
ABSTRACT
Porphyromonas. gingivalis (P. gingivalis)
is an obligate, asaccharolytic, gram-negative bacteria commonly associated with increased periodontal and systemic inflammation. P. gingivalis is known to survive and persist within the host tissues as it modulates the entire ecosystem by either engineering its environment or modifying the host’s immune response. It interacts with various host receptors and alters signaling pathways of inflammation, complement system, cell cycle, and apoptosis. P. gingivalis is even known to induce suicidal cell death of the host and other microbes in its vicinity with the emergence of pathobiont species. Recently, new molecular and immunological mechanisms and virulence factors of P. gingivalis that increase its chance of survival and immune evasion within the host have been discovered. Thus, the present paper aims to provide a consolidated update on the new intricate and unique molecular mechanisms and virulence factors of P. gingivalis associated with its survival, persistence, and immune evasion within the host.
Introduction
Periodontitis is a multifactorial immuno-inflammatory disease initiated by the interaction of the host to the pathogenic microorganisms in the oral cavity [Citation1–Citation5]. The host-microbial interaction activates a cascade of signaling pathways that cause release of proinflammatory cytokines and tissue destructive enzymes. The increased inflammatory response in the periodontal tissues alters the composition of the entire microbiome that shifts the gram-positive aerobic cocci to gram-negative anaerobic rods and motile spirochetes [Citation2,Citation6–Citation9]. Some of the common gram-negative anaerobic species that predominate the oral biofilm at the later stages of periodontitis include Porphyromonas gingivalis (P. gingivalis), Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans), Tanerella forsythia (T. forsythia), Fusobacterium nucleatum (F. nucleatum), Treponema denticola (T. denticola), Camplyobacter rectus (C. rectus), Prevotella intermedia (P. intermedia), etc [Citation3–Citation9]. Amongst all these pathogens, the role of P. gingivalis in exaggerating periodontal inflammation and dysbiosis is very unique and intricate [Citation10–Citation17]
P. gingivalis, formerly known as Bacteroides gingivalis, is a gram-negative non-motile, asaccharolytic, obligate, capnocytophagic, anaerobic rod-shaped bacteria. P. gingivalis is considered as a prime etiological agent in the pathogenesis and progression of periodontal inflammation and alveolar bone loss [Citation2,Citation7,Citation12–Citation15]. Approximately, 10%–25% of healthy subjects and 79%–90% of subjects with periodontitis have micro-colonies of P. gingivalis in their oral cavity [Citation16,Citation17]. A positive correlation between the depth of the periodontal pocket and the presence of P. gingivalis has also been established [Citation17]. Even at low abundance, i.e. <0.01% of the total bacterial count, P. gingivalis has the inherent ability to transform the entire microbial community and amplify the disease process [Citation10]. It has been referred to as a ‘keystone pathogen’ and a ‘master of polymicrobial synergy, dysbiosis and immune subversion’, as it exploits several sabotage tactics to evade, weaken, or deceive the host’s immune system [Citation10–Citation15]. P. gingivalis is also known to gain entry into the host cell and systemic circulation to reach distant organ systems [Citation15–Citation19]. Numerous studies have explained and discovered the novel virulence factors and pathogenic mechanisms that help P. gingivalis to survive and persist in the host [Citation12–Citation15]. However, the knowledge about how P. gingivalis modulates the immune response with activation of newly discovered virulence factors, host proteins, and signaling pathways is limited. No paper to our knowledge has provided a consolidated update on the recent immunological receptors, signaling pathways, and molecular and cellular mechanisms associated with P. gingivalis invasion and persistence within the host tissue. Furthermore, the knowledge about the cellular and molecular mechanisms linked with the interaction of P. gingivalis with other members of the oral microbiome is not fully explored. It is important to understand how P. gingivalis causes of suicidal death of other members of the oral microbiome along with the emergence of pathobionts and virulent commensals in the host.
Therefore the present review aims to provide a critical and consolidated update on the new pathogenic mechanisms and survival strategies of P. gingivalis within host associated with periodontal inflammation and dysbiosis. A thorough understanding of these key virulence factors and pathogenic mechanisms is crucial not only to understand the pathogenesis of periodontal diseases and systemic inflammation but also to develop novel therapeutic modalities that to prevent the onset and progression of periodontal disease.
Mechanism of P. gingivalis invasion and persistence within the host
P. gingivalis is a virulent subgingival periodontal pathogen that can orchestrate the inflammatory response within the host by altering various inter-bacterial and host-bacterial interactions [Citation10–Citation20]. P. gingivalis survives within the host by adhering to a wide range of oral substrates and molecules like extracellular matrix proteins, oral epithelial cells, and other commensal bacteria such as Streptococci gordonii (S. gordinii), Filifactor alocis (F. alocis), F. nucleatum, and Actinomyces viscosus [Citation21–Citation26]. P. gingivalis impairs various components of the innate immune response: alters the functions of the complement system, Toll-like receptors (TLRs), neutrophils, macrophages, dendritic cells, and, T cells (Th1/Th2/Th17) [Citation14–Citation17]. It can even modulate its virulence factors and nutritional demand to survive and persist at the times of crisis and nutritional deficiency [Citation12–Citation23]. Gingipains (Cysteine proteases), capsule, lipopolysaccharide (LPS), fimbriae, nucleoside diphosphate kinase (NDK), ceramide, and outer membrane proteins (OMs) are some of the important virulence factors that help P. gingivalis to invade and survive with the periodontal tissues and gain entry into the systemic circulation () [Citation21–Citation25]. The ‘inflammophilic’ nature of P. gingivalis even helps it to utilize the tissue destructive byproducts, enzymes, and proinflammatory cytokines produced during inflammation as its nutrients [Citation26–Citation28]. P. gingivalis can even indirectly modulate the maturation and growth of other microorganisms within the biofilm growth by indirectly sending the short-range paracrine signaling molecules into its environment and enhancing quorum sensing [Citation3,Citation23,Citation28–Citation30]. The interaction with other microorganisms not only facilitates its growth and survival but also favors the growth and development of other bystanders, commensal, and pathobiont species [Citation29–Citation32]. Some of the other unique and intricate molecular mechanisms associated with its interaction of other members of the biofilm and host that increases its growth, survival, and persistence can be summarized as follows ():
Table 1. Novel Pathogenic mechanisms adopted by P. gingivalis to exaggerate periodontal inflammation.
