
ABSTRACT
Pancreatic stellate cells (PSCs) are the primary cell components of pancreatic cancer (PC) and are involved in tumor growth, metastasis and resistance. However, the role and the mechanism of PSCs in gemcitabine (GEM) resistance to PC still need more investigation. We found that CXCL12 mRNA and secreted CXCL12 protein were higher in PSCs after GEM treatment. The conditioned medium (CM) from GEM-treated PSCs reduced the GEM sensitivity of PC cells. Blocking of CXCL12 in CM by anti-CXCL12 antibody partly restored the GEM sensitivity of PC cells. Blocking of CXCL12 decreased glucose consumption, lactate production, ECAR, and glycolysis-related gene expression in PC cells. The PI3K/AKT/mTOR pathway was activated by the binding of CXCL12 and CXCR4. Moreover, CXCR4 mRNA and protein expressions in PC cells were increased after GEM treatment. Our results indicated the cross-talk between PSCs and PC cells during GEM chemotherapy. CXCL12 secreted by PSCs reduces GEM sensitivity of PC cells by binding to CXCR4 and activating PI3K/AKT/mTOR-glycolysis pathway in PC. Our findings would lay the foundation for solving GEM resistance in PC.
Introduction
Pancreatic cancer (PC) is usually asymptomatic in its earliest stages of development which leads to a late diagnosis in patients with PC (Moutinho-Ribeiro et al. Citation2018). PC patients have the shortest survival time of all cancer patients, with 62210 new cases and 49,830 estimated deaths in 2022 (Siegel et al. Citation2022). PC is predicted to turn into the 2nd leading cause of cancer death by 2030 (Latenstein et al. Citation2020). So far, complete surgical resection has been regarded as the only curative treatment for PC (Kamisawa et al. Citation2016). The clinical efficacy of gemcitabine (GEM), the first-line mainstay chemotherapy in PC, is still not very ideal (Sarvepalli et al. Citation2019).
The high mortality of PC is mainly caused by the absence of effective treatment strategies. Mainstream treatment strategies for PC are to target tumor cells themselves. However, the stroma/desmoplastic reaction is a major feature of PC. As the primary cell components of PC, Pancreatic stellate cells (PSCs) can generate abundant extracellular matrix (ECM) (Pothula et al. Citation2016). Activated PSCs could promote the production of ECM which would lead to damaged drug delivery and acquired chemoresistance (Zhao et al. Citation2018; Amrutkar et al. Citation2019). PSCs also promote tumor metabolism by providing nutrients (Sousa et al. Citation2016). Therefore, understanding the function and regulation mechanism of PSCs in PC during GEM chemotherapy may help alleviate GEM resistance.
CXCL12, attached to the CXC family of chemokines, is released by stromal cells. CXCL12 binds to the CXCR4 expressed cells and regulates cell directional migration (Teicher and Fricker Citation2010). CXCL12 has a tumor protective effect and could accelerate tumor growth (Righi et al. Citation2011; Feig et al. Citation2013; Sleightholm et al. Citation2017). The secretion of CXCL12 from PSCs by inhibiting cytotoxic T cell infiltration (Garg et al. Citation2018). CXCL12-CXCR4 signaling axis is implicated in GEM resistance in PC (Singh et al. Citation2010). However, the role and mechanism of CXCL12 in GEM resistance still need more exploration.
In the sections below, we describe the role of PSCs on GEM sensitivity in PC and explored the underlying mechanisms. Our study indicated that enhanced CXCL12 secreted by PSCs after GEM treatment decreased GEM sensitivity of PC cells by inducing glycolytic reprogramming via activating the CXCR4-PI3K/AKT/mTOR signaling.
Materials and methods
Reagents and antibodies
GEM (cat. no. S1149) was obtained from Selleck Chemicals. Antibodies against CXCR4 (ab181020), PI3K (ab40776), and beta-actin (ab8226) were obtained from Abcam. Antibodies against p-PI3K (cat. no. 4228S), AKT (cat. no. 4685S), p-AKT (cat. no. 4060S), p-mTOR (cat. no. 2974), and mTOR (cat. no. 2983) were purchased from Cell Signaling Technology, Inc.
