
ABSTRACT
Polyunsaturated fatty acids (PUFAs) have important functions in biological systems. The beneficial effects of dietary PUFAs against inflammatory diseases, cardiovascular diseases, and metabolic disorders have been shown. Studies using cancer cells have presented the anti-tumorigenic effects of docosahexaenoic acid (DHA), an n-3 PUFA, while arachidonic acid (AA), an n-6 PUFA, has been shown to elicit both pro- and anti-tumorigenic effects. In the current study, the anti-tumorigenic effects of AA were evaluated in HT-29 human colon cancer cells. Upon adding AA in the media, more than 90% of HT-29 cells died, while the MCF7 cells showed good proliferation. AA inhibited the expression of SREBP-1 and its target genes that encode enzymes involved in fatty acid synthesis. As HT-29 cells contained lower basal levels of fatty acid synthase, a target gene of SREBP-1, than that in MCF7 cells, the inhibitory effects of AA on the fatty acid synthase levels in HT-29 cells were much stronger than those in MCF-7 cells. When oleic acid (OA), a monounsaturated fatty acid that can be synthesized endogenously, was added along with AA, the HT-29 cells were able to proliferate. These results suggested that HT-29 cells could not synthesize enough fatty acids for cell division in the presence of AA because of the suppression of lipogenesis. HT-29 cells may incorporate more AA into their membrane phospholipids to proliferate, which resulted in ER stress, thereby inducing apoptosis. AA could be used as an anti-tumorigenic agent against cancer cells in which the basal fatty acid synthase levels are low.
Introduction
Polyunsaturated fatty acids (PUFAs) of the n-3 and n-6 series are components of membrane phospholipids, precursors of signal molecules such as prostaglandins and leukotrienes, and regulators of various transcription factors. The health benefits of PUFAs in inflammatory diseases, autoimmune diseases (Simopoulos Citation2002), cardiovascular diseases (Simopoulos Citation2002; Harris et al. Citation2013), and metabolic disorders (Zivkovic et al. Citation2007) have been actively studied. In general, n-3 PUFAs such as eicosapentaenoic acid (EPA, C20:5, n-3) and docosahexaenoic acid (DHA, C22:6, n-3) have been shown to reduce the risks of cardiovascular disease and inhibit inflammatory reactions (Morland et al. Citation2016; Calder Citation2017). The roles of n-6 PUFA metabolites in such diseases have been suggested to be mainly pro-inflammatory and pro-allergenic (Patterson et al. Citation2012); however, n-6 PUFAs also have effects similar to those of n-3 PUFAs when they act as regulators of transcription factors (Hannah et al. Citation2001; Patterson et al. Citation2012).
N-3 PUFAs have shown protective effects against cancers and are expected to reduce the incidence of cancer (Hall et al. Citation2008; Sawada et al. Citation2012; Song et al. Citation2017). Multiple mechanisms involving changes in membrane properties, eicosanoid profiles, and ROS generation have been proposed (Das Citation1999; Larsson et al. Citation2004; Tanaka et al. Citation2017). Through the regulation of various transcription factors including sterol regulatory element-binding proteins (SREBPs) (Hannah et al. Citation2001), peroxisome proliferation activating receptors (PPARs) (Bordoni et al. Citation2006), and liver X receptor (LXR) (Howell et al. Citation2009), they affect cellular metabolic pathways that may be critical in cancer cell metabolism.