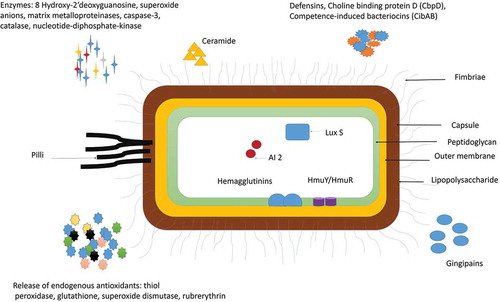
Figure 1. Virulence factors of P. gingivalis: P gingivalis has various virulence factors that help it to invade the host cell and evade the defense mechanisms of the host. Some of the most important adhesions of P. gingivalis include Lipopolysaccharide (LPS), Capsule, outer Membrane protein, Peptidoglycan, Major, and minor Fimbriae and Pilli. P. gingivalis also release enzymes and cytotoxic molecules such as defensins, autoinducer proteins (AI-2); 8 hydroxy 2ʹ deoxyguanosine, superoxide anions, matrix metalloproteinases, caspase-3, catalase, nucleotide-diphosphate-kinase. These enzymes help to invade the host cell, increase oxidative stress, and enhance biofilm formation. P. gingivalis is also known to release endogenous antioxidants such as thiol, peroxidase, glutathione, superoxide dismutase, rubrerythrin to protect itself from the surrounding free radicals.
2.a. Synergistic interaction of P. gingivalis with other microorganisms and development of pathobiont species
P. gingivalis interacts with other microorganisms to elicit its full range of pathogenicity and virulence factors, as in germ-free conditions, it cannot survive or produce any disease () [Citation10]. The interspecies synergy of P. gingivalis has been observed with Streptococci mitis (S. mitis), S. gordonii, A. actinomycetomcomitans, F. nucleatum, T. denticola, P. intermedia, F. alocis [Citation8–Citation10,Citation24,Citation30]. P. gingivalis modulates the phenotypic profile of microorganisms by interacting with their surface adhesins and regulating their virulence factors, nutritional demands, and growth [Citation25–Citation28]. The most common synergistic interaction of P. gingivalis is observed with F. nucleatum, S gordonii and F. alocis (Figure 2–) [Citation30–Citation37]
Figure 2. P. gingivalis modulates the host immune response by facilitating the growth of pathobionts species and altering the function of various immune cells of the host. The weakened immune response enhances biofilm formation and oxidative stress that in turn increases the periodontal inflammation and favors the growth of P. gingivalis [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; ROS-Reactive oxygen species; MMPS- matrix metalloproteinase; IL- interleukin; RANKL- Receptor activator of nuclear factor-kappa beta; TNF- Tumor Necrotic Factor-alpha; Th – T helper cells]. [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; ROS-Reactive oxygen species; MMPS- matrix metalloproteinase; IL- Interleukin; RANKL- Receptor activator of nuclear factor-kappa beta; TNF- Tumor Necrotic Factor-alpha; Th – T helper cells; NOD-nucleotide-binding oligomerization domain; NF-Kb – Nuclear Factor kappa Beta].
![Figure 2. P. gingivalis modulates the host immune response by facilitating the growth of pathobionts species and altering the function of various immune cells of the host. The weakened immune response enhances biofilm formation and oxidative stress that in turn increases the periodontal inflammation and favors the growth of P. gingivalis [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; ROS-Reactive oxygen species; MMPS- matrix metalloproteinase; IL- interleukin; RANKL- Receptor activator of nuclear factor-kappa beta; TNF- Tumor Necrotic Factor-alpha; Th – T helper cells]. [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; ROS-Reactive oxygen species; MMPS- matrix metalloproteinase; IL- Interleukin; RANKL- Receptor activator of nuclear factor-kappa beta; TNF- Tumor Necrotic Factor-alpha; Th – T helper cells; NOD-nucleotide-binding oligomerization domain; NF-Kb – Nuclear Factor kappa Beta].](/cms/asset/e7174048-18ff-43d2-b58e-847c46a08305/zjom_a_1801090_f0002_oc.jpg)
Recently, P. gingivalis’ is discovered to favor the development of many pathobiont species in the oral biofilm. A pathobiont is a harmless symbiont that becomes pathogenic under altered environmental conditions or immunosuppression [Citation15,Citation30–Citation38]. The pathobionts outcompete the other anaerobic species to become the major etiological agent for the onset of periodontitis [Citation23,Citation32,Citation33]. A recent metatranscriptomic and metagenomic analysis has confirmed a positive correlation between P. gingivalis and development of several underappreciated pathobionts species like Peptostreptococcus stomatis, F. alocis, Megasphaera, Selenomonas, and Desulfobulbus [Citation13,Citation15,Citation30–Citation32]. The pathobiont species help P. gingivalis to acquire essential nutrients, enhance virulence factors, and activate the release of cytokines and enzymes in the oral environment [Citation30–Citation34]. The pathobionts even accumulate at the sites of damaged periodontal tissues and selectively bind to the intracellular nucleotide-binding oligomerization domain 1 [Nod1] like receptors that indirectly activates the Nuclear Factor-kappa Beta (NF-Kb) mediated proinflammatory cytokines production () [Citation34–Citation39].
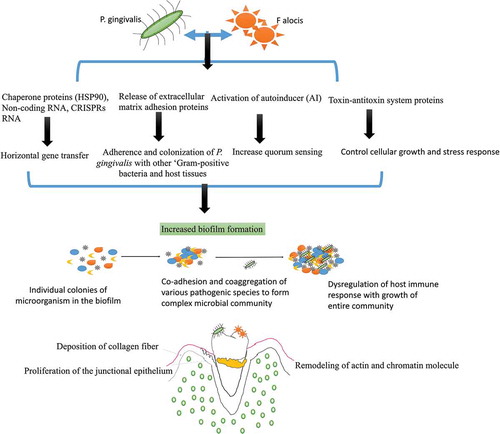
The most common pathobiont that favors P. gingivalis’ growth and survival is F. alocis. Studies have shown that P. gingivalis co-cultured with F. alocis can modulate the innate immune response of the host as both these species auto-aggregate and express unique gene expression () [Citation31–Citation33]. P. gingivalis-F. alocis is linked with the remodeling of actin and chromatin molecule, activation of autoinducer (AI) associated quorum sensing, the proliferation of the junctional epithelium, and deposition of collagen fibers in the gingival epithelial cells [Citation30–Citation33]. The co-infection also upregulate the production of extracellular matrix adhesion proteins like actin, vinculin, vimentin, plectin, transgelin, profilin, endoplasmin proteins (filamin B and filamin C), chaperone proteins (HSP90), non-coding RNA, CRISPRs RNA, ‘microbial surface component-recognizing adhesion matrix molecules’ (MSCRAMMs), and toxin-antitoxin system proteins [Citation31]. These novel proteins are required for the adherence and colonization of P. gingivalis with other ‘Gram-positive bacteria and host tissues’, and are directly linked with increased biofilm formation [Citation31–Citation40]. The increased production of CRISPR-RNA is also associated with triggering the stress response, chaperone formation, and horizontal gene transfer among oral bacteria [Citation41–Citation43]. The CRISPRs RNA even helps P. gingivalis to induce genomic rearrangements and intercellular recombination that helps to acquire the useful DNA sequences for its survival, limit transposition of insertion sequences and acquire resistance to foreign RNA and DNA [Citation44–Citation48]. Watanabe et al. (2017) also observed a highly ‘expressed transcripts of CRISPR regulatory small non-coding regulatory RNA (sRNA) in the intergenic sequences (IGS) upstream of CRISPR-associated (cas) gene arrays in P. gingivalis that help to limit the genetic exchange within the biofilm and prevent mutation [Citation42,Citation45–Citation47]. Another highly upregulated hypothetical proteins, known as ‘HMPREF0389_00967, containing a CHASE3 extracellular sensory domain’, has been observed during F. alocis – P. gingivalis co-infection [Citation31,Citation48]. The HMPREF0389_00967 is found to regulate the production of histidine kinase, adenylate cyclases, and other chemotaxis proteins that helps P. gingivalis to evade the process of phagocytosis [Citation31,Citation45–Citation48]. F. alocis – P. gingivalis co-infection is also known to regulate other proteins such as ‘histone cluster proteins and PPIA-peptidyl-prolyl isomerase, transferrin receptor protein 1 (involved in iron transport), transmembrane emp24 domain-containing protein 10, dynein (involved in vesicular protein trafficking), surfeit 4, and solute carrier family proteins’ in the host [Citation48]. The alteration in the production of these proteins indirectly weakens the innate immune response of the host and favors microbial growth within the tissues. P. gingivalis can even acquire and utilize some of these proteins, nucleosides, and nucleobases for its nutrition, growth, maturation, and survival [Citation49].
