phylogenetic relationship
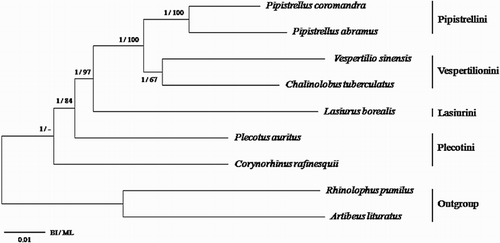
The monophyly of the subfamily Vespertilioninae is supported by high bootstrap values in the BI analysis, but by relatively lower bootstrap values in the ML analyses (). The two Pipistrellus species, P. coromandra and P. abramus, are well grouped. The two species of the tribe Vespertilionini, V. sinensis and C. tuberculatus, also form a sister group. As in a previous study (Koubínová et al. Citation2013), the tribes Pipistrellini and Vespertilionini are sisters, which in turn are sisters to the tribe Lasiurini. The tribe Plecotini is basal to the other tribes. In the ML analysis, however, phylogenetic position of C. rafinesquii (Plecotini) and the sister relationship between the two species of the Vespertilionini (V. sinensis and C. tuberculatus) are unclear. Therefore, it is required inclusion of mitogenomes from further taxa for resolving the exact relationship between its members.
Figure 3. Higher phylogeny of the subfamily Vespertilioninae. The phylogenetic relationships among the four tribes (Pipistrellini, Vespertilionini, Lasiurini, and Plecotini) were inferred from concatenated nucleotide dataset of 13 mitochondrial PCGs, 2 rRNA genes, and 22 tRNA genes using Bayesian (BI) analysis and maximum likelihood (ML) method. Numbers on the nodes represent BI posterior probability/ML bootstrap value, respectively.
4. Conclusions
We sequenced the completed mitogenome of P. coromandra and characterized a typical set of 37 mitochondrial genes and a CR on the mitogenome. In the mitogenome comparison across seven species of the subfamily Vespertilioninae, the general features of the mitogenomes, including gene region size, base composition, and codon usage for translation initiation and termination, were relatively well conserved within Vespertilioninae. In the phylogenetic analysis, P. coromandra was placed within the tribe Pipistrellini, forming a sister clade to P. abramus. The phylogenetic relationship of the four tribes was relatively well resolved by the BI analysis based on the mitogenome sequences. However, since the current results depend on only seven mitogenome sequences, more mitogenome sequences need to be included for understanding higher phylogeny of the subfamily Vespertilioninae.
Acknowledgements
The authors would like to thank the anonymous reviewers for their valuable comments and suggestions to improve the quality of the paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Adachi J, Hasegawa M. 1996. Model of amino acid substitution in proteins encoded by mitochondrial DNA. J Mol Evol. 42:459–468. doi: 10.1007/BF02498640
- Agnarsson I, Zambrana-Torrelio CM, Flores-Saldana NP, May-Collado LJ. 2011. A time-calibrated species-level phylogeny of bats (Chiroptera, Mammalia). PLOS Currents Tree of Life. Edition 1. doi:10.1371/currents.RRN1212.
- Anderson S, Bankier AT, Barrell BG, De Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature. 209:457–465. doi: 10.1038/290457a0
- Annex BH, Williams RS. 1990. Mitochondrial DNA structure and expression in specialized subtypes of mammalian striated muscle. Mol Cell Biol. 10:5671–5678. doi: 10.1128/MCB.10.11.5671
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580. doi: 10.1093/nar/27.2.573
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319. doi: 10.1016/j.ympev.2012.08.023
- Bi SG, Sokouri DP. 2015. Nucleotide variation and selective pressure in the mitochondrial genome of African elephants. Int J Sci Res (IJSR). 4:1733–1742.
- Boore JL. 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27:1767–1780. doi: 10.1093/nar/27.8.1767
- Castellana S, Vicario S, Saccone C. 2011. Evolutionary patterns of the mitochondrial genome in metazoa: exploring the role of mutation and selection in mitochondrial protein-coding genes. Genome Biol Evol. 3:1067–1079. doi: 10.1093/gbe/evr040
- Fernandez-Silva P, Enriquez JA, Montoya J. 2003. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 88:41–56. doi: 10.1113/eph8802514
- da Fonseca RR, Johnson WE, O'Brien SJ, Ramos MJ, Antunes A. 2008. The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics. 9:119. doi: 10.1186/1471-2164-9-119
- Gibson A, Gowri-Shankar V, Higgs PG, Rattray MA. 2005. Comprehensive analysis of mammalian mitochondrial genome base composition and improved phylogenetic methods. Mol Biol Evol. 22:251–264. doi: 10.1093/molbev/msi012
- Gissi C, Iannelli F, Pesole G. 2008. Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity. 101:301–320. doi: 10.1038/hdy.2008.62
- Hassanin A, Leger N, Deutsch J. 2005. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst Biol. 54:277–298. doi: 10.1080/10635150590947843
- Jeon MG, Park YC. 2015. The complete mitogenome of the wood-feeding cockroach Cryptocercus kyebangensis (Blattodea: Cryptocercidae) and phylogenetic relations among cockroach families. Anim Cells Syst. 19:432–438. doi: 10.1080/19768354.2015.1105866
- Kilpert F, Podsiadlowski L. 2006. The complete mitochondrial genome of the common sea slater, Ligia oceanica (Crustacea, Isopoda) bears a novel gene order and unusual control region features BMC. Genomics. 7:241.