In contrast to the strong anti-tumorigenic effects of n-3 PUFAs, the roles of n-6 PUFAs in cancers have been shown to be pro-tumorigenic (Rose and Connolly Citation1990; Williams et al. Citation2011). Arachidonic acid (AA, C20:4, n-6) is metabolized to produce prostaglandins (PG) by cyclooxygenase (COX). Elevated levels of COX-2 and the accompanying elevation of the PGE2 levels have often been observed in colorectal cancer cases (Sano et al. Citation1995; Brown and DuBois Citation2005), and various studies have shown that COX-2 inhibition by conventional non-steroid anti-inflammatory drugs (NSAIDs) inhibits intestinal polyposis and cancer cell growth in rodent models (Oshima et al. Citation1996; Kawamori et al. Citation1998) and also reduces the risk of colorectal cancer in humans (Chan et al. Citation2007, Citation2009). It has been proposed that in DLD-1 colorectal adenocarcinoma cells, PGE2 binds to the EP2 receptor, which activates the PI-3 K/AKT and β-catenin signaling pathways, thereby stimulating tumor growth (Castellone et al. Citation2005). AA has been reported to stabilize β-catenin in a PG-independent manner through direct interactions with Fas-associated factor 1 (FAF1), which inhibits the degradation of β-catenin and stimulates the growth of tumors in clear cell renal cell carcinoma (Kim et al. Citation2015).
The aim of the current study was to evaluate the effects of AA on cancer cells using HT-29 and MCF7 cells. Endogenous de novo lipogenesis is essential for cancer cells to generate cell membranes for proliferation. AA inhibits the activities of transcription factors that regulate fatty acid synthesis (Hannah et al. Citation2001; Moon et al. Citation2009). In this study, we observed the anti-tumorigenic effects of AA in HT-29 human colon cancer cells but not in MCF7 cells. In HT-29 cells, AA inhibited the endogenous synthesis of fatty acids through the inhibition of SREBP-1, which rendered the cells highly dependent on exogenously supplied fatty acids. The cells treated with AA exhibited endoplasmic reticulum (ER) stress, leading to apoptosis and cell death.
Materials and methods
Cell culture
HT-29 (human colorectal adenocarcinoma) cells and MCF7 cells (human breast adenocarcinoma) were obtained from the ATCC (Manassas, VA). The MCF7 cells were cultured in Dulbecco’s modified Eagle medium (DMEM, GIBCO, Waltham, MA) supplemented with 10% fetal bovine serum (FBS) and 1 × antibiotics (GIBCO). The HT-29 cells were maintained in RPMI-1640 (GIBCO) medium supplemented with 10% FBS and 1 × antibiotics (GIBCO). Fatty acids were obtained from Nu-Chek Prep, Inc. (Elysian, MN); the BSA-bound fatty acids were prepared as described in a previous study (Moon et al. Citation2001) and added to the culture medium. Delipidated FBS (DLFBS) was prepared as previously described (Hannah et al. Citation2001). The cultures were incubated in a humidified atmosphere of 5% CO2 at 37°C. Cerulenin (Sigma, St. Louis, MO) was dissolved in ethanol and added to the medium at 12.5 µM.
MTT assay
The cells were seeded into 96-well plates at a density of 2 × 104 cells/well on day 0. On day 1, the culture media were changed to media supplemented with either 10% FBS or 10% DLFBS. The indicated fatty acids were added to the media at a concentration of 100 µM. On day 4, MTT assay was performed using the Cell Titer 96® AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI), as described in the manufacturer’s protocol.
Detection of apoptosis
The cells were seeded into 60 mm dishes at a density of 4 × 105 cells/dish on day 0. On day 1, the cells were treated with the fatty acids as described above. On day 3, the cells were detached and stained with FITC-Annexin V and propidium iodide using an FITC/Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA). The cells were separated using the BD Accuri C6 flow cytometer (BD Biosciences).
Measurement of ROS
The cells were seeded into 96-well plates at a density of 2 × 104 cells/well on day 0. On day 1, the cells were treated with the fatty acids. On day 2, 10 µl of carboxy-H2DCFDA (Thermo Fisher Scientific) was added to the cells. After incubation for 30 min, the fluorescence from the cells was measured using a Spark microplate reader (Tecan, Thermo Fisher Scientific).