Figure 3. Interaction of P. gingivalis with F. alocis and its effects on biofilm formation and periodontal inflammation: P. gingivalis interaction with F. alocis can modulate the innate immune response of the host as both these species auto-aggregate and express unique genes expression. P. gingivalis-F. alocis remodeling the actin and chromatin molecule, activation of autoinducer (AI) associated quorum sensing, the proliferation of the junctional epithelium, and deposition of collagen fibers in the gingival epithelial cells. The co-infection also upregulates the production of extracellular matrix adhesion proteins, CRISPRs RNA, and toxin-antitoxin system proteins that increase the adherence and colonization of P. gingivalis with other ‘Gram-positive bacteria and host tissues’, and are directly linked with increased biofilm formation. The CRISPR-RNA also helps with triggering the stress response, chaperone formation, and horizontal gene transfer among oral bacteria that favor microbial community development.
P. gingivalis is known to modulate its nutritional demand and virulence factors and according to the changes in its environment, immune response, co-infection, and temperature and nutrient supply [Citation12,Citation47,Citation50–Citation60]. Studies have confirmed that the addition of P. gingivalis to a healthy multispecies oral biofilm model rearranges the DNA profiles of other commensal species, alters the functions of the complement system, subvert phagocytosis, protease production, alter its adhesions, and cause cytotoxicity [Citation12,Citation20,Citation50–Citation54]. P. gingivalis also changes the genetic expression of other commensal and symbiotic bacteria according to changes in the level of hemin, polyphosphate, rhein, temperature, oxidative stress, etc. in the periodontal tissues [Citation47,Citation54–Citation59]. Studies have shown that high levels of hemin and rise in environmental temperature cause P. gingivalis’ to alter its Pentaacylated LPS 1690 isoform to the Tetraacylated LPS 1435/1449 isoform [Citation47,Citation51–Citation59]. This alteration in the LPS structure inhibits the process of phagocytosis by inhibiting the expression of LPS binding proteins (LBP) and the production of key chemoattractants like IL8 and IL6 [Citation53]. High levels of hemin and rise in temperature also upregulate the expression of mRNA associated with the growth, maturation, cellular differentiation, alteration of chaperones, ABC transport systems, and transposases formation () [Citation33,Citation47,Citation55–Citation61]. Duran-Pinedo et al., (2014) observed that P. gingivalis invasion upregulate the expression of putative transposases in S. mitis and Lactobacillus casei microcolonies that enhance biofilm formation [Citation61–Citation68].
Figure 4. Modulation of immunoinflammatory response by two forms of P. gingivalis lipid A depending on the microenvironment (hemin level) and their interference in TLR4 receptor signaling downstream activation. It can regulate the inflammatory response according to changes in the environmental conditions by changing its LPS moiety [Abbreviation: LPS: lipopolysaccharide; p65: nuclear factor NF-κB protein p65 subunit; p50: nuclear factor NF-κB protein p50 subunit; TLR4: Toll-like receptor-4; TRAF 6: tumor necrosis factor receptor-associated factor 6; TRIF: TIR-domain-containing adapter-inducing interferon-β; TRAM: TRIF-related adaptor molecule].
![Figure 4. Modulation of immunoinflammatory response by two forms of P. gingivalis lipid A depending on the microenvironment (hemin level) and their interference in TLR4 receptor signaling downstream activation. It can regulate the inflammatory response according to changes in the environmental conditions by changing its LPS moiety [Abbreviation: LPS: lipopolysaccharide; p65: nuclear factor NF-κB protein p65 subunit; p50: nuclear factor NF-κB protein p50 subunit; TLR4: Toll-like receptor-4; TRAF 6: tumor necrosis factor receptor-associated factor 6; TRIF: TIR-domain-containing adapter-inducing interferon-β; TRAM: TRIF-related adaptor molecule].](/cms/asset/30926e1f-2e89-48fa-a0e4-bb4543026300/zjom_a_1801090_f0004_oc.jpg)
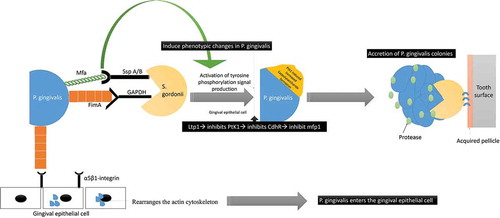
P. gingivalis’ interaction with S. gordonii is also confirmed to have a profound effect on the phenotypic profile of the entire microbial community with activation of several genetic functions before the ‘mutualistic communities’ development [Citation21,Citation62–Citation66]. S. gordonii facilitates the colonization of P. gingivalis even in the absence of any bridging species like F. nucleatum [Citation10,Citation61–Citation72]. P. gingivalis adherence to S. gordonii is by a ‘multimodal’ and ‘coordinated adhesion system’ comprising of the major (FimA) and minor (Mfa1) fimbriae [Citation65–Citation68]. FimA and Mfa1 of P. gingivalis bind to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and streptococcal SspA/B adhesins (SspA/B termed BAR antigen I/II) of S. gordonii, respectively () [Citation67–Citation74] Moreover, it has been confirmed that community development with P. gingivalis does not occur with Streptococci species that lacks the BAR motif, such as S. mutans and S. intermedius [Citation63,Citation64,Citation75]. Therefore, even though, all of the oral streptococci express antigen I/II, P. gingivalis selectively binds to S. gordonii and the related oralis group of Streptococci [Citation76]. Additionally, studies have shown that before P. gingivalis adheres to S. gordonii, it accretes to form ‘tower-like microcolonies’ that are separated by fluid-filled channels [Citation76,Citation77]. P. gingivalis – S gordonii co-infection even favors the ‘heterotypic community formation with F. nucleatum. P. gingivalis-F. nucleatum coaggregation is mediated by a galactoside moiety on P. gingivalis’ surface and a lectin adhesion on F. nucleatum [Citation10,Citation16,Citation21,Citation26–Citation29,Citation33]. Studies have observed that F. nucleatum increases the invasion of P. gingivalis into the gingival epithelial cells by 2–20‐folds, along with an increase in the osteoclastic activity, immune modulation with enhanced proinflammatory cytokine production and apoptosis [Citation34–Citation36]. The adaptation of a community lifestyle provides physiologic support to its constituent species, lower levels of environmental stress, and promotes the chance of survival [Citation75]. S. gordonii is considered as a homeostatic commensal that is capable of mitigating the activity of P. gingivalis and modulating the host signaling [Citation78–Citation82].
