
ABSTRACT
Few studies have evaluated the relationship of oral microbiome with obesity. We investigated the oral microbiome among 647 obese and 969 non-obese individuals from the Southern Community Cohort Study, through 16S rRNA gene sequencing in mouth rinse samples. We first investigated 16 taxa in two probiotic genera, Bifidobacterium and Lactobacillus. Among them, eight showed nominal associations with obesity (P < 0.05). Especially, Bifidobacterium (odds ratio [OR] = 0.67, 95% confidence interval [CI]:0.54, 0.83) and Bifidobacterium longum (OR = 0.57, 95% CI: 0.45, 0.73) were significantly associated with decreased obesity prevalence with false-discovery rate (FDR)-corrected P of 0.01 and 5.41 × 10−4, respectively. Multiple other bacterial taxa were also significantly associated with obesity prevalence at FDR-corrected P < 0.05. Among them, five in Firmicutes and two respectively in Actinobacteria and Proteobacteria were significantly associated with increased obesity prevalence. Significant associations with decreased obesity prevalence were observed for two taxa respectively in Actinobacteria and Firmicutes. Most of these taxa were associated with body mass index at study enrollment and weight gain during adulthood. Also, most of these associations were observed in both European- and African-Americans. Our findings indicate that multiple oral bacterial taxa, including several probiotic taxa, were significantly associated with obesity.
Obesity has become a domestic and global pandemic [Citation1], with adult overweight and obesity rates at nearly 36.5% in the USA [Citation2]. Obesity is a major risk factor for a wide range of chronic diseases, including type 2 diabetes, cardiovascular diseases, some cancers, renal diseases, and irritable bowel disease [Citation3]. Host genetic factors [Citation4], high-calorie diets [Citation5] and physical inactivity [Citation3] are associated with increased risk of obesity.
Humans are supraorganisms with approximately 90% of cells being commensal microorganisms known as microbiota [Citation6]. The gut microbiome has been associated with obesity [Citation5]. Based on these findings, multiple bacterial taxa in the genera Bifidobacterium (belonging to the phylum Actinobacteria) and Lactobacillus (belonging to the phylum Firmicutes) [Citation7], have already been used as probiotics to decrease body weight in human clinical trials [Citation8,Citation9].
The oral microbiota is the second most complex microbial community in the human body after the colon [Citation10]. Oral microbes play important roles in maintaining oral and systemic health through colonization resistance [Citation10], nutrient digestion [Citation11] and immunity response [Citation12]. It has been suggested that worse oral health may be associated with increased risk of obesity [Citation13,Citation14]. Multiple studies, including ours [Citation15], have showed that the oral microbiome was associated with various diseases, such as pancreatic cancer [Citation16], head and neck squamous cell cancer [Citation17] and colorectal cancer [Citation15,Citation18]. However, evidence linking the oral microbiome to obesity remains scarce. Previous studies have used DNA probe hybridization technique to profile saliva/subgingival bacterial community among overweight/obese and non-obese individuals. They found multiple oral bacteria showing significant associations with overweight/obesity [Citation19,Citation20], suggesting that the oral microbiome may play a role in the pathophysiology of obesity. However, because of the limited availability of bacteria-specific probes, only dozens of microbes were investigated in these studies though hundreds of oral microbes were observed. In addition, these studies were conducted with a small sample size (N < 350) and primarily among European-ancestry populations. In the present study, we evaluated the relationship between oral microbial composition and obesity via deep sequencing in a large low-income and African American populations from the Southern Community Cohort Study (SCCS), in which the prevalence of obesity is high.
Materials and methods
Study population and data collection
The SCCS is designed to investigate health disparities in low-income populations, predominantly African Americans. Details of the study have been described elsewhere [Citation21,Citation22]. In short, more than 85,000 adults aged 40 to 70 were recruited from 12 southeastern states between 2002 ~ 2009. Approximately 86% of the enrollment occurred in community health centers (CHCs), institutions mainly in underserved areas to provide basic health care and disease prevention. Recruitment of the remaining 14% of individuals were fulfilled through mail-based general population sampling. At the time of enrollment, mouth rinse samples were obtained from ~34,100 participants. The institutional review boards at Vanderbilt University Medical Center and Meharry Medical College reviewed and approved the study, and all participants provided written informed consent.