RNA extraction and quantitative real-time PCR
The cells were plated in 60 mm dishes at a density of 4 × 105 cells/dish on day 0. On day 1, the cells were treated with the fatty acids as described above. After 24 h, the total RNA from the cells was extracted using TRizol (Thermo Fisher Scientific), as described in the manufacturer’s protocol. The isolated RNA was treated with DNaseI using a DNA-free kit (Invitrogen), and cDNA was synthesized from the total RNA samples (2 μg) using the iScript™ cDNA Synthesis kit (Bio-Rad, Hercules, CA). Quantitative real-time PCR (qPCR) was performed using the CFX connect™ Optic Module PCR kit (Bio-Rad) and iQ™ SYBRⓇ Green Supermix PCR reagents (Bio-Rad). The results were evaluated using the CFX Maestro™ software (Bio-Rad). The mRNA expression levels of the tested genes were normalized with those of cyclophilin.
Preparation of proteins and immunoblotting analysis
The cells were plated in 100 mm dishes at a density of 2 × 106cells/dish on day 0. On day 1, the cells were treated with the fatty acids as described above. After 24 h, whole-cell lysates were prepared using RIPA lysis buffer (Thermo Fisher Scientific) containing Complete Protease Inhibitor Cocktail Tablets (Roche, Indianapolis, IN). The total protein samples were loaded onto SDS-PAGE gels immunoblotting was performed using rabbit anti-SREBP1 clone 20B12 (Sigma), anti-calnexin (Enzo, Farmingdale, NY), anti-CREB, anti-eIF2α, anti-phospho-eIF2α (Cell Signaling Technology, Danvers, MA), anti-caspase-3, anti-β-actin (Santa Cruz Biotechnology, Dallas, TX). The signals were developed using an enhanced chemiluminescence (ECL) kit (Bio-Rad) and the bands were visualized using the ChemiDoc™ Touch Image system (Bio-Rad).
Determination of XBP1 splicing
The unprocessed and the processed forms of X-box binding protein 1 (XBP1) were examined by PCR using the cDNA prepared as described above. The primers used for the reaction were sense 5′-AAACAGAGTAGCAGCTCAGACTGC-3′ and antisense 5′-TCCTTCTGGGTAGACCTCTGGGAG-3′. The PCR products were separated on a 2% agarose gel.
Results
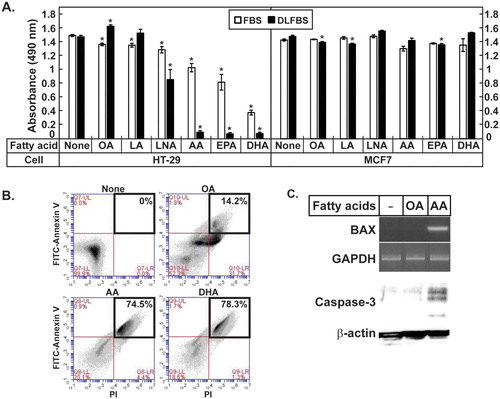
HT-29 and MCF7 cells were cultured in the media supplemented with 10% FBS or 10% DLFBS, and fatty acids of various chain lengths and degrees of desaturation, including oleic acid (OA, C18:1, n-9), linoleic acid (LA, C18:2, n-6), linolenic acid (LNA, C18:3, n-3), AA (C20:4, n-6), EPA (C20:5, n-3), and DHA (C22:6, n-3), were added to the media. Upon the addition of AA, EPA, or DHA to the media supplemented with 10% FBS, 20–60% of the HT-29 cells died, while the HT-29 cells that were not treated with the fatty acids and those treated with OA, LA, or LNA grew well (A). When AA, EPA, or DHA was added to the media supplemented with 10% DLFBS, more than 90% of the HT-29 cells died, while the cells that were not treated with the fatty acids and those that were treated with OA, LA, or LNA grew well (A). MCF7 cells cultured in either the medium containing 10% FBS or that containing 10% DLFBS were not greatly affected by the treatment with any of the fatty acids tested (A). The HT-29 cells incubated without the fatty acids and those that were treated with OA, AA, or DHA were stained with FITC-Annexin V and propidium iodide to determine the degree of apoptosis. The proportions of AA- and DHA-treated HT-29 cells that were stained positively after the FITC-Annexin V/propidium iodide staining were 74.5% and 78.3%, respectively, which indicated the occurrence of apoptosis. In contrast, among the cells not treated with the fatty acids or those treated with OA, 0% and 14.2% of the cells were positively stained, respectively (B). Proapoptotic proteins such as BAX and Caspase-3 were induced in the HT-29 cells treated with AA (C). These results suggested that very-long-chain PUFAs such as AA, EPA, and DHA led to HT-29 cell death, especially when the amounts of exogenous lipids from the serum were limited.