Figure 5. Schematic representation of the interaction between P. gingivalis and S. gordonii: P. gingivalis interacts with S. gordonii by utilizing its major (FimA) and minor fimbriae (Mfa1). FimA and Mfa1 of P. gingivalis binds to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and streptococcal SspA/B adhesins (SspA/B termed BAR antigen I/II) of S. gordonii, respectively The Mfa1 interactions with SspB lead to phenotypic changes with subsequent activation of tyrosine phosphorylation signal production (PTK). The increased PTK signaling causes exopolysaccharide formation that causes accretion of P. gingivalis colonies. The FimA also interacts to the α5β1-integrin receptors on gingival epithelial to facilitate bacterial entry.
P. gingivalis-F. nucleatum- S. gordonii consortia is known to cause extensive alterations in the cell envelope and cell wall protein, especially the outer membrane of P. gingivalis [Citation78]. Kuboniwa et al., 2006, showed that 84 proteins of cell envelope were detected during the co-infection, out of which 40 showed reduced abundance during the three species community formation [Citation78,Citation79]. Some of the common proteins affected during the co-infection include proteins regulating thiamine, cobalamin, and pyrimidine synthesis; OmpH proteins, two lipoproteins, MreB protein (a bacterial actin homolog) that play an important role in determining cell shape; and DNA repair proteins. The HmuR, a TonB dependent outer membrane receptor, was up-regulated in the community with an increase in protein synthesis. Some of the other encoded proteins that affected by this interaction include proteins for maintenance of cell wall integrity (murE), intercellular signaling (cbe), regulation of redox state (spxB and msrA), ribosomal proteins and translation elongation and initiation proteins. A decrease in vitamin B1, B 12, Biotin, pyrimidine synthesis was also observed during this co-infection [Citation78,Citation79] A community derived P. gingivalis biofilm also showed a significant reduction in DNA repair proteins along with an up-gradation of DNA repair genes [Citation80].
Apart from the up-regulation of these genes, P. gingivalis regulates the expression of Zinc finger E-box-binding homeobox (ZEB2), a transcription factor, which controls epithelial-mesenchymal transition and inflammatory responses within the periodontal tissues [Citation80,Citation81]. P. gingivalis mediated ZEB2 regulation occurs through pathways involving β-catenin and Forkhead box protein O1 (FOXO1). S. gordonii can antagonize ZEB2 expression, and thereby control P. gingivalis mediated inflammation response. Furthermore, P. gingivalis and S. gordonii coinfection is even found to enhance the expression of Protein Tyrosine Phosphatase (Ltp1) and AI-2 in the periodontal tissues [Citation60–Citation64,Citation69,Citation77,Citation81–Citation91]. The increased Ltp1 expression subsequently dephosphorylates P. gingivalis tyrosine kinase 1 (Ptk1) receptors and increases the transcription of Complementarity Determining Region (CDrR) to promote colony formation [Citation68,Citation82,Citation83,Citation91] (). S. gordonii also inhibits the production of CdhR (Community development and hemin Regulator), a negative regulator of Mfa1, and increases the co-adhesion and co-aggregation of P. gingivalis with other microorganisms [Citation60–Citation62,Citation84,Citation85]. However, once the colony has been established, it dampens the micro-colony development by utilizing the Ltp1 receptors and reducing Mfa1 expression [Citation90]. The Ltp1 phosphatase activity is also known to down-regulate the transcription of luxS and several other genes involved in exopolysaccharide synthesis and transport of nutrients [Citation90] Ltp1 also promotes the transcription of the hmu operon involved in haem uptake and dephosphorylation of the gingipain proteases. The increased CDrR expression even help P. gingivalis to acquire nutrients such as peptides and heam from its environment at the times of crisis [Citation92]. Since P. gingivalis is unable to synthesize its porphyrin, it relies on exogenous porphyrin and heam biosynthesis intermediates, from host sources and other bacteria. S. gordonii helps P. gingivalis to acquire heam by promoting met- Hemoglobin formation in the presence of Oxy-Hemoglobin. The heme-binding protein receptor (Hmu-Y) of P. gingivalis’ captures and extracts the Iron III-Protoporphyrin IX using the Met-Hemoglobin obtained from S. gordonii [Citation89–Citation97]. Hmu-Y along with its cognate outer-membrane receptor, HmuR, aid in the transportation of the heam molecule through the outer membrane of P. gingivalis. This process of acquiring heam by P. gingivalis, also dependent upon the protein TonB, which helps to transduce energy for the passage of heam and other ligands into the periplasm. Furthermore, the Hmu-Y receptors help P. gingivalis to increase the extracellular polymeric substances (EPS) production that mediates cohesion among the microbial community and prevents injury from any external physical forces [Citation90–Citation97]. Under the iron-limited conditions, P. gingivalis expresses a haemophore-like protein, HusA, to mediate the uptake of essential porphyrin and support its survival within the host [Citation92–Citation94]. The Hmu-Y receptor of P. gingivalis can even interact with the Interpain-A (InpA) of P. intermedia and promote community development [Citation90,Citation98,Citation99].
Additionally, since P. gingivalis is devoid of LuxI/LuxR-based signaling mechanisms, it utilizes the LuxI/LuxR receptors on S. gordonii to enhance the gene expression responsible for the acquisition of hemin, extracellular polysaccharide formation, and accretion into micro-colonies [Citation91–Citation94]. The LuxS in S. gordonii regulate the levels of the ‘Ssp’ adhesins and influence its ability to adhere to micro-colonies and autoaggregation in P. gingivalis [Citation50].
Recently, two novel proteins named ‘internalin pro-Clptein (InlJ)’ along with the ‘expression of the short fimbriae and the universal stress protein UspA’ have been linked with monospecies P. gingivalis biofilms formation and initial attachment [Citation100–Citation106]. Furthermore, the loss of several genes has been observed within the P. gingivalis colonies to regulate the growth of the biofilm and promotes its chance of survival. Some of the inhibitors of homotypic biofilm accumulation include Caseinolytic Mitochondrial Matrix Peptidase Chaperone Subunit X (ClpXP) along with ClpC, and GalE (UDP‐galactose 4‐epimerase) have been found in P. gingivalis microcolonies [Citation99,Citation103–Citation107]. P. gingivalis is also observed to cause lysine acetylation in colonies, and this is considered as novel mechanism of metabolic regulation, adaptation, and survival during infection [Citation105]. The acetylation of RprY, a response regulator of P gingivalis during the oxidative stress, has been considered a key factor regulating gene expression. Lysine acetylation reduces the promoter DNA binding ability of RprY, which consequently alters the gene expression of the host [Citation105–Citation107].