Isolation and identification of PSCs
This study was approved by the Ethics Committee of Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China. The paper form that is patient's informed consent is acquired from all patients. PSCs were prepared by the enzymatic digestion-gradient centrifugation method (Liu et al. Citation2019) and were employed in their second-fourth passages. Under cyto-immunochemistry examination, a high level of α-SMA and GFAP were observed in the cytoplasm of PSCs.
Cell culture
The PANC-1 and CFPAC-1 human PC cell lines (Procell, Wuhan, China) were incubated in DMEM added with penicillin–streptomycin and 10% FBS.
Conditioned medium
The PSCs were treated with 2 nM GEM for 24 h and then replaced with the fresh complete medium. After another twenty-four hours of incubation, the conditioned medium (CM) was harvested. After filtering by a 0.2 μM SFCA membrane (Corning), CM was used at a 1:5 dilution.
CCK-8
Cell viability was evaluated using CCK-8 (MedChemExpress, Monmouth Junction, USA). A total of 4 K PC cells were inoculated per well in 96-well plates overnight. The medium was changed to CM and added different concentrations of GEM. Cell viability was evaluated after culture for 0, 24, 48, and 72 h. Absorbance was detected at 450 nm.
Flow cytometry
PC cells were seeded in 6-well plates at a density of 5×104 cells/well for overnight incubation. The medium was changed to CM and added 8nM GEM for 48 h incubation. Then, cell apoptosis was assessed using an Annexin V-FITC/PI Apoptosis Detection Kit (Yeasen Biotechnology Co., Ltd. Shanghai, China) and flow cytometry (Beckman Coulter, Miami, USA).
TUNEL assay
The cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% sodium citrate and 0.1% Triton X. DNA fragmentation was detected by TdT-mediated dUTP nick end labeling (TUNEL) following the manufacturer's instructions (Beyotime, Shanghai, China).
Bioinformatics analysis
Microarray analysis was used to filter abnormally expressed genes in PSCs with or without 2 nM GEM treatment. Abnormal genes were filtered based on the fold change and adjusted p-values. The inclusion criteria were set as follows: adjusted p-values < 0.05 and fold change >4 or fold change <0.2.
ELISA
The levels of CXCL12 in the culture supernatant were detected by the Human CXCL12 alpha ELISA Kit (ab100637, Abcam) according to the manufacturer's instructions.
RNA extraction and RT-qPCR
RT-qPCR was used as described previously (Liu et al. Citation2022). The data were normalized by β-actin and relative expression was calculated by the 2-ΔΔCt values. The following primers were used: CXCR4, forward 5′-GTA AAACGACGGCCAGT-3′ and reverse 5′-CAGGAAACAGCTATGAC-3′; CXCL12, forward 5′-GTCAGCCTGAGCTACAGATGC-3′ and reverse 5′-CTTTAGCTTCGGGTCAATGC-3; GLUT1, forward 5’-TGTCGTGTCGCTGTTTGTGGTGGA-3’ and reverse 5’-TGAAGAACAGAACCAGGAGCACAG-3’; HK2, forward 5’-TGATGTGGCTGTGGATGAGCT-3’ and reverse 5’-GCCAGGCAGTCACTCTCAATCTG-3’; LDHA, forward 5’-AGGCTGGGAGTTCACCCATTAAGC-3’ and reverse 5’-GAGTCCAATAGCCCAGGATGTG-3’; β-actin, forward 5’-CTACGTCGCCCTGGACTTCGAGC-3’ and reverse 5’-GATGGAGCCGCCGATCCACACGG-3’.
Western blotting
Cells were lysed in RIPA buffer (Solarbio, China) supplemented PMSF (Solarbio, China). The protein concentration was tested by a BCA protein assay kit (Solarbio, China). The proteins were separated by SDS-PAGE and blotted onto PVDF membranes (Millipore, USA). After blocking, the membranes were incubated with the indicated primary antibodies and secondary antibodies. The blots were developed with a BeyoECL Star kit (Beyotime, Shanghai, China) and visualized using X-ray films.