Figure 1. AA induced apoptosis in HT-29 cells. (A) HT-29 and MCF7 cells were treated with the indicated fatty acids, then the MTT assay was performed. The values represent the mean ± SE of 4 wells. * indicates p < 0.01 (Student’s t-test; compared to the untreated cells). (B) The HT-29 cells treated with the indicated fatty acids were stained with FITC-Annexin V/propidium iodide and separated by FACS. The portions of the apoptotic cells were boxed. (C) Expression of BAX were analyzed by reverse transcription-PCR using RNA prepared from the HT-29 cells treated with the indicated fatty acids in the media containing DLFBS. Caspase-3 was analyzed by western blotting. GAPDH and β-actin are the loading controls.
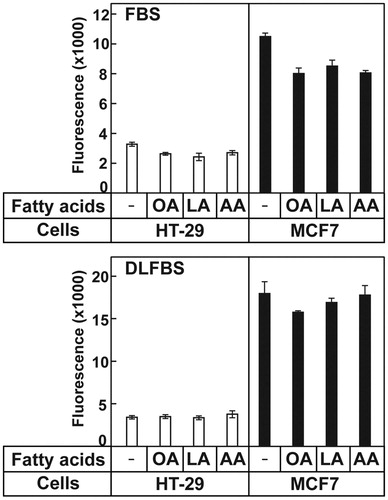
High concentrations of fatty acids can generate ROS in cells, which can be toxic and result in cell death. To ascertain whether the addition of very-long-chain PUFAs increased the ROS levels in the cells, the ROS levels were measured in the cells that were not treated with fatty acids and those that were treated with OA, LA, or AA. The ROS levels in the MCF7 and HT-29 cells did not increase following the addition of either of these fatty acids to both the medium containing 10% FBS and that containing DLFBS (). These results suggested that ROS were not the reason for the death of the AA-treated HT-29 cells.
Figure 2. AA did not affect ROS generation in the cells. HT-29 and MCF7 cells were cultured and treated with the indicated fatty acids, then ROS production was measured from the cells. The values represent the mean ± S.E. of 4 wells.
Cells can synthesize long-chain saturated and monounsaturated fatty acids, such as palmitic (C16:0), stearic (C18:0), and oleic (C18:1, n-9) acids, which are major fatty acids of membrane lipids. The enzymes responsible for de novo fatty acid synthesis are regulated by the transcription factor SREBP-1 (Morton and Shimomura Citation1999; Horton et al. Citation2002). Unsaturated fatty acids including OA and AA are known inhibitors of SREBP-1 (Hannah et al. Citation2001), and thus, its target genes. To know whether OA and AA inhibited SREBP-1 and lipogenesis, the SREBP-1 levels in the HT-29 and MCF-7cells were determined (A). The mRNA level of SREBP-1 and the precursor (inactive) and the nuclear (active) forms of SREBP-1 were remarkably reduced by OA and AA treatment in MCF7 cells. The mRNA level of SREBP-1 in HT-29 was lower than that in MCF-7 cells and inhibition by OA and AA was not as big as in MCF7 cells, although the nuclear form SREBP-1 was reduced by OA and AA treatment (A). The SREBP-1 target genes, including fatty acid synthase (FAS) and stearoyl-CoA desaturase (SCD)-1, in the HT-29 cells and MCF7 cells were found to be reduced by OA and AA (B). The basal mRNA expression levels of FAS and SCD-1 in HT-29 cells were lower than those in MCF7 cells, which were 16% and 19% of those in MCF7 cells, respectively; the expression of these genes was further inhibited by OA and AA (B). These results suggested that the addition of unsaturated fatty acids to the cells suppressed the expression of lipogenic genes, which might inhibit de novo fatty acid synthesis. The low levels of SREBP-1, FAS, and SCD-1 could make HT-29 cells highly susceptible to the suppression of fatty acid synthesis by unsaturated fatty acids, given their dependence on the fatty acids supplied exogenously.