- Kim SG, Morishimab K, Arai K. 2010. Genetic structure of wild brown sole inferred from mitochondrial DNA analysis. Anim Cells Syst. 14:197–206. doi: 10.1080/19768354.2010.506267
- Koubínová D, Irwin N, Hulva P, Koubek P, Zima J. 2013. Hidden diversity in Senegalese bats and associated findings in the systematics of the family Vespertilionidae. Front Zool. 10:48. doi: 10.1186/1742-9994-10-48
- Lavrov DV, Brown WM, Boore JL. 2000. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc Natl Acad Sci. 97:13738–13742. doi: 10.1073/pnas.250402997
- Lei R, Shorea GD, Brennemana RA, Engberga SE, Sitzmanna BD, Baileya CA, Kimmela LM, Randriamampionona R, Ranaivoarisoab JF, Louis Jr EE. 2010. Complete sequence and genome organization of the mitochondrial genome for Hubbard's sportive lemur (Lepilemur hubbardorum). Gene. 464:44–49. doi: 10.1016/j.gene.2010.06.001
- Lin Y, Penny D. 2001. Implications for bat evolution from two new complete mitochondrial genomes. Mol Biol Evol.18:684–688.
- Lobry JR. 1996. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964. doi: 10.1093/nar/25.5.0955
- Meganathan PR, Heidi JTP, McCulloch SE, Richard DS, David AR. 2012. Complete mitochondrial genome sequences of three bats species and whole genome mitochondrial analyses reveal patterns of codon bias and lend support to a basal split in Chiroptera. Gene. 492:121–129. doi: 10.1016/j.gene.2011.10.038
- Nikaido M, Kawai K, Cao Y, Harada M, Tomita S, Okada N. 2001. Maximum likelihood analysis of the complete mitochondrial genomes of eutherians and a reevaluation of the phylogeny of bats and insectivores. J Mol Evol. 53:508–516. doi: 10.1007/s002390010241
- Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics. 14:817–818. doi: 10.1093/bioinformatics/14.9.817
- Pumo DE, Finamore PS, Franek WR, Phillips CJ, Tarzami S, Balzarano D. 1998. Complete mitochondrial genome of a neotropical fruit bat, Artibeus jamaicensis, and a new hypothesis of the relationships of bats to other eutherian mammals. J Mol Evol. 47:709–717. doi: 10.1007/PL00006430
- Rand DM. 1993. Endotherms, ectotherms, and mitochondrial genome-size variation. J Mol Evol. 37:281–295. doi: 10.1007/BF00175505
- Reyes A, Gissi C, Pesole G, Saccone C. 1998. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 15:957–966. doi: 10.1093/oxfordjournals.molbev.a026011
- Roehrs ZP, Lack JB, Van Den Bussche RA. 2010. Tribal phylogenetic relationships within Vespertilioninae (Chiroptera: Vespertilioninae) based on mitochondrial and nuclear sequence data. J Mammal. 91:1073–1092. doi: 10.1644/09-MAMM-A-325.1
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. Mrbayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542. doi: 10.1093/sysbio/sys029
- Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. 2004. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 303:223–226. doi: 10.1126/science.1088434
- Ruokonen M, Kvist L. 2002. Structure and evolution of the avian mitochondrial control region. Mol Phylogenet Evol. 23:422–432. doi: 10.1016/S1055-7903(02)00021-0
- Schnitzler HU, Kalko EKV. 2001. Echolocation by insect-eating bats. Bioscience. 51:557–569. doi: 10.1641/0006-3568(2001)051[0557:EBIEB]2.0.CO;2
- Selosse MA, Albert B, Godelle B. 2001. Reducing the genome size of organelles favours gene transfer to the nucleus. Trends Ecol Evol. 16:135–141. doi: 10.1016/S0169-5347(00)02084-X
- Seutin G, Lang BF, Mindell DP, Morais R. 1994. Evolution of the WANCY region in amniote mitochondrial DNA. Mol Biol Evol. 11:329–340.
- Simmons NB. 2005. Order Chiroptera. In: Wilson DE, Reeder DM, editors. Mammal species of the world. Baltimore, MD: The Johns Hopkins University Press; p. 312–529.
- Sueoka N. 1962. On the genetic basis of variation and heterogeneity of DNA base composition. Proc Natl Acad Sci USA. 48:582–592. doi: 10.1073/pnas.48.4.582
- Wilkinson GS, Mayer F, Kerth G, Petri B. 1997. Evolution of repeated sequence arrays in the D-loop region of bat mitochondrial DNA. Genetics. 146:1035–1048.
- Wolstenholme DR. 1992. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 141:173–216. doi: 10.1016/S0074-7696(08)62066-5
- Yoon KB, Lee JH, Cho JY, Park YC. 2016. The complete mitochondrial genome of the Asian particolored bat Vespertilio sinensis (Chiroptera: Vespertilionidae) in Korea. Mitochondrial DNA. 27:299–300.