All participants took the baseline survey through a comprehensive questionnaire during enrollment. Information about lifestyle factors, disease history, anthropometric characteristics, medication use, oral health, socioeconomic status and dietary intake was collected. Study participants’ weight at study enrollment, weight at 21 years and maximal lifetime weight were obtained at the baseline survey with the questions: ‘How much do you weigh?’, ‘What was your weight at age 21?’ and ‘What is the most you have ever weighed?’, respectively. Height was collected by either taking a measurement at enrollment or through the self-reported baseline survey question: ‘How tall are you?’. Body mass index (BMI) at enrollment was calculated according to the formula: weight (kg)/height2 (m2). Participants were categorized to two groups: obese (BMI of ≥ 30) and non-obese (BMI < 30). Weight change was calculated as: weight (kg) at enrollment (aged 40 to 70) – weight (kg) at 21 years. Total energy intake (kcal/day) was derived from the food frequency questionnaire. Alcohol consumption was classified into ‘None’, ‘Light’, ‘Moderate’ and ‘Heavy’ based on the answers to a series of questions about quantity and frequency of alcohol consumption. Tabaco smoking was reported as ‘current’, ‘former’, ‘never’. Oral health was assessed by tooth loss, which was grouped to ‘None’, ‘1 to 4’, ‘5 to 10’, ‘More than 10 but not all of them’, and ‘All of them’ through answering the question ‘About how many adult teeth you have lost in your lifetime due to tooth decay or gum disease?’.
After recruitment, participants follow-up was performed through record linkage and surveys via mail or telephone. Major health outcomes such as cancer incidence were ascertained via linkage with state cancer registries and/or from the National Death Index mortality records. In the present study, we included participants who had been selected for four nested case-control studies to investigate the oral microbiome and incident cases (diagnosed after mouth rinse sample collection) of type 2 diabetes, lung cancer, upper aerodigestive tract cancer and colorectal cancer. In these case-control studies, controls were matched to cases by age (± 2 years), race, sex, smoking status (current, former or never), date of mouth rinse sample collection (± 90 days), recruitment source and the community health center recruitment site. After removing individuals who reported a history of antibiotics usage during the year before sample collection, totally 1,616 individuals, of which 647 were obese and 969 non-obese, were included in the current analysis. All these participants were disease-free at the study enrollment when the mouth rinse sample was collected.
DNA extraction and 16S rRNA gene sequencing
Qiagen’s QIAmp DNA kit (Qiagen Inc., Germantown, MD) was used to isolate DNA from mouth rinse samples and NEXTflex 16S V4 Amplicon-Seq Kit (Bioo Scientific, Austin, TX), designed to sequence approximately 253 bp of bacterial 16S rRNA’s fourth hypervariable (V4) domain [Citation23,Citation24] used for sequencing the library preparation, guided by the manufacturer’s protocol. Sequencing was performed for two batches. For the first batch, 150 bp pair-end data were generated using Illumina MiSeq 300 (Illumina, San Diego, CA) at the VANderbilt Technologies for Advanced Genomics Core. For the 2nd batch, sequencing was performed at pair-end 250 bp using Illumina HiSeq System at the BGI Americas (Cambridge, MA). For both batches, on each 96-well plate, two duplicated quality control samples (QCs) and one negative control sample were included. In total, samples from the same single participant were sequenced six times and the microbiome profiles were very similar. For example, for the Shannon index and the Simpson index, i.e. measurements of alpha diversity within each sample, the coefficient of variability among the six samples were 1.7% and 0.3%, respectively. In addition, the correlations of phylum-level microbial relative abundance among these six samples were very high, with Pearson correlation coefficients range from 97.8% to 99.9%.
Sequence data processing and quality controls
De-multiplexed raw sequencing was subjected to QC using Sickle [Citation25] to remove low-quality bases and reads. The clean pair-end reads were then processed by BayesHammer [Citation26] to go through sequencing error correction and PANDAseq [Citation27] to be stitched together [Citation28]. Assembled reads were processed by the Quantitative Insights Into Microbial Ecology (QIIME; v1.9.1) [Citation29] pipeline using the default parameters. Chimeric sequences were identified and removed using ChimeraSlayer, implemented in identify_chimeric_seqs.py and excluded using filter_fasta.py. Non-chimeric sequences were then clustered into operational taxonomic units (OTUs) at 97% sequence identity using the closed reference-based strategy with UCLUST against the Human Oral Microbiome Database (HOMD) [Citation29]. OTU tables of samples from two studies were rarefied to 5,000 and 20,000 sequencing reads per sample, respectively, to account for any variation in sequencing depth using single_rarefaction.py in QIIME. Different depths were used for rarefaction because of the significant difference of overall sequencing depth between the two studies. Those OTUs observed less than two times were excluded using filter_otus_from_otu_table.py. Rarefied and singleton-removed OTU tables were merged together using merge_otu_tables.py. Finally, we estimated the correlation network based on the combined OTU table by utilizing the SparCC software [Citation30] with the bootstrapping of 1,000 repetitions, during which only OTUs observed in at least 25% of the samples were used. The significant correlations, with an absolute correlation coefficient of ≥ 0.5 and a two-sided pseudo P < 0.001, were used for community structure detection, using the greedy optimization of a modularity algorithm [Citation31], implemented in the R package ‘igraph’.