Figure 3. AA inhibited the expression of SREBP-1c and its target genes, and OA could rescue the AA mediated cell death in HT-29 cells. (A) The relative mRNA expression of SREBP-1c was determined by qPCR. The precursor form (pSREBP-1) and the active form of SREBP-1 (nSREBP-1) were determined using proteins prepared from the indicated cells. Calnexin and CREB are the loading controls for pSREBP-1 and nSREBP-1, respectively. (B) The relative expression levels of FAS and SCD-1 were determined by qPCR. The expression levels of these genes in MCF7 cells that were not treated with the fatty acids are defined as 1. The values represent the mean ± SE of 3 plates. * indicates p < 0.05 (Student’s t-test; compared to the untreated cells). (C) OA was added to the cells at the indicated concentrations in the presence or absence of AA and the MTT assay was performed. The values represent the mean ± SE of 4 wells. * and ** indicate p < 0.05 and p < 0.01, respectively (Student’s t-test; compared to cells cultured in DLFBS with 100 µM AA). (D) MCF7 cells were incubated with OA and AA in the presence of cerulenin and the MTT assay was performed. The values represent the mean ± SE of 4 wells. ** indicates p < 0.01 of Student’s t-test by comparison of cerulenin alone and cerulenin with AA).
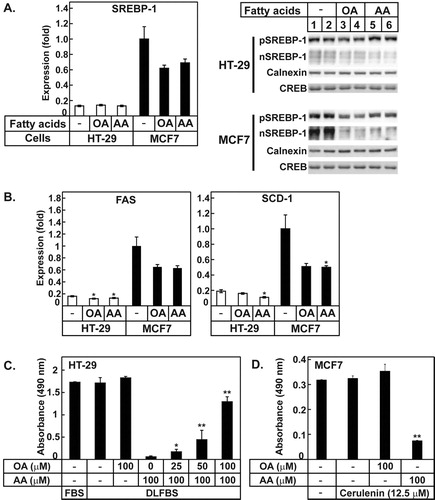
The HT-29 cells were partially protected from cell death by AA when they were cultured in the media supplemented with normal serum, while more than 90% of HT-29 cells died following the addition of AA into the media supplemented with DLFCS (A). In contrast to AA, OA, one of the major fatty acids synthesized endogenously in animal tissues, did not greatly affect the growth of HT-29 cells cultured both in the medium supplemented with FBS and that supplemented with DLFBS despite inhibition of FAS (A and 3B). To determine whether OA could rescue the effect of AA, OA was added to the medium containing DLFBS at various concentrations in the presence of AA. The AA-mediated death of HT-29 cells was rescued by the addition of OA in a dose-dependent manner; 80% of cell death was rescued by the addition of 100 µM OA (C). These results suggested that OA is a possible component that may have been lacking in the HT-29 cells cultured in the media containing AA and DLFBS. In a similar vein, MCF7 cells were treated with cerulenin, an FAS inhibitor, to limit endogenous lipogenesis. Cerulenin is a known anti-cancer reagent (Shiragami et al. Citation2013), however, was added to the media below the concentration of cytotoxicity. MCF7 cells under 12.5 µM cerulenin grew well but died with the addition of AA (D). This result also suggested that the low level of FAS activity could make cancer cells susceptible to AA.