Apart from regulating the genetic expression and release of important proteins, P. gingivalis can even facilitate the growth of other bacterial species by acting as a ‘bridging species’ [Citation99,Citation108,Citation109]. P. gingivalis facilitates the entry of pathogens into deep periodontal pockets. Studies have proven that the outer membrane vesicle of P. gingivalis (Omp A) facilitate co-aggregation and piggybacking of T. denticola [Citation109–Citation112]. Furthermore, P. gingivalis promotes the attachment of Lachnoanaerbaculum. saburreum to T. denticola by acting as a ‘bridge’ and facilitating complex microbial community formation [Citation109–Citation111]. The piggybacking of T. denticola and Lachnoanaerbaculum saburreum favors the migration of bacteria to the anaerobic sub-gingival niches and changing the ecology of the entire sub-gingival sulcus. The cooperation and co-existence of P. gingivalis with T. denticola can also increase the production of gingipains (Kgp, RgpA, HagA) and dentilisism from T. denticola [Citation99]. The increased ‘gingipain’ production inhibits blood coagulation, escalates nutrient accumulation, and in turn favors attachment of P. gingivalis to the other microorganisms, epithelial cells, and fibroblast [Citation113]. P. gingivalis is also known to ‘cheat’ its community as it retains some of the energy-consuming mechanisms that are discarded by other membranes of the community. This mechanism of retaining certain discarded features helps P. gingivalis to survive in times of crisis and support the growth and functioning of the entire microbial community [Citation15].
An important metabolite that contributes regulate the metabolic interactions of P. gingivalis with other accessory pathogens and prevents its attachment to the host is arginine. Studies have shown that arginine deaminase (ArcA) of Streptococcus cristatus and S. intermedius impairs biofilm formation by inhibiting the production of FimA surface proteins [Citation113–Citation116]. The down-regulation of FimA catalyzes the conversion of arginine to citrulline. Reduced levels of extracellular arginine and/or citrulline accumulation inhibits FimA expression and biofilm formation [Citation24,Citation117]. An in vitro study on a mouse model has even confirmed that colonization of ArcA-expressing S. cristatus followed by P. gingivalis infection decrease the colonization of P. gingivalis [Citation115]. Although the presence of ArcA from S. gordonii is yet to be confirmed, it is well characterized that S. gordonii ArcA has a well-developed arginine deaminase system (ADS), which catalyzes the intracellular conversion of arginine to ammonia and CO2, along with concomitant production of ATP [Citation117]. Therefore, the distinctive response of P. gingivalis to S. gordonii is due to its ability to transform arginine into citrulline in an extracellular manner. Therefore, targeting ArcA surface proteins could form a ‘potential anti-biofilm agent to fight P. gingivalis infections’ [Citation114–Citation118]. Arginine and its derivatives also affect the interaction of F. nucleatum to other microorganisms in the biofilm [Citation119]. F. nucleatum harbors an adhesin that is inhibited by arginine (RadD) [Citation117,Citation119]. Therefore high concentrations of arginine can inhibit cell-to-cell contact (i.e., coaggregation) between F. nucleatum and other bacterial species [Citation120]. Another, novel cross-feeding interaction between S. gordonii and F. nucleatum is that involving ornithine, which is exported by the arginine-ornithine antiporter ArcD in the ADS of S. gordonii, has been reported [Citation119]. In-vitro studies confirmed that ‘deletion of ArcD attenuates the accumulation of F. nucleatum in S. gordonii biofilm, while ornithine supplementation restored the bio-volume of F. nucleatum in mono-species and dual-species biofilms with the S. gordonii DarcD mutant’. This proves that ArcD-exported ornithine supports the growth of F. nucleatum and sustains the development of its biofilm. F. nucleatum is also known to increase the levels of ornithine decarboxylase (ODC), an enzyme responsible for the conversion of ornithine/arginine to putrescine, in the community biofilms formed with S. gordonii [Citation37]. F. nucleatum utilizes the ornithine released by S. gordonii ArcD as a substrate of ODC and enhance the overall community development by mediating cross-feeding of ornithine, and reinforcing coaggregation between S. gordonii and F. nucleatum. These findings suggest that sustained delivery of ornithine from accessory pathogens induce a state of dysbiosis, by sustaining the growth of the entire microbial community [Citation116]. Furthermore, S. gordonii produces a distinct pattern of protein in communities with F. nucleatum or P. gingivalis, especially with the ADS component enzymes ArcA, ArcB (catabolic ornithine carbamoyltransferase), ArcC (carbamate kinase), and ArcD has been observed [Citation61–Citation68,Citation115,Citation116,Citation118]. The levels of ArcA, ArcB, and ArcC of S. gordonii were reduced in community biofilms formed with P. gingivalis as compared to mono-species biofilms. On the other hand, the interaction of S. gordonii with F. nucleatum showed a marked increase in the levels of ArcA, ArcB, and ArcC despite a significant reduction in the level of ArcD in S. gordonii [Citation62–Citation65,Citation121].
2b. Persistence and survival in the host cells by facilitating tissue invasive and immune modulation strategies
Neutrophil dysfunction and increase cytokine production
Apart from inter-bacterial interaction, P. gingivalis survives and persist within the host by evading the immune response and promoting cellular invasion [Citation10–Citation16,Citation26,Citation27,Citation52,Citation121]. The type II fimbriae of P. gingivalis adheres to the α5β1-integrin in the gingival epithelium [Citation34,Citation52,Citation53,Citation121–Citation130] The fimbriae-integrin binding rearranges the actin cytoskeleton and exploits the cellular endocytosis to get internalized by encapsulation within an early endosome [Citation25–Citation27,Citation46]. Upon entry into the gingival epithelium, P. gingivalis accumulates around the nucleus and remains viable before disseminating into the other parts of the cells [Citation128–Citation130]. It rapidly locates itself at the most anoxic part of the cytoplasm and utilizes the cellular machinery for its growth and survival. It even remodels the cytoskeletal microtubule dynamics of the cell by interacting with the integrin-dependent signaling pathway and actin molecules. The α5β1-integrin on epithelial cells is correlated with the S phase of the cell cycle, and P. gingivalis persistence may be associated with the ability to preferentially target dividing cells [Citation47]. P. gingivalis is also associated with degradation of focal adhesion kinase and paxillinin the host cells, which could be linked with periodontal tissue degradation, poor wound healing, and regeneration in periodontitis [Citation57,Citation58]. The altered cytoskeleton disrupts the morphology and function of the actin molecule and allows further bacterial entry [Citation128,Citation131]. Once it exits from the invaded cells, it subsequently infects the ‘neighboring gingival cells’ that further invasion and persistent within the host [Citation128]. P. gingivalis also release a functionally versatile compound, known as Serine phosphatase protein (SerB), which can modify the function of the actin molecule [Citation131–Citation137]. SerB can dephosphorylate the actin-depolymerizing molecule ‘cofilin’, a major actin-binding protein that regulates nuclear translocation, mitosis, chemotaxis, and actin-mediated endocytosis ( & ). SerB even alters the functioning of the actin microfilament and stability of the microtubules [Citation96]. Therefore, SerB inactivated cofilin favors P. gingivalis invasion and promotes its survival within the cell [Citation138–Citation153].