Statistical analyses
The microbial richness and evenness, known as alpha diversity, was evaluated using Shannon, Simpson, and phylogenetic diversity (PD) whole-tree indices. The differences between obese and non-obese participants were estimated using two-sample t-tests at greatest rarefaction depth, implemented in compare_alpha_diversity.py in QIIME. The relationship between overall oral microbiota composition and obesity was evaluated using MiRKAT [Citation32], which utilized regression-based kernel association tests based on a calculation of three distance metrics: Bray-Curtis dissimilarity, unweighted UniFrac and weighted UniFrac.
Probiotics bacteria have been increasingly used in obesity prevention and treatment [Citation9]. Several clinical trials [Citation8,Citation9,Citation33–Citation39] had suggested the anti-obesity or weight-losing potential of multiple probiotic bacterial taxa in the genera Bifidobacterium and Lactobacillus [Citation7]. Hence, to estimate the relationship between bacterial taxa and obesity, we first investigated these two probiotic genera, i.e. Bifidobacterium and Lactobacillus, and all the species belonging to them.
Then, we investigated other bacterial taxa in association with obesity. We focused on the ‘common taxa’ with median relative abundance of ≥ 0.10% among the non-obese participants. In total, five phyla, 15 families, 15 genera and 28 species were analyzed. Each taxon was categorized into three groups (tertile) based on the relative abundance in non-obese individuals. Then, a logistic regression analysis was performed to test the association for the 2nd and 3rd tertile with the 1st tertile as a reference. We also tried the compositional analysis methodology [Citation40], in which the taxon abundance was assessed via the sequencing read counts instead of relative abundance. Read count for each taxon was added by 1 and centered log-ratio transformation was used for normalization [Citation41]. Then logistic regression analyses were conducted. The results were very similar to those observed in the analyses based on categorizing the data to tertiles. For microbial communities with at least five OTUs, which were identified in the community structure analyses, we also investigated them in association with obesity risk. Briefly, the relative abundance of each community was calculated as the sum of the relative abundance of all OTUs belonging to it. Then, the associations of the relative abundance of microbial communities with the risk of obesity were estimated using logistic regression following the same strategy as used for the analyses of common taxa.
For those ‘rare taxa’ with median relative abundance < 0.10% in non-obese individuals, we investigated the prevalence of these bacteria. Study participants were grouped into two groups, carriers and non-carriers, and then logistic regression analyses were conducted. Some taxa were very rare and were observed in only few individuals. There is no statistical power to investigate these bacteria. Thus, in the present study, we only analyzed those with prevalence > 30% among non-obese individuals. In total, three phyla, 16 families, 37 genera and 92 species were investigated for their prevalence in association with obesity.
During all analyses, potential confounders such as age, sex, race, smoking, alcohol consumption, total energy intake, oral health, disease status during the follow-up and study batch were additionally adjusted. For association analysis of individual taxon and obesity, odds ratio (OR), 95% confidence interval (CI) and P value were estimated using ‘glm’ of R. We used false discovery rate (FDR) to perform multiple testing correction for common taxa and rare taxa (including probiotic taxa) separately. For taxa showing an association with obesity with an FDR-corrected P < 0.05, we further investigated their associations with BMI at enrollment and weight change (between enrollment and 21 years old) via linear regression. The potential confounders included in the analyses for obesity were included in the analyses for BMI and weight change. For the analyses of weight change, weight at 21 years old was also included in the model as a covariate. We also conducted stratified analyses by African-Americans and European-Americans and the heterogeneity was estimated by the Cochran’s Q test.
Results
Characteristics of the study participants
The distribution of demographic characteristics for study participants is shown in . A total of 1,616 participants (647 obese and 969 non-obese) were included in the present investigation. Among them, 1,058 were of African-ancestry and 558 were of European-ancestry. The average weight change between weight at enrollment and weight at 21 years old was 32.52 kg among obese and 8.55 kg among non-obese participants. Obese participants had slightly lower total energy intake (an average of 2,250 vs. 2,664; kcal/day), lower smoking rate (percentage of never smokers: 42.8% vs. 27.9%) and lower alcohol consumption rate (percentage of never-drinkers: 55.8% vs. 41.2%) than non-obese participants. Oral health status is comparable between obese and non-obese participants.