Glycolysis process assay
The glucose assay kit (ab65333, Abcam) and lactate assay kit (ab65330, Abcam) were respectively used to detect the level of glucose and lactate in culture supernatant according to the manufacturer's instructions. For the glycolytic function assay, ECAR was detected using the Seahorse XF Glycolysis Stress Test kit (Agilent Technologies, Santa Clara, USA) according to the manufacturer's instructions. The basal ECAR was measured under the basal condition, followed by the sequential addition to each well of glucose (10 mM), oligomycin (2 mM), and 2-deoxyglucose (100 mM).
Statistical analysis
The data were presented as mean ± standard deviation. Two-tailed Student’s t-test was used to compare the differences between the two groups. For multiple groups, one-way ANOVA followed by Tukey’s test was used. p < 0.05 was considered statistically significant.
Results
The CM of GEM-treated PSCs decreases GEM sensitivity in PC cells
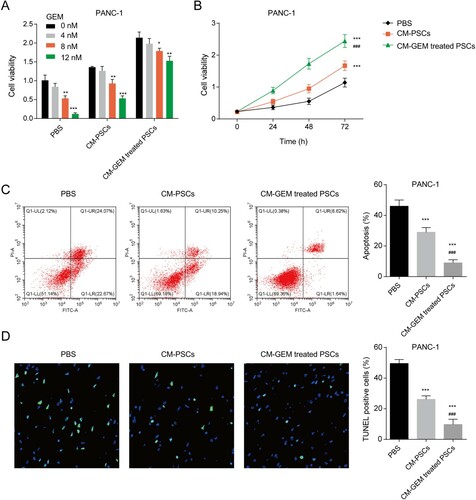
The PANC-1 and CFPAC-1 cells co-cultured with CM of PSCs with or without GEM treatment were treated with 0, 4, 8, 12 nM GEM and then the cell viability was measured. The GEM sensitivity of PANC-1 and CFPAC-1 cells decreased significantly after being incubated with CM-PSCs ((A) and Figure S1A). After incubating with CM from GEM-treated PSCs, the sensitivity of PANC-1 and CFPAC-1 cells to GEM was weaker when compared with CM from PSCs. Moreover, the reduction of GEM sensitivity induced by CM was time-dependent ((B)). The apoptosis assay showed that both CM from PSCs or GEM-treated PSCs inhibited the apoptosis of PANC-1 and CFPAC-1 cells induced by GEM ((C) and Figure S1B). CM from GEM-treated PSCs has a stronger reversal effect. The results of the TUNEL assay further verified the reversal effect of CM to GEM in PANC-1 and CFPAC-1 cells ((D) and Figure S1C). All the data indicated that CM form GEM-treated PSCs could decrease the sensitivity of PANC-1 and CFPAC-1 cells to GEM.
Figure 1. The CM of GEM-treated PSCs regulated the GEM sensitivity of PANC-1 cells. (A) The cell viability of PANC-1 cells with CM and various concentrations of GEM treatment for 48 h. *p < 0.05, **p < 0.01, ***p < 0.001 vs. 0 nM. (B) The cell viability of PANC-1 cells with CM and 8 nM GEM treatment for 0, 24, 48, and 72 h. (C) The apoptosis of PANC-1 cells was detected by a flow cytometer. (D) The apoptosis of PANC-1 cells was detected by a TUNEL assay. ***p < 0.001 vs. PBS; ###p < 0.001 vs. CM-PSCs. CM-PSCs, the PC cells were co-cultured with the conditioned medium from PSCs; CM-GEM-treated PSCs, the PC cells were co-cultured with the conditioned medium from GEM-treated PSCs.