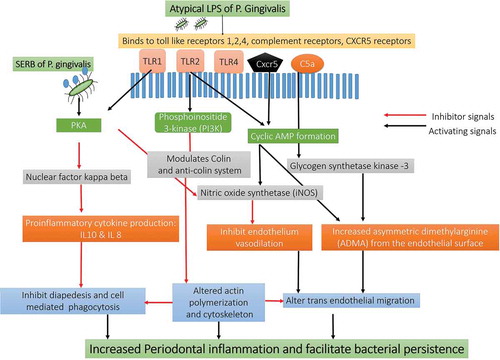
Figure 6. Schematic representation Immune response pathways triggered by the activation of TLR2/TLR4/CXCR5/C5aR receptors by gingipains P. gingivalis: The gingipains of P. gingivalis degrade the C5 and C3 from the complement system and degrade them C5a and C3a. The C5a interacts with the receptors on the neutrophils, epithelial cells, and endothelium in the host to Impairs phagocytosis and increase proinflammatory cytokine. C3a also inhibits the caspase 11–dependent non-canonical inflammasome pathway and prevents the apoptosis of the cell and allows P. gingivalis to use the host cell for its growth. The C5aR-TLR2 cross-talk activated by P. gingivalis pili induced degradation of MyD88 in neutrophils. In the absence of MyD88, the co-association of C5aR-TLR2 promotes P. gingivalis infection v activation of the TIRAP-dependent PI3 K signaling pathway. This, in turn, causes an inflammatory cytokine TNF-α along with inhibition of RhoA activation and actin polymerization that impairs the process of the maturation of phagosomes and P. gingivalis phagocytosis [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; CXCR4: C-X-C chemokine receptor type 4; cAMP: cyclic adenosine monophosphate; iNOS: inducible nitric oxide synthase; Mal: MyD88 adapter-like; p38MAPK: mitogen-activated protein kinase p38; PKA: protein kinase A; PI3 K: phosphoinositide-3-kinase; RhoA: Ras homolog gene family, member A; sTREM-1 – sTREM-1 – salivary Triggering Receptor Expressed on Myeloid cells 1ʹ Protease-activated receptors [PAR]-2].
![Figure 6. Schematic representation Immune response pathways triggered by the activation of TLR2/TLR4/CXCR5/C5aR receptors by gingipains P. gingivalis: The gingipains of P. gingivalis degrade the C5 and C3 from the complement system and degrade them C5a and C3a. The C5a interacts with the receptors on the neutrophils, epithelial cells, and endothelium in the host to Impairs phagocytosis and increase proinflammatory cytokine. C3a also inhibits the caspase 11–dependent non-canonical inflammasome pathway and prevents the apoptosis of the cell and allows P. gingivalis to use the host cell for its growth. The C5aR-TLR2 cross-talk activated by P. gingivalis pili induced degradation of MyD88 in neutrophils. In the absence of MyD88, the co-association of C5aR-TLR2 promotes P. gingivalis infection v activation of the TIRAP-dependent PI3 K signaling pathway. This, in turn, causes an inflammatory cytokine TNF-α along with inhibition of RhoA activation and actin polymerization that impairs the process of the maturation of phagosomes and P. gingivalis phagocytosis [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; CXCR4: C-X-C chemokine receptor type 4; cAMP: cyclic adenosine monophosphate; iNOS: inducible nitric oxide synthase; Mal: MyD88 adapter-like; p38MAPK: mitogen-activated protein kinase p38; PKA: protein kinase A; PI3 K: phosphoinositide-3-kinase; RhoA: Ras homolog gene family, member A; sTREM-1 – sTREM-1 – salivary Triggering Receptor Expressed on Myeloid cells 1ʹ Protease-activated receptors [PAR]-2].](/cms/asset/a45cf5e1-ceca-4546-b260-32a3f2c4ac3b/zjom_a_1801090_f0006_oc.jpg)
Figure 7. Schematic presentation of the immunological pathways triggered by LPS of P.gingivalis.
Apart from using the host cell to gain entry, inactivation of cofilin by SerB impairs the process of phagocytosis, chemotaxis, and transendothelial migration of neutrophils [Citation134–Citation144]. It is also known to upregulate P-21 Activated Kinase (PAK) and Rho-Associated-Kinase (ROCK) receptors that indirectly increase pro-inflammatory cytokines production in the host [Citation90–Citation92]. SerB dephosphorylates the serine residues of the Nuclear factor-kappa Beta (NF-κB) and inhibits CXCL8 induced IL8 production [Citation15,Citation93–Citation98,Citation154-Citation158]. Reduced IL8 levels impair the process of phagocytosis and allow P. gingivalis to use the epithelial cells as an autophagosome for its growth and survival [Citation96,Citation149–Citation157]. The SerB even remains functionally viable within the cells by interacting with other bacteria proteins. For example, SerB of P. gingivalis binds to the Heat Shock Protein (HSP) and GAPDH (glyceraldehyde 3 phosphate dehydrogenase) of Streptococcus oralis within the periodontal tissues to increase the inflammatory response [Citation80,Citation83,Citation100] P. gingivalis also inhibit, TLRs, E-selectin and intercellular adhesion molecular (ICAM) expression on the endothelial cell surface and binds to the non-chemotactic methionyl peptides receptor to inhibits the neutrophil chemotactic gradient, leukocyte adhesion, and recruitment of neutrophil to the site of inflammation [Citation150–Citation160]. The reduced chemotaxis decreases the rate of phagocytosis and facilitates the growth of the entire microbial community. The activation of TLR2 is required for bacterial persistence and its deficiency has been linked with inhibition of alveolar bone resorption [Citation161]. Apart from SerB, lipopolysaccharide (LPS), FimA fimbriae, Lipoteichoic acid of P. gingivalis modulate the immunoinflammatory response by inhibiting the production of antimicrobial molecules, complement system, and recruitment of leukocytes and favoring colonization of microorganisms in the periodontal tissues [Citation95–Citation98,Citation100,Citation151,Citation155,Citation160–Citation162] P. gingivalis release of various antimicrobial peptides (βeta defensins 2), activate the coagulation cascade and the kallikrein/kinin cascade to increase the inflammation in the host [Citation150,Citation160].