Table 1. Characteristics of the participants in the southern community cohort study.
Association of overall microbial composition with obesity
Regarding the alpha-diversity, no significant differences were observed between obese and non-obese participants, with P values of 0.60, 0.48 and 0.84 for Shannon, Simpson and PD whole-tree index, respectively. We did not find any significant difference between obese and non-obese participants in beta-diversity either, as estimated by Bray-Curtis dissimilarity, unweighted UniFrac and weighted UniFrac distances with an omnibus P value of 0.36 as calculated by MiRKAT.
Associations of pre-identified and potential probiotic bacteria taxa with obesity
Multiple bacterial species belonging to the genera Bifidobacterium and Lactobacillus have been used as probiotics to decrease the risk of obesity in both animal studies and human clinical trials [Citation8,Citation9,Citation33–Citation39]. In this study, four species of Bifidobacterium and 12 species of Lactobacillus were observed. All these bacterial taxa showed lower prevalence in obese than in non-obese participants, with eight taxa reaching nominal significance (P < 0.05) (). The genera Bifidobacterium (OR = 0.67, 95% CI: 0.54, 0.83) and Lactobacillus (OR = 0.78, 95% CI: 0.63, 0.97) themselves were associated with decreased obesity prevalence with P values of 2.99 × 10−4 and 0.03, respectively. Within the genus Bifidobacterium, the species Bifidobacterium longum, observed among 25.5% obese and 35.8% non-obese participants, showed the strongest association (OR = 0.57, 95% CI: 0.45, 0.73) with P = 4.13 × 10−6. Additionally, two other species within the genus Bifidobacterium, including Bifidobacterium scardovii (OR = 0.74, 95% CI: 0.59, 0.92) and Bifidobacterium subtile (OR = 0.55, 95% CI: 0.35, 0.86), were also associated with decreased obesity prevalence with P of 8.18 × 10−3 and 9.85 × 10−3, respectively. Within the genus Lactobacillus, three species, including Lactobacillus fermentum (OR = 0.72, 95% CI: 0.56, 0.93), Lactobacillus panis (OR = 0.74, 95% CI: 0.55, 0.98) and Lactobacillus ultunensis (OR = 0.61, 95% CI: 0.39, 0.95), were associated with decreased prevalence of obesity. After adjusted for 166 comparisons, including 18 probiotic taxa and 148 ‘rare’ taxa, using FDR, the genus Bifidobacterium and the species B. longum still showed an association with decreased prevalence of obesity, with FDR-corrected P of 0.01 and 5.41 × 10−4, respectively.
Table 2. Probiotic bacterial taxa showing a significantly higher prevalence in non-obese than in obese individuals.
Associations of other bacterial taxa with obesity
As shown in , of the 63 ‘common taxa’ investigated, six taxa were associated with increased obesity prevalence after FDR correction. In Firmicutes, five taxa were associated with increased obesity prevalence. Among them, the family Carnobacteriaceae along with two taxa within it, including the genus Granulicatella and the species Granulicatella adiacens, were associated with obesity with P of 5.79 × 10−5, 1.90 × 10−4 and 3.68 × 10−5, respectively. Two other taxa in this phylum, the genus Gemella and the species Streptococcus oligofermentans, were also positively associated with the prevalence of obesity, with P of 2.25 × 10−3 and 5.50 × 10−3, respectively. In the phylum Actinobacteria, the species Actinomyces sp. oral taxon 180 was associated with increased prevalence of obesity, with P = 4.38 × 10−3. All of these six taxa were still associated with obesity prevalence after FDR correction for 63 tests with an adjusted P < 0.05.
Table 3. Common bacterial taxaa showing a significantly higher prevalence in obese than in non-obese individuals.
The community structure analyses led to the identification of 14 microbial communities (), with five communities composed of at least five OTUs. These five communities included 23, 13, 10, six, and seven OTUs. Among them, we found that the community which was comprised of seven OTUs, including Lactobacillus crispatus, Lactobacillus reuteri, L. panis, L. fermentum, Lactobacillus gasseri, Lactobacillus oris, and Enterococcus casseliflavus, was associated with a decreased risk of obesity (OR = 0.93, 95% CI: 0.88, 0.98) with a P = 6.56 × 10−3.
Figure 1. Co-abundance network of OTUs among all study participants. OTUs that were classified to a same community were marked by the same color and different communities were differentiated by different colors.