GEM promotes the secretion of CXCL12 in PSCs
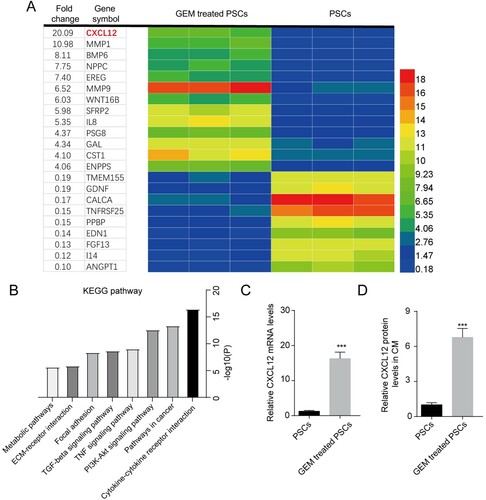
To explore the underlying mechanisms for the change of GEM sensitivity induced by CM from GEM-treated PSCs, microarray analysis was performed to filter abnormally expressed genes in PSCs with or without GEM treatment. Total 22 genes were screened out (p-values < 0.05 and fold change > 4 or fold change <0.2) ((A)). CXCL12 was found significantly increased in GEM-treated PSCs among the abnormally expressed genes. The abnormally expressed genes were mainly enriched in the cytokine-cytokine receptor interaction, pathways in cancer, PI3K-Akt signaling pathway, TNF signaling pathway, and TGF-beta signaling pathway ((B)). The mRNA level of CXCL12 was enhanced in GEM-treated PSCs ((C)). Moreover, the protein level of CXCL12 in the culture supernatant of GEM-treated PSCs was also found to be increased ((D)). All these results showed that GEM promoted the expression and secretion of CXCL12 in PSCs.
Figure 2. GEM promotes the secretion of CXCL12 in PSCs. (A) The abnormally expressed genes in PSCs after 2 nM GEM treatment. (B) Metascape database was then employed to analyze the enrichment of screened genes in the KEGG pathway. (C) The mRNA level of CXCL12 in PSCs. (D) The protein levels of CXCL12 in the culture supernatant of PSCs. ***p < 0.001 vs. PSCs.
CXCL12 secreted by PSCs decreases GEM sensitivity in PC cells
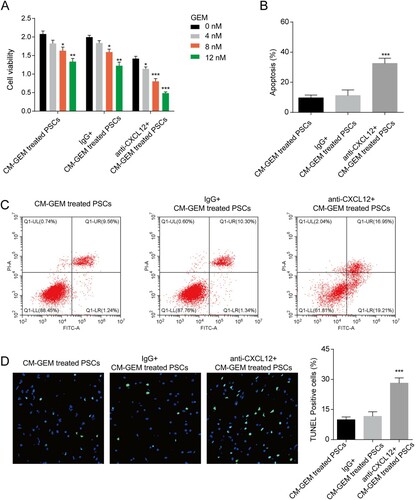
We next explored the role of CXCL12 in CM on the GEM sensitivity of PC cells. The anti-CXCL12 antibody was used to block the CXCL12 in CM. The results showed that PANC-1 and CFPAC-1 cells with anti-CXCL12 antibody incubation showed decreased cell viability ((A) and Figure S1D). The results of the apoptosis assay and TUNEL assay indicated the treatment of the anti-CXCL12 antibody enhanced the apoptosis induced by GEM ((B–D) and Figure S1E-F). In short, blocking CXCL12 in CM could reverse the effect of CM on GEM sensitivity, indicating the key role of CXCL12 on GEM sensitivity.
Figure 3. CXCL12 regulated GEM sensitivity of PANC-1 cells. (A) The effect of CXCL12 in CM on GEM sensitivity of PANC-1 cells. *p < 0.05, **p < 0.01, ***p < 0.001 vs. 0 nM. (B–C) The effect of CXCL12 in CM on GEM-induced apoptosis of PANC-1 cells was detected by a flow cytometer. ***p < 0.001 vs. IgG + CM-GEM-treated PSCs. (D) The effect of CXCL12 in CM on GEM-induced apoptosis of PANC-1 cells was detected by a TUNEL assay. ***p < 0.001 vs. IgG + CM-GEM-treated PSCs. CM-GEM-treated PSCs, the PC cells were co-cultured with the conditioned medium from GEM-treated PSCs; IgG + CM-GEM-treated PSCs, the PC cells were co-cultured with IgG and the conditioned medium from GEM-treated PSCs; anti-CXCL12 + CM-GEM-treated PSCs, the PC cells were co-cultured with anti-CXCL12 antibody and the conditioned medium from GEM-treated PSCs.