The LPS of P. gingivalis (4 acyl mono-phosphorylated lipid A moiety) with its two isoforms (Penta-acylated LPS 1690 and Tetra-acylated LPS 1435/1449 is even responsible for the dual inflammatory response by modulating the TLR4 in the periodontal tissues [Citation23,Citation50,Citation150,Citation155–Citation163]. P. gingivalis LPS shifts its Penta-acylated 1690 isoform to tetraacylated 1435/1449 according to the changes in the environmental condition to survive and maintain its growth [Citation158,Citation159]. Moreover, the shift of LPS moiety disrupts the activity of the LPS binding protein (LBP) and downregulates LBP induced IL8 and IL6 production [Citation157,Citation164]. The shift of LPS moiety even modulates the complement system, subvert leukocytes phagocytosis, activate protease production, enhance virulence factors expression, and induce cytotoxicity [Citation158–Citation169]. Furthermore, the binding of LBP to the TLR4 receptor by using a membrane-bound or soluble cluster of differentiation molecule (CD14), triggers the release of various pro-inflammatory and anti-inflammatory mediators in the periodontal tissues [Citation150]. The LPS of P. gingivalis even alters the functioning of cementoblast and osteoclast cells by modulating the RANKL/Osteoprotegerin (OPG) receptors. The LPS induced activation of RANKL receptors is associated with resorption and destruction of alveolar bone destruction [Citation165,Citation169,Citation170].
P. gingivalis LPS also activates the TLR 2-CXC chemokine receptor 4 (CXCR4) and Complement receptors (C5aR) on the neutrophil surface [Citation150–Citation153,Citation170]. The Arg-specific gingipains (RgpB and HRgpA) co-activate and mediate the cross-talk between complement receptor (C5aR), TLR2, and CXCR4 [Citation157]. TLR2-C5aR activations increase the ubiquitin via E3 ubiquitin ligase Smurf1 dependent proteasomal degradation and ubiquitination of Myeloid differentiation primary response gene 88 (MyD 88). The inhibition of MyD 88 receptor blocks the signals from the TLR2-PI3 K pathway and activate IL1 receptors with increased production of IL 1 [Citation95,Citation100,Citation110,Citation156–Citation159]. The crosstalk between C5aR with TLR2 activates the PI3 K signaling pathway and indirectly increases the inflammatory response [Citation36,Citation126,Citation153]. The PI3 K activation prevents phagocytosis, inhibits RhoA activation, and actin polymerization. The inhibition of TLR2-PI3 K pathway even impairs the process of phagocytosis by inhibiting the RhoA GTPase dependent actin polymerization [Citation95–Citation98,Citation100-Citation102,Citation161–Citation163]. The inhibition of LR2/MyD88 signaling causes the death of infected neutrophils and blocks the process of phagocytosis in the host [Citation29,Citation96]. P. gingivalis can also inhibit all the components of the complement system (classical, alternative and lectin pathways) and degrades the byproducts of the complement system [Citation139,Citation163] P. gingivalis inhibits the Mannose-Binding Lectin (MBL), Ficolins (FCN), C3, C3b, and C4 components of the complement system and prevent the formation of the ‘Membrane Attack Complex’ (MAC) [Citation161,Citation163–Citation168] The inhibition of MAC and other components of the complement system favors P. gingivalis to evade the complement-mediated phagocytosis and survive within the host [Citation139–Citation143]. Moreover, P. gingivalis utilizes its gingipain to entrap the circulating C4b-binding protein and defend itself from being phagocytized by the molecules of the complement system [Citation109,Citation141]. The Arg specific gingipains even possess C5 convertase like properties that increase the concentration of C5a in the gingival crevicular fluid [Citation108]. The C5a binds to the C5a receptor (C5aR) on the leukocytes and impair leukocyte killing capacity [Citation151,Citation170]. The cross-talk between upon P. gingivalis with TLR2-CXCR4 receptors causes a sustained release of cAMP [Citation171]. The increase in cAMP production weakens the macrophages induced nitric oxide synthase [iNOS] dependent phagocytosis and facilitate the growth of the entire microbial community () [Citation20,Citation114,Citation126]. The increased c-AMP production along with a decrease in the IL10 levels reduces the production of nitric oxide (NO) from the endothelial cells. P. gingivalis also evade the nitric oxide synthetase expression by modifying the structure of the Lipid A moiety of its LPS that indirectly inhibit TLR4 activation [Citation15,Citation21]. Additionally, RgpB and HRgpA gingipains of P. gingivalis along with karilysin enzyme secreted from Tannerella forsythia and InpA molecules from P. intermedia activate the release of C5a from C5 and degrade the central component C3 and Immunoglobulin G [Citation172]. This synergistic interaction of gingipains with Interpain A and karilysin weakens the host immune response and allows persistence of the microbe within the host [Citation90].
The LPS of P. gingivalis also increases the production of thrombospondin-1 (TSP), an extracellular matrix protein secreted by monocytic cells in the periodontal tissues. TSP stimulates the movement of the macrophages and increases the macrophage-mediated inflammation in the periodontal tissues [Citation173]. Additionally, P. gingivalis increases the production of IL17 to synergistically enhance the production of TSP1 and plasminogen activator inhibitor type I in the gingival and periodontal tissues. The increased production of TSP1 and plasminogen activator inhibitor type I contribute to the persistence of the microorganisms within the host that leads to the progression and chronicity of periodontal disease [Citation174,Citation175]. P. gingivalis persistence within the gingival epithelial cells is known to induce the production of several microRNAs, such as miR-105 and miR-203 and short-chain fatty acids. The miR-105 and miR-203 can suppress TLR2 function and inhibit the release of Suppressor of Cytokine Signaling 3 (SOCS3) and SOCS6 respectively [Citation148,Citation149]. The long-term cohabitation of P. gingivalis within the host cell favors the establishment of an ‘inter-kingdom’ whereby it impairs the important defense mechanisms of the host () [Citation171,Citation176,Citation177].