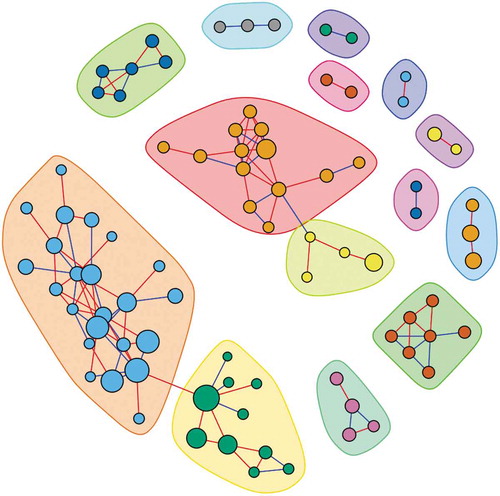
Of the 148 ‘rare’ taxa evaluated, prevalence of three taxa were associated with obesity after FDR correction (). In the phylum Actinobacteria, the genus Alloscardovia was observed among 21.6% obese and 31.4% non-obese participants. Carrying this taxon was associated with decreased obesity prevalence (OR = 0.64, 95% CI: 0.50, 0.81) with P = 3.05 × 10−4. In the phylum Firmicutes, the genus Anaeroglobus was associated with a decreased prevalence of obesity (OR = 0.66, 95% CI: 0.52, 0.84) with P = 6.04 × 10−4. In the phylum Proteobacteria, the species Aggregatibacter sp. oral taxon 512 showed an association with increased obesity prevalence (OR = 1.47, 95% CI: 1.16, 1.87) with P = 1.50 × 10−3. All of these three associations retained significance (FDR P < 0.05) after correction for 166 tests (18 probiotic taxa and 148 ‘rare’ taxa).
Table 4. Rare taxa a showing a significantly higher prevalence in obese or non-obese individuals.
Most of these associations were observed in both African-Americans and European-Americans (Table S1-S3) and none of them showed a significant heterogeneity between these two ethnic groups. The sample size in African-Americans (N = 1,058) was almost double of that in European-Americans (N = 558). Thus, the statistic power was higher in African-Americans than in European-Americans. It is expected that some bacteria shoed more significant associations in African-Americans, e.g. the genus Bifidobacterium and the species B. scardovii and L. ultunensis (Table S1), the species S. oligofermentans and Actinomyces sp. oral taxon 180 (Table S2), the genera Alloscardovia and Anaeroglobus, and the species Aggregatibacter sp. oral taxon 512 (Table S3).
For all bacterial taxa showing an association with obesity, the associations with BMI (Table S4-S6) were very similar to the associations with obesity (–). In addition, most taxa that were associated with increased risk of obesity and higher BMI were also associated with increased weight gain (Table S7-S9). These results suggested that these taxa were associated with BMI at enrollment (aged 40 to 70) but not at 21 years old. However, there are some exceptions. Four potential probiotic bacteria, B. scardovii and B. subtile, genus Lactobacillus and species L. panis were associated with obesity at study enrollment but not with weight change ( and Table S7). Two rare genera, Alloscardovia and Anaeroglobus, showed more significant associations with obesity at enrollment with P values of 3.05 × 10−4 and 6.04 × 10−4 than the associations with weight change, with P values of 0.01 and 0.06, respectively (Table S9).
Discussion
In the present study, we investigated the oral microbiome in relationship with obesity, BMI and weight gain using deep sequencing technology. We discovered multiple bacterial taxa, including several pre-identified and potential probiotic bacteria, significantly associated with obesity, BMI and weight gain.
Over the past several decades, probiotics bacteria have been increasingly used in the prevention and treatment of different disorders, including obesity [Citation9]. Multiple bacterial taxa within the genera Bifidobacterium and Lactobacillus have been suggested to have anti-obesity or weight-losing potential [Citation8,Citation9]. These bacteria may inhibit dietary fat absorption, increase fat excretion [Citation42], promote calories and fat burning [Citation43], and decrease fat storage [Citation44]. In the present study, we found that all taxa within the genera Bifidobacterium and Lactobacillus were associated with decreased obesity prevalence, lower BMI, and lower weight gain between aged at 40 − 70 and the age of 21, among both African-Americans and European-Americans. Among them, B. longum, the anti-obesity and weight loss effects of which had been indicated in both rats [Citation45] and human studies [Citation46], showed the strongest association in our study. Within the genus Bifidobacterium, we also found associations for another two species, B. scardovii and B. subtile, which had not been reported for anti-obesity and weight loss in any literature. In the genus Lactobacillus, three species, L. fermentum, L. panis and L. ultunensis, were also associated with a decreased prevalence of obesity. Consuming yogurt containing the species L. fermentum has been suggested to be associated with body fat loss and an increase of the abundance of Lactobacillus in the gut microflora [Citation47]. However, there is no data available regarding the associations for L. panis and L. ultunensis in the literatures. Within the genus Lactobacillus, two other species, L. gasseri and Lactobacillus rhamnosus have been reported to induce weight loss or have anti-obesity potential [Citation42,Citation48,Citation49]. In our study, both of these two taxa were associated with decreased obesity prevalence, although the associations did not reach statistical significance. However, given that the probiotic potential of these species was mostly found in the gut, since we do not have gut microbiome data, the associations we identified in oral samples may not directly reflect the role of these species in the gut.