CXCL12 secreted by PSCs enhances the glycolysis of PC cells
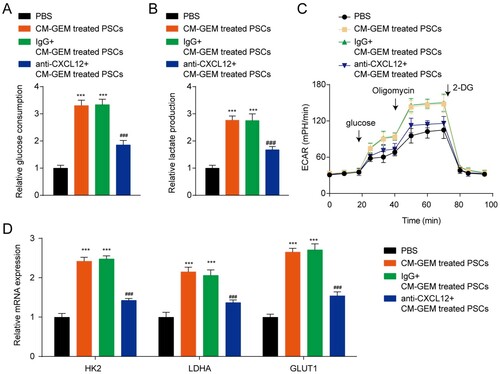
Metabolism reprogramming is a hallmark of many cancers and is closely related to drug resistance. We then detected the glycolysis of PC cells after incubated with CM. The results indicated that CM from GEM-treated PSCs enhanced the glucose consumption, lactate production, and ECAR of PANC-1 cells ((A–C)). The mRNA level of glycolysis-related genes (HK2, LDHA, and GLUT1) in PANC-1 cells also was increased after incubated with CM from GEM-treated PSCs ((D)). Blocking of CXCL12 in CM by anti-CXCL12 antibody decreased glucose consumption, lactate production, ECAR, and glycolysis-related gene expression, indicating the key role of CXCL12 in glycolysis. Similar results were also confirmed in CFPAC-1 cells (Figure S2). In a word, the CXCL12 decreased the GEM sensitivity of PC cells by promoting the glycolysis of PC cells.
Figure 4. CXCL12 secreted by PSCs promotes the glycolysis of PANC-1 cells. (A) Detection of glucose consumption in PANC-1 cells. (B) Detection of lactate excretion in PANC-1 cells. (C) Detection of glycolytic capacity (ECAR) in PANC-1 cells. (D) The mRNA levels of HK2, LDHA, and GLUT1 in PANC-1 cells. ***p < 0.001 vs. PBS; ###p < 0.001 vs. IgG + CM-GEM-treated PSCs.
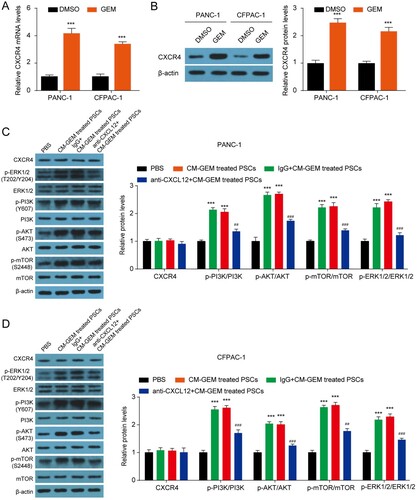
CXCL12 secreted by PSCs activates PI3K/AKT/mTOR pathway by binding CXCR4
CXCL12 binds to CXCR4 to activate PI3K/AKT/mTOR pathway and thus regulates tumor growth, metastasis, and chemoresistance (Braun et al. Citation2016; Yang et al. Citation2019; Yi et al. Citation2019). We then explored whether the CXCR4-PI3K/AKT/mTOR pathway was implicated in the CXCL12 mediated the change of GEM sensitivity of PC cells. Firstly, we found that the treatment of GEM in PANC-1 and CFPAC-1 cells significantly enhanced the mRNA and protein expression of CXCR4 ((A–B)). The incubation of CM had no effect on CXCR4, ERK1/2, PI3K, AKT, and mTOR protein levels, but obviously increased the levels of p-ERK1/2, p-PI3K, p-AKT, and p-mTOR in both PANC-1 and CFPAC-1 cells ((C–D)). Blocking of CXCL12 in CM by anti-CXCL12 antibody partly restored the levels of p-ERK1/2, p-PI3K, p-AKT, and p-mTOR in PANC-1 and CFPAC-1 cells induced by CM, indicating that the activation of PI3K/AKT/mTOR signaling pathway was partly induced by CXCL12. In a word, PSCs inhibited the GEM sensitivity of PC cells by secreting CXCL12 and activating the PI3K-AKT-mTOR pathway.