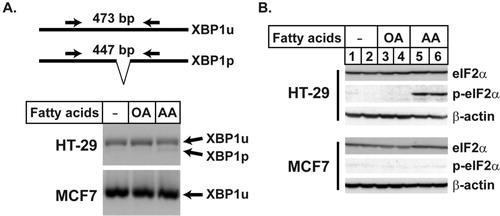
When de novo lipogenesis is inhibited and cells cannot synthesize enough fatty acids, the supplemented fatty acids would be incorporated into the membranes of proliferating cells. Changing the fatty acid composition of the ER membrane would result in ER stress, and thus, lead to apoptosis. Therefore, the ER stress in HT-29 cells incubated with AA was determined by analyzing XBP1 processing and phosphorylation of eIF2α, which are known changes associated with ER stress (Calfon et al. Citation2002; Chen and Brandizzi Citation2013). HT-29 cells treated with AA showed an increase in the level of the processed form of XBP1 and phosphorylated eIF2α, while the MCF7 cells did not show any change in these molecules (). These results suggested that the HT-29 cells with AA became more dependent on the fatty acids supplied exogenously and incorporated more AA supplied into their membrane lipids, which changed the membrane properties, resulted in ER stress, and triggered apoptosis.
Figure 4. AA induced ER stress in HT-29 cells. The cDNAs prepared for the experiment described in were used to determine XBP1 processing. Equal amount of the PCR reaction of the individual samples in each group were pooled and separated on a 2% agarose gel. The unprocessed (XBP1u) and processed (XBP1p) forms of XBP1 generated 473 and 447 bp bands, respectively. (B) Total eIF2α and the phosphorylated eIF2α were determined by immunoblotting in the cells treated with the indicated fatty acids. β-actin is used as the loading control.
Discussion
The aim of the current study was to evaluate the effects of AA on cancer cells using HT-29 and MCF7 cells. AA induced apoptosis in HT-29 cells, while the MCF7 cells showed good proliferation. In both cells, AA inhibited the expression of SREBP-1 and its target genes that encode enzymes involved in fatty acid synthesis. As the HT-29 cells showed lower basal levels of FAS and SCD-1 than those in the MCF7 cell, HT-29 cells could not synthesize enough fatty acids for cell division in the presence of AA, and became more dependent on fatty acids supplied exogenously. HT-29 cells may incorporate more AA into their membrane phospholipids during their proliferation, which resulted in ER stress, ultimately causing apoptosis.
The effects of AA on cancer cells have been evaluated in various studies. The majority of such studies have shown pro-tumorigenic effects of AA by producing more PGE2, which activates growth signals in the cells. The stabilization of β-catenin by AA has been shown to promote the proliferation of renal cell carcinoma cells independent on PGE2. However, depending on the cancer cell type, AA also exhibited anti-tumorigenic effects via the generation of ROS. The current study shows that the HT-29 colon cancer cells were susceptible to AA, while the MCF7 human breast cancer cells were protected. The ability of growing cells to synthesize endogenous fatty acids for the maintenance of membrane lipids seems to be a critical factor that decides the anti-tumorigenic effects of AA in these cells. Both OA and AA inhibited the SREBP-1 activity and the expression of its target genes in HT-29, as well as MCF7 cells. However, HT-29 cells treated with OA, even when OA was added in the presence of AA, proliferated well; this strongly suggested that OA could compensate for the lack of fatty acids produced by de novo lipogenesis, but AA could not. A continuous supply of long-chain saturated or monounsaturated fatty acids, such as palmitic acid, stearic acid, and OA, is critical for the synthesis of membrane phospholipids and maintenance of membrane functions, especially in proliferating cells. The supplied OA would be used by the proliferating cells for forming membranes. AA has 4 cis-double bonds, and its incorporation into the membrane phospholipids as a major component may generate an abnormal membrane structure, which triggers apoptotic reactions.