We also found multiple other taxa that were significantly associated with obesity. Previous gut microbiome studies had shown that obese people/animals tend to have a higher portion of the phylum Bacteroidetes and a lower portion of the phylum Firmicutes. However, subsequent studies failed to replicate these findings, and some even reported opposite associations [Citation5]. Our study did not find a significant association between oral Bacteroidetes or Firmicutes with obesity. We found five taxa within Firmicutes, one family (Carnobacteriaceae), two genera (Gemella and Granulicatella) and two species (G. adiacens and S. oligofermentans) were more abundant among obese participants than among non-obese participants. These taxa have not been reported in any literatures for their association with obesity. However, interestingly, most of these taxa, especially G. adiacens [Citation50], S. oligofermentans [Citation51] and several species of the genus Gemella [Citation52,Citation53], have been previously linked to infective endocarditis. In the phylum Actinobacteria, the species Actinomyces sp. oral taxon 180 was associated with increased obesity prevalence, while no studies have inspected its association with obesity. We also found three ‘rare taxa’ associated with obesity, BMI and weight change. Similar with many common taxa, no studies have reported their relationships with obesity.
Stratification analyses by race showed that most associations were observed among both African-Americans and European-Americans, however some taxa showed more significant associations in the former than in the latter. This may be due to the larger sample size of African-Americans in the present study. We also observed more significant associations in European-Americans than in African-Americans for some bacterial taxa. This may be due to the racial difference in the oral microbiome. Such racial difference has been reported in earlier studies of the vaginal microbiome [Citation54,Citation55].
This study, as far as we know, is the largest one to investigate the association between the oral microbiome and obesity. In the present study, 16S rRNA sequencing techniques were used to profile the oral microbiome, which has a higher resolution compared with the checkerboard DNA-DNA hybridization used in previous studies. A wide range of underlying covariates such as age, race, gender, tobacco smoking, alcohol consumption, oral health, and total energy intake were adjusted in all the subsequent analyses, which, to a great extent, minimized the effect of these confounders. In the present study, both African-Americans and European-Americans were included, which helped the investigation of racial disparity of the oral microbiome. However, there are several limitations in the present study. First, the present study has a cross-sectional design, hence we could not determine the causality of associations. On the other hand, body weight and height of the study participants were self-reported. However, in the SCCS, clinic-recorded weights were available for over 20% of the participants. Among them, the correlation between self-reported and clinic-recorded weight was greater than 0.95 [Citation21]. In the analyses of the oral microbiome with BMI at age 21 and weight change between enrollment and age 21, we did not control the memory bias regarding weight at age 21, which is another limitation. We found strong associations, and especially, most of the bacterial taxa, which were associated with BMI at age 21, were also associated with weight gain during adulthood. Further, lacking a systematic assessment of oral health at the baseline examination when the samples were collected is a limitation as well.
In summary, we found that multiple bacterial taxa, including pre-identified and potential probiotic bacteria, in mouth rinse samples were associated with obesity. These results suggest that the oral microbiome may play an important role in obesity.
Availability of data and material
Data used in the present study can be requested through the SCCS online request system (https://www.southerncommunitystudy.org/research-opportunities.html).
Acknowledgments
The authors wish to thank all the individuals who took part in the study, and all the researchers, clinicians, technicians, and administrative staff who enabled this work to be carried out. We thank Regina Courtney, Jie Wu, and Marshal Younger for their help with sample preparation, statistical analysis and technical support for the project. The data analyses were conducted using the Advanced Computing Center for Research and Education (ACCRE) at Vanderbilt University.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Ng M, Fleming T, Robinson M, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the global burden of disease study 2013. Lancet. 2014;384:766–9.