Figure 5. CXCL12 secreted by PSCs activates the PI3K-AKT-mTOR signaling pathway by binding CXCR4. (A–B) The mRNA and protein level of CXCR4 in PANC-1 and CFPAC-1 cells after 8 nM GEM treatment for 48 h. ***p < 0.001 vs. DMSO. (C–D) The protein level of CXCR4, p-ERK1/2, ERK1/2, p-PI3K, PI3K, p-AKT, AKT, p-mTOR and mTOR in PANC-1 and CFPAC-1 cells. ***p < 0.001 vs. PBS; ##p < 0.01, ###p < 0.001 vs. IgG + CM-GEM-treated PSCs.
Discussion
Here, we have confirmed the role of PSCs during GEM chemotherapy in PC. During GEM chemotherapy, the secretion of CXCL12 is increased. The increased CXCL12 then binds CXCR4 and activates PI3K/AKT/mTOR pathway, leading to metabolic reprogramming and GEM resistance in PC cells. Targeting both PC cells and PSCs may be an effective treatment strategy for PC and potential means to address GEM resistance.
Metabolism reprogramming is ubiquity in PC, and the abnormal glycolysis confers tumor growth, metastasis, and resistance to PC (Zhang et al. Citation2017; Zheng et al. Citation2020; Yan et al. Citation2021). Xu et al indicate that HIF1A-AS1 aggravates GEM resistance in PC by promoting glycolysis by activating the AKT pathway (Xu et al. Citation2021). Feng et al show that LAT2 promotes the activation of mTOR to enhance glycolysis and reduce GEM sensitivity in PC (Feng et al. Citation2018). hENT1 is reported to reverse the chemoresistance of GEM by influencing glycolysis in PC (Xi et al. Citation2020). Cancer cell death is obviously increased after glycolysis inhibition and GEM sensitivity of PC cells was enhanced after the inhibition of glycolysis (Dai et al. Citation2020). In this study, we found that the CM from PSCs or PSCs with GEM treatment could restrain the GEM sensitivity by enhancing the glycolysis of PC cells.
CXCL12 could induce a series of signaling events in PC cells and neutralizes the cytotoxic effects of GEM (Singh et al. Citation2010). CXCL12-CXCR4 signaling axis is closely related to GEM resistance in PC cells (Morimoto et al. Citation2016). Cancer-associated fibroblasts enhance GEM resistance in PC by the CXCL12/CXCR4 pathway (Wei et al. Citation2018). In our data, we found that CXCR4 was enhanced in PCs and the secretion of CXCL12 in PSCs was increased after GEM treatment. PSCs decreased GEM sensitivity of PC cells by secreting CXCL12 under GEM treatment. Our results further revealed the mechanism for the GEM resistance of PC.
The PI3K/AKT/mTOR pathway plays a vital role in tumor growth and metastasis. The PI3K/AKT/mTOR pathway is implicated in glycolysis and promotes PC growth (Hu et al. Citation2019). ARHGAP25 suppresses the growth of PC cells by inhibiting glycolysis via the AKT/mTOR pathway (Huang et al. Citation2021). TRIM59 is reported to promote PC progression through the PI3K/AKT/mTOR-glycolysis signaling axis (Li et al. Citation2020). PI3K/AKT/mTOR signaling also participates in GEM resistance of PC cells by regulating the Warburg effect (Cui et al. Citation2021). Moreover, PI3K/AKT/mTOR signaling is reported to be the downstream signaling pathway after the activation of CXCR4 (Yi et al. Citation2019; Yi and Gao Citation2019; Liao et al. Citation2021). In this study, we found that CXCL12 secreted by PSCs could bind to CXCR4 and then activate PI3K/AKT/mTOR signaling in PC cells. CXCL12 decreased the sensitivity of PC cells to GEM by inhibiting glycolysis via PI3K/AKT/mTOR pathway.
In conclusion, our findings reveal the mechanism of GEM resistance in PC and find that PSCs promote GEM resistance in PC by the secretion of CXCL12, which further bind to CXCR4 and activate the PI3K/AKT/mTOR-glycolysis signaling axis. All of these findings would facilitate the progression of treatment strategies for PC and improve GEM resistance.
Supplemental Material
Download MS Word (585.9 KB)Acknowledgements
Author contributions: XYL, YLW and JG have given substantial contributions to the conception and the design of the manuscript, RC, XJY and JG to acquisition, analysis and interpretation of the data. All authors have participated to drafting the manuscript, JG, XYL and YLW revised it critically. All authors read and approved the final version of the manuscript. This work was supported by Sichuan Science and Technology Support Plan Project (No. 2020YJ0462).