Recently, P. gingivalis is found to increase the production of butyrate and propionate and decrease the release of cytokine-induced neutrophil chemoattractant (CINC) 2αβ, another powerful chemoattractant and inhibitor of chemokines production [Citation132,Citation175]. The outer membrane components of P. gingivalis is proven to possess ‘porin-like activity’ that can depolarize the electrochemical potential on the neutrophil membrane and prevents its migration in response to the chemotactic stimuli [Citation132,Citation171]. A study done by Chen et al. (2011) showed that the outer membrane protein (PG0027) is ‘essential for the O-deacylation of LPS, secretion of gingipain to the cell surface, and attachment of P. gingivalis’ to host cells [Citation171]. The hemaagglutinins and gingipains released from P. gingivalis help to create a nutrient-rich but oxygen-deficient environment that protects P. gingivalis from the immune response of the host and promotes its survival [Citation22,Citation113]. P. gingivalis along with T. denticola and T. forsythia degrade and inactivate various antimicrobial molecules and enzymes secreted by the neutrophils to survive in the inflammatory milieu [Citation177–Citation180]. Recently, the gingipains from P. gingivalis are found to degrade the salivary Triggering Receptor Expressed on Myeloid cells 1 (sTREM-1) on the neutrophils and impede the process of phagocytosis and chemotaxis (). P. gingivalis utilize its Arg-gingipain and sTREM1 receptors to create an inflammation rich environment for its acquiring nutrients. However, when the inflammation starts to compromise its existence, P. gingivalis changes from its Arg-gingipain to Lys-gingipain and attenuates the periodontal inflammation. This ‘twin regulation’ of sTREM 1 by Lys and Arg gingipains is a unique mechanism adopted by P. gingivalis for its survival and persistence [Citation179–Citation183]. Nylund et al., (2017) evaluated the association of the Salivary TREM-1 (sTREM- 1)/its Ligand Peptidoglycan Recognition Protein 1 (PGLYRP1) in periodontitis patients with renal diseases and concluded that PGLYRP1 and sTREM-1 are strongly associated with increased proinflammatory cytokine production in patients with periodontitis [Citation183]. Patients with deep and active periodontal pockets have more salivary sTREM-1 and PGLYRP1 concentration as compared to individuals with shallow probing depth. Further research has shown that P. gingivalis modulates sTREM-1 receptor by acting as a decoy receptor for TLR activation and inhibiting neutrophil migration. The simultaneous activation of TLR4 and sTREM1 synergistically increase the production of TNF αlpha, IL1β, and decreased the production of anti-inflammatory cytokines like IL10. The TLR-sTREM1 activation is also known to activate various receptors associated with increased production of proinflammatory cytokines such as IRAK-1 (IL1 R-associated kinases), MAPK, p38 MAPK, Jun N-terminal Kinase (JNK), PI3 K, ERK1/2, NF-kB ( & Figure 7) [Citation183]. TREM-1 molecule is known to activate nucleotide-binding oligomerization receptors (NOD1 and NOD2) and indirectly increase the activation of caspase and NF-kB receptors that can exaggerate the production of proinflammatory cytokine in an autocrine manner [Citation184].
Inhibition of apoptosis increased oxidative stress and activation of the inflammasome
P. gingivalis is also known to inhibit the apoptotic pathways of the infected host cell and utilizes the cellular machinery for its survival. The inhibition of apoptosis contributes to the chronicity of the periodontal disease by promoting the growth of other bacterial species () [Citation17,Citation91]. The LPS of P. gingivalis activates caspase 11 dependent non-canonical inflammasome and initiates the process of pyroptosis and lytic cell death within the macrophages [Citation135,Citation184–Citation188]. P. gingivalis LPS, specifically the O-antigen region and HmuY is known to affect the ‘viability and apoptosis of gingival epithelial cells’ [Citation54,Citation62,Citation155,Citation187–Citation190]. The HmuY protein of P. gingivalis can induce apoptosis in the gingival epithelial cells by increasing FAS Ligand expression and NFκB receptor activation. The Fas ligand, also referred to as CD95 L or CD178, is a type-II transmembrane protein belonging to the TNF family, which initiates the process of apoptosis. A study by Meghill et al., 2019 elucidates the underlying mechanisms by which P. gingivalis manipulates dendritic cell signaling to perturb both autophagy and apoptosis. The results of their study showed that the minor (Mfa1) fimbriae of P. gingivalis induce Akt nuclear localization and activate the Akt/mTOR axis required for autophagosome formation and maturation [Citation190]. P. gingivalis also increase the mitotic cell cycle and suppress apoptosis by inhibiting Janus A Kinase (JAK), Phosphoinositide 3 Kinase (PI3 K), Signal Transducer of Activation (STAT), alpha-serine/threonine-protein kinase (Akt) and purinoceptor (P2X7) pathways () [Citation91] Furthermore, P. gingivalis reduce the renewal capacity of cells and inhibit apoptosis by activating p38, mitogen-activated protein kinase (MAPK), and extracellular-signal-regulated kinase (Erk1/2) pathways [Citation184–Citation186] The activation of p38, MAPK, and Erk1/2 pathways decrease the expression of cyclin D and inhibit cellular proliferation by arresting the cell cycle at the G1 phase [Citation72,Citation73]. Studies have confirmed the deregulation of apoptosis-related genes, such as Bax, Bcl2, Nlrp3, or Smad2, in the gingival tissues of patients with periodontitis [Citation181,Citation189]
P. gingivalis has also confirmed to possess cell-specific modulation of apoptosis receptors and signaling pathways such as Apoptotic protease activating factor (APAF 1), B-cell lymphoma Associated X (Bax1) and Caspase and reduce B-cell lymphoma (BCL 2) in the epithelial cells and fibroblasts [Citation184]. In the epithelial cells, P. gingivalis blocks the epithelial cell death by decreasing the APAF-1 expression, reducing the enzymatic activity of caspase enzyme, and increasing the expression of X-linked inhibitor of apoptosis protein (XIAP). However, in the fibroblast, P. gingivalis stimulates the APAF-1 pathway and reduces XIAP expression, increases caspase expression and apoptosis. The oligomerization of APAF1, induced by its binding to cytochrome C, helps apoptosome formation, a structure that recruits and activates a caspase initiator, caspase 9. The activation of caspase 9 cleaves and activates caspase effector caspase 3 and caspase 7, leading to apoptosis [Citation184]. The increased caspase expression following P. gingivalis infection increases the process of pyroptosis, hypoxia, and exaggerate the inflammatory burden in the periodontal tissues and systemic circulation [Citation190–Citation195].
P. gingivalis can even inhibit F. nucleatum induced activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome and modulates the NLRP3 inflammasome cytokine secretion from the macrophages [Citation189]. NLRP3 inflammasome is associated with the release of cytokines (IL1b and IL18), apoptosis, phagocytosis, and pyroptosis. NLRP3 regulates the process of apoptosis in the osteoblasts cells infected with P. gingivalis and A. actinomycetemcomitans [Citation188]. Additionally, P. gingivalis interacts with the CR3 receptors on the macrophages and decreases the IL12p70 induced bacterial clearance by the macrophages [Citation190–Citation193]. The enzyme sialidase in P. gingivalis has been lately discovered as novel mechanisms to inhibit macrophage activity. Studies have shown that sialidase-deficiency in P. gingivalis can attenuate CR3 activation in macrophages, reduce inhibition of lncRNA GAS5, with less miR-21 and more IL12 production. Inhibition of sialidase in P. gingivalis would render P. gingivalis more easily cleared by macrophages [Citation193]. The compromised macrophage function impairs the process of antigen presentation, lymph node activation, and delays the onset of cellular immunity in the host [Citation189].
Additionally, P. gingivalis increases oxidative stress and extracellular ATP production by activating the pannexin-1 receptors (P2X4, P2X7) and NADPH-oxidase family members (NOX 4) [Citation194–Citation199]. NOX4 is a transmembranous