- Ogden CL, Carroll MD, Kit BK, et al. Prevalence of childhood and adult obesity in the USA, 2011-2012. JAMA. 2014;311:806–814.
- Smith KB, Smith MS. Obesity statistics. Prim Care. 2016;43:121–135.
- Wardle J, Carnell S, Haworth CM, et al. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87(2):398–404.
- Zhao L. The gut microbiota and obesity: from correlation to causality. Nat Rev Microbiol. 2013;11:639.
- Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133.
- Hill C, Guarner F, Reid G, et al. Expert consensus document: the international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11:506.
- Ouwehand AC, Salminen S, Isolauri E. Probiotics: an overview of beneficial effects. In: lactic acid bacteria: genetics, metabolism and applications. Dordrecht: Springer; 2002. p. 279–289.
- Sáez-Lara MJ, Robles-Sanchez C, Ruiz-Ojeda FJ, et al. Effects of probiotics and synbiotics on obesity, insulin resistance syndrome, type 2 diabetes and non-alcoholic fatty liver disease: a review of human clinical trials. Int J Mol Sci. 2016;17:928.
- Wade WG. The oral microbiome in health and disease. PharmacolRes. 2013;69:137–143.
- Moye ZD, Zeng L, Burne RA. Fueling the caries process: carbohydrate metabolism and gene regulation by Streptococcus mutans. J Oral Microbiol. 2014;6.
- Slocum C, Kramer C, Genco C. Immune dysregulation mediated by the oral microbiome: potential link to chronic inflammation and atherosclerosis. J Intern Med. 2016;280(1):114–128.
- A-L Ö, Bengtsson C, Lissner L, et al. Oral health and obesity indicators. BMC Oral Health. 2012;12:50.
- Cinar AB, Murtomaa H. Interrelation between obesity, oral health and life-style factors among Turkish school children. Clin Oral Invest. 2011;15:177–184.
- Yang Y, Cai Q, Shu XO, et al. Prospective study of oral microbiome and colorectal cancer risk in low-income and African American populations. Int J Cancer. 2019;144(10):2381–2389.
- Fan X, Alekseyenko AV, Wu J, et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2018;67:120–127.
- Hayes RB, Ahn J, Fan X, et al. Association of oral microbiome with risk for incident head and neck squamous cell cancer. JAMA Oncol. 2018;4(3):358–365.
- Flemer B, Warren RD, Barrett MP, et al. The oral microbiota in colorectal cancer is distinctive and predictive. Gut. 2018;67:1454–1463.
- Goodson J, Groppo D, Halem S, et al. Is obesity an oral bacterial disease? J Dent Res. 2009;88:519–523.
- Zeigler CC, Persson GR, Wondimu B, et al. Microbiota in the oral subgingival biofilm is associated with obesity in adolescence. Obesity. 2012;20:157–164.
- Signorello LB, Hargreaves MK, Blot WJ. The southern community cohort study: investigating health disparities. J Health Care Poor Underserved. 2010;21(1 Suppl):26–37.
- Signorello LB, Hargreaves MK, Steinwandel MD, et al. Southern community cohort study: establishing a cohort to investigate health disparities. J Natl Med Assoc. 2005;97:972.
- Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4516–4522.
- Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme J. 2012;6(8):1621–1624.
- Joshi N, Fass J Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. Available from: github.com/najoshi/sickle. 2011.
- Nikolenko SI, Korobeynikov AI, Alekseyev MA. BayesHammer: bayesian clustering for error correction in single-cell sequencing. BMC Genomics. 2013;14(Suppl 1):S7.
- Masella AP, Bartram AK, Truszkowski JM, et al. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31.
- Schirmer M, Ijaz UZ, D’Amore R, et al. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015;43(6):e37.
- Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336.
- Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. Plos Comput Biol. 2012;8:e1002687.
- Palla G, Derényi I, Farkas I, et al. Uncovering the overlapping community structure of complex networks in nature and society. Nature. 2005;435:814.
- Zhao N, Chen J, Carroll IM, et al. Testing in microbiome-profiling studies with MiRKAT, the microbiome regression-based kernel association test. Am J Hum Genet. 2015;96(5):797–807.
- Oishi K, Yokoi W, Yoshida Y, et al. Effect of probiotics, bifidobacterium breve and lactobacillus casei, on bisphenol A exposure in rats. Biosci Biotechnol Biochem. 2008;72(6):1409–1415.