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Amrutkar M, Aasrum M, Verbeke CS, Gladhaug IP. 2019. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer. 19:596.
- Braun M, Qorraj M, Buttner M, Klein FA, Saul D, Aigner M, Huber W, Mackensen A, Jitschin R, Mougiakakos D. 2016. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leukemia. 30:1788–1792.
- Cui J, Guo Y, Wu H, Xiong J, Peng T. 2021. Everolimus regulates the activity of gemcitabine-resistant pancreatic cancer cells by targeting the Warburg effect via PI3K/AKT/mTOR signaling. Mol Med. 27:38.
- Dai S, Peng Y, Zhu Y, Xu D, Zhu F, Xu W, Chen Q, Zhu X, Liu T, Hou C, et al. 2020. Glycolysis promotes the progression of pancreatic cancer and reduces cancer cell sensitivity to gemcitabine. Biomed Pharmacother. 121:109521.
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. 2013. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 110:20212–20217.
- Feng M, Xiong G, Cao Z, Yang G, Zheng S, Qiu J, You L, Zheng L, Zhang T, Zhao Y. 2018. LAT2 regulates glutamine-dependent mTOR activation to promote glycolysis and chemoresistance in pancreatic cancer. J Exp Clin Cancer Res. 37:274.
- Garg B, Giri B, Modi S, Sethi V, Castro I, Umland O, Ban Y, Lavania S, Dawra R, Banerjee S, et al. 2018. NFkappab in pancreatic stellate cells reduces infiltration of tumors by cytotoxic T cells and killing of cancer cells, via Up-regulation of CXCL12. Gastroenterology. 155:880–891.e888.
- Hu LP, Zhang XX, Jiang SH, Tao LY, Li Q, Zhu LL, Yang MW, Huo YM, Jiang YS, Tian GA, et al. 2019. Targeting purinergic receptor P2Y2 prevents the growth of pancreatic ductal adenocarcinoma by inhibiting cancer cell glycolysis. Clin Cancer Res. 25:1318–1330.
- Huang WK, Chen Y, Su H, Chen TY, Gao J, Liu Y, Yeh CN, Li S. 2021. ARHGAP25 inhibits pancreatic adenocarcinoma growth by suppressing glycolysis via AKT/mTOR pathway. Int J Biol Sci. 17:1808–1820.
- Kamisawa T, Wood LD, Itoi T, Takaori K. 2016. Pancreatic cancer. Lancet. 388:73–85.
- Latenstein AEJ, van der Geest LGM, Bonsing BA, Groot Koerkamp B, Haj Mohammad N, de Hingh I, de Meijer VE, Molenaar IQ, van Santvoort HC, van Tienhoven G, et al. 2020. Nationwide trends in incidence, treatment and survival of pancreatic ductal adenocarcinoma. Eur J Cancer. 125:83–93.
- Li R, Weng L, Liu B, Zhu L, Zhang X, Tian G, Hu L, Li Q, Jiang S, Shang M. 2020. TRIM59 predicts poor prognosis and promotes pancreatic cancer progression via the PI3K/AKT/mTOR-glycolysis signaling axis. J Cell Biochem. 121:1986–1997.
- Liao YX, Lv JY, Zhou ZF, Xu TY, Yang D, Gao QM, Fan L, Li GD, Yu HY, Liu KY. 2021. CXCR4 blockade sensitizes osteosarcoma to doxorubicin by inducing autophagic cell death via PI3KAktmTOR pathway inhibition. Int J Oncol. 59:49.
- Liu S, Hu C, Li M, An J, Zhou W, Guo J, Xiao Y. 2022. Estrogen receptor beta promotes lung cancer invasion via increasing CXCR4 expression. Cell Death Dis. 13:70.
- Liu SL, Cao SG, Li Y, Sun B, Chen D, Wang DS, Zhou YB. 2019. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol Lett. 17:2057–2062.
- Morimoto M, Matsuo Y, Koide S, Tsuboi K, Shamoto T, Sato T, Saito K, Takahashi H, Takeyama H. 2016. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: effect of CXCR4 antagonists. BMC Cancer. 16:305.