The FAS level in the cells could be one of the factors for the different responses of HT-29 cells and MCF7 cells to AA. Because MCF7 cells show high expression levels of FAS and SCD-1, they could maintain enough lipogenesis even under conditions when the SREBP-1 activity was inhibited by AA. By screening the FAS levels of the cancer tissues, AA can be used to inhibit tumor growth. Rapidly proliferating cancer cells would be affected to a greater degree by AA, because normal cells do not divide fast. More cancer cells where AA exhibits anti-tumorigenic effects should be studied to propose AA as an anti-cancer therapy. Even though AA alone cannot kill cancer cells completely, combining it with other therapeutic agents will help kill the cancer cells. Thus, local administration of AA may be useful for the treatment of some cancers, especially colon cancers, which are accessible from the outside of the body via endoscopy.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bordoni A, Di Nunzio M, Danesi F, Biagi PL. 2006. Polyunsaturated fatty acids: from diet to binding to ppars and other nuclear receptors. Genes Nutr. 1(2):95–106. doi: 10.1007/BF02829951
- Brown JR, DuBois RN. 2005. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 23(12):2840–2855. doi: 10.1200/JCO.2005.09.051
- Calder PC. 2017. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem Soc Trans. 45(5):1105–1115. doi: 10.1042/BST20160474
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 415(6867):92–96. doi: 10.1038/415092a
- Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. 2005. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-ß-catenin signaling axis. Science. 310(5753):1504–1510. doi: 10.1126/science.1116221
- Chan AT, Ogino S, Fuchs CS. 2007. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med. 356(21):2131–2142. doi: 10.1056/NEJMoa067208
- Chan AT, Ogino S, Fuchs CS. 2009. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 302(6):649–658. doi: 10.1001/jama.2009.1112
- Chen Y, Brandizzi F. 2013. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 23(11):547–555. doi: 10.1016/j.tcb.2013.06.005
- Das UN. 1999. Essential fatty acids, lipid peroxidation and apoptosis. Prostaglandins Leukot Essent Fatty Acids. 61(3):157–163. doi: 10.1054/plef.1999.0085
- Hall MN, Chavarro JE, Lee I-M, Willett WC, Ma J. 2008. A 22-year prospective study of fish, n-3 fatty acid intake, and colorectal cancer risk in men. Cancer Epidemiol Biomarkers Prev. 17(5):1136–1143. doi: 10.1158/1055-9965.EPI-07-2803
- Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS. 2001. Unsaturated fatty acids down-regulate SREBP isoforms 1a and 1c by two mechanisms in HEK-293 cells. J Biol Chem. 276(6):4365–4372. doi: 10.1074/jbc.M007273200
- Harris WS, Dayspring TD, Moran TJ. 2013. Omega-3 fatty acids and cardiovascular disease: new developments and applications. Postgrad Med. 125(6):100–113. doi: 10.3810/pgm.2013.11.2717
- Horton JD, Goldstein JL, Brown MS. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 109(9):1125–1131. doi: 10.1172/JCI0215593
- Howell G, Deng X, Yellaturu C, Park EA, Wilcox HG, Raghow R, Elam MB. 2009. N-3 polyunsaturated fatty acids suppress insulin-induced SREBP-1c transcription via reduced trans-activating capacity of LXRα. Biochim Biophys Acta. 1791(12):1190–1196. doi: 10.1016/j.bbalip.2009.08.008
- Kawamori T, Rao CV, Seibert K, Reddy BS. 1998. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res. 58(3):409–412.