- Matsuda F, Chowdhury M, Saha A, et al. Evaluation of a probiotics, Bifidobacterium breve BBG-01, for enhancement of immunogenicity of an oral inactivated cholera vaccine and safety: a randomized, double-blind, placebo-controlled trial in Bangladeshi children under 5 years of age. Vaccine. 2011;29:1855–1858.
- Kim JY, Choi YO, Ji GE. Effect of oral probiotics (Bifidobacterium lactis AD011 and Lactobacillus acidophilus AD031) administration on ovalbumin-induced food allergy mouse model. J Microbiol Biotechnol. 2008;18(8):1393–1400.
- Lee H-Y, Park J-H, Seok S-H, et al. Human originated bacteria, Lactobacillus rhamnosus PL60, produce conjugated linoleic acid and show anti-obesity effects in diet-induced obese mice. Biochim Biophys Acta. 2006;1761(7):736–744.
- Bernardeau M, Vernoux JP, Gueguen M. Safety and efficacy of probiotic lactobacilli in promoting growth in post-weaning Swiss mice. Int J Food Microbiol. 2002;77(1–2):19–27.
- Larsen N, Vogensen FK, Gøbel RJ, et al. Effect of Lactobacillus salivarius Ls-33 on fecal microbiota in obese adolescents. Clin Nutrit. 2013;32:935–940.
- Zarrati M, Salehi E, Mofid V, et al. Relationship between probiotic consumption and IL-10 and IL-17 secreted by PBMCs in overweight and obese people. Iran J Allergy Asthma Immunol. 2013;12(4):404–406.
- Gloor GB, Macklaim JM, Pawlowsky-Glahn V, et al. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 2017;8:2224.
- Peters BA, Wu J, Pei Z, et al. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res. 2017;77:6777–6787.
- Ogawa A, Kobayashi T, Sakai F, et al. Lactobacillus gasseri SBT2055 suppresses fatty acid release through enlargement of fat emulsion size in vitro and promotes fecal fat excretion in healthy Japanese subjects. Lipids Health Dis. 2015;14:20.
- Yadav H, Lee J-H, Lloyd J, et al. Beneficial metabolic effects of a probiotic via butyrate induced GLP-1 secretion. J Biol Chem. 2013: jbc. M113. 452516.
- Aronsson L, Huang Y, Parini P, et al. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4). PloS One. 2010;5:e13087.
- An HM, Park SY, Lee DK, et al. Antiobesity and lipid-lowering effects of bifidobacterium spp. in high fat diet-induced obese rats. Lipids Health Dis. 2011;10:116.
- Osterberg KL, Boutagy NE, McMillan RP, et al. Probiotic supplementation attenuates increases in body mass and fat mass during high‐fat diet in healthy young adults. Obesity. 2015;23:2364–2370.
- Omar JM, Chan Y-M, Jones ML, et al. Lactobacillus fermentum and Lactobacillus amylovorus as probiotics alter body adiposity and gut microflora in healthy persons. J Function Foods. 2013;5:116–123.
- Miyoshi M, Ogawa A, Higurashi S, et al. Anti-obesity effect of Lactobacillus gasseri SBT2055 accompanied by inhibition of pro-inflammatory gene expression in the visceral adipose tissue in diet-induced obese mice. Eur J Nutr. 2014;53:599–606.
- Sanchez M, Darimont C, Drapeau V, et al. Effect of Lactobacillus rhamnosus CGMCC1. 3724 supplementation on weight loss and maintenance in obese men and women. Br J Nutr. 2014;111:1507–1519.
- Woo PC-Y, Fung AM-Y, Lau SK-P, et al. Granulicatella adiacens and Abiotrophia defectiva bacteraemia characterized by 16S rRNA gene sequencing. J Med Microbiol. 2003;52:137–140.
- Matta M, Gousseff M, Monsel F, et al. First case of Streptococcus oligofermentans endocarditis determined based on sodA gene sequences after amplification directly from valvular samples. J Clin Microbiol. 2009;47:855–856.
- Mosquera J, Zabalza M, Lantero M, et al. Endocarditis due to Gemella haemolysans in a patient with hemochromatosis. Clin Microbiol Infect. 2000;6:566–568.
- Elsayed S, Zhang K. Gemella bergeriae endocarditis diagnosed by sequencing of rRNA genes in heart valve tissue. J Clin Microbiol. 2004;42:4897–4900.
- Gliga S, Devaux M, Woimant MG, et al. Actinomyces graevenitzii pulmonary abscess mimicking tuberculosis in a healthy young man. Can Resp J. 2014;21:e75–e77.
- Zhou X, Brown CJ, Abdo Z, et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. Isme J. 2007;1:121–133.