- Moutinho-Ribeiro P, Macedo G, Melo SA. 2018. Pancreatic cancer diagnosis and management: has the time come to prick the bubble? Front Endocrinol (Lausanne). 9:779.
- Pothula SP, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. 2016. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 381:194–200.
- Righi E, Kashiwagi S, Yuan J, Santosuosso M, Leblanc P, Ingraham R, Forbes B, Edelblute B, Collette B, Xing D, et al. 2011. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 71:5522–5534.
- Sarvepalli D, Rashid MU, Rahman AU, Ullah W, Hussain I, Hasan B, Jehanzeb S, Khan AK, Jain AG, Khetpal N, et al. 2019. Gemcitabine: a review of chemoresistance in pancreatic cancer. Crit Rev Oncog. 24:199–212.
- Siegel RL, Miller KD, Fuchs HE, Jemal A. 2022. Cancer statistics, 2022. CA Cancer J Clin. 72:7–33.
- Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. 2010. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 103:1671–1679.
- Sleightholm RL, Neilsen BK, Li J, Steele MM, Singh RK, Hollingsworth MA, Oupicky D. 2017. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol Ther. 179:158–170.
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. 2016. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 536:479–483.
- Teicher BA, Fricker SP. 2010. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 16:2927–2931.
- Wei L, Ye H, Li G, Lu Y, Zhou Q, Zheng S, Lin Q, Liu Y, Li Z, Chen R. 2018. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 9:1065.
- Xi Y, Yuan P, Li T, Zhang M, Liu MF, Li B. 2020. hENT1 reverses chemoresistance by regulating glycolysis in pancreatic cancer. Cancer Lett. 479:112–122.
- Xu F, Huang M, Chen Q, Niu Y, Hu Y, Hu P, Chen D, He C, Huang K, Zeng Z, et al. 2021. LncRNA HIF1A-AS1 promotes gemcitabine resistance of pancreatic cancer by enhancing glycolysis through modulating the AKT/YB1/HIF1alpha pathway. Cancer Res. 81:5678–5691.
- Yan L, Tu B, Yao J, Gong J, Carugo A, Bristow CA, Wang Q, Zhu C, Dai B, Kang Y, et al. 2021. Targeting glucose metabolism sensitizes pancreatic cancer to MEK inhibition. Cancer Res. 81:4054–4065.
- Yang F, Takagaki Y, Yoshitomi Y, Ikeda T, Li J, Kitada M, Kumagai A, Kawakita E, Shi S, Kanasaki K, et al. 2019. Inhibition of dipeptidyl peptidase-4 accelerates epithelial-mesenchymal transition and breast cancer metastasis via the CXCL12/CXCR4/mTOR axis. Cancer Res. 79:735–746.
- Yi J, Gao ZF. 2019. MicroRNA-9-5p promotes angiogenesis but inhibits apoptosis and inflammation of high glucose-induced injury in human umbilical vascular endothelial cells by targeting CXCR4. Int J Biol Macromol. 130:1–9.
- Yi L, Zhou X, Li T, Liu P, Hai L, Tong L, Ma H, Tao Z, Xie Y, Zhang C, et al. 2019. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J Exp Clin Cancer Res. 38:339.
- Zhang Q, Lou Y, Zhang J, Fu Q, Wei T, Sun X, Chen Q, Yang J, Bai X, Liang T. 2017. Hypoxia-inducible factor-2alpha promotes tumor progression and has crosstalk with Wnt/beta-catenin signaling in pancreatic cancer. Mol Cancer. 16:119.
- Zhao W, Ajani JA, Sushovan G, Ochi N, Hwang R, Hafley M, Johnson RL, Bresalier RS, Logsdon CD, Zhang Z, et al. 2018. Galectin-3 mediates tumor cell-stroma interactions by activating pancreatic stellate cells to produce cytokines via integrin signaling. Gastroenterology. 154:1524–1537.e1526.
- Zheng Y, Wu C, Yang J, Zhao Y, Jia H, Xue M, Xu D, Yang F, Fu D, Wang C, et al. 2020. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer. Signal Transduct Target Ther. 5:53.