- Kim H, Rodriguez-Navas C, Kollipara Rahul K, Kapur P, Pedrosa I, Brugarolas J, Kittler R, Ye J. 2015. Unsaturated fatty acids stimulate tumor growth through stabilization of β-catenin. Cell Rep. 13(3):495–503. doi: 10.1016/j.celrep.2015.09.010
- Larsson SC, Kumlin M, Ingelman-Sundberg M, Wolk A. 2004. Dietary long-chain n−3 fatty acids for the prevention of cancer: a review of potential mechanisms. Am J Clin Nutr. 79(6):935–945. doi: 10.1093/ajcn/79.6.935
- Moon Y-A, Hammer RE, Horton JD. 2009. Deletion of ELOVL5 leads to fatty liver through activation of SREBP-1c in mice. J Lipid Res. 50(3):412–423. doi: 10.1194/jlr.M800383-JLR200
- Moon Y-A, Shah NA, Mohapatra S, Warrington JA, Horton JD. 2001. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J Biol Chem. 276(48):45358–45366. doi: 10.1074/jbc.M108413200
- Morland SL, Martins KJB, Mazurak VC. 2016. N-3 polyunsaturated fatty acid supplementation during cancer chemotherapy. J Nutr Intermed Metab. 5:107–116. doi: 10.1016/j.jnim.2016.05.001
- Morton JD, Shimomura I. 1999. Sterol regulatory element-binding proteins. Curr Opin Lipidol. 10:143–150. doi: 10.1097/00041433-199904000-00008
- Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. 1996. Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell. 87(5):803–809. doi: 10.1016/S0092-8674(00)81988-1
- Patterson E, Wall R, Fitzgerald GF, Ross RP, Stanton C. 2012. Health implications of high dietary omega-6 polyunsaturated fatty acids. J Nutr Metab. 2012:1–16. doi: 10.1155/2012/539426
- Rose DP, Connolly JM. 1990. Effects of fatty acids and inhibitors of eicosanoid synthesis on the growth of a human breast cancer cell line in culture. Cancer Res. 50(22):7139–7144.
- Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, Kimura S, Kato H, Kondo M, Hla T. 1995. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 55(17):3785–3789.
- Sawada N, Inoue M, Iwasaki M, Sasazuki S, Shimazu T, Yamaji T, Takachi R, Tanaka Y, Mizokami M, Tsugane S. 2012. Consumption of n-3 fatty acids and fish reduces risk of hepatocellular carcinoma. Gastroenterology. 142(7):1468–1475. doi: 10.1053/j.gastro.2012.02.018
- Shiragami R, Murata S, Kosugi C, Tezuka T, Yamazaki M, Hirano A, Yoshimura Y, Suzuki M, Shuto K, Koda K. 2013. Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. Int J Oncol. 43(2):431–438. doi: 10.3892/ijo.2013.1978
- Simopoulos AP. 2002. Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr. 21(6):495–505. doi: 10.1080/07315724.2002.10719248
- Song M, Zhang X, Meyerhardt JA, Giovannucci EL, Ogino S, Fuchs CS, Chan AT. 2017. Marine ω-3 polyunsaturated fatty acid intake and survival after colorectal cancer diagnosis. Gut. 66(10):1790–1796. doi: 10.1136/gutjnl-2016-311990
- Tanaka A, Yamamoto A, Murota K, Tsujiuchi T, Iwamori M, Fukushima N. 2017. Polyunsaturated fatty acids induce ovarian cancer cell death through ROS-dependent MAP kinase activation. Biochem Biophys Res Commun. 493(1):468–473. doi: 10.1016/j.bbrc.2017.08.168
- Williams CD, Whitley BM, Hoyo C, Grant DJ, Iraggi JD, Newman KA, Gerber L, Taylor LA, McKeever MG, Freedland SJ. 2011. A high ratio of dietary n-6/n-3 polyunsaturated fatty acids is associated with increased risk of prostate cancer. Nutr Res. 31(1):1–8. doi: 10.1016/j.nutres.2011.01.002
- Zivkovic AM, German JB, Sanyal AJ. 2007. Comparative review of diets for the metabolic syndrome: implications for nonalcoholic fatty liver disease. Am J Clin Nutr. 86(2):285–300. doi: 10.1093/ajcn/86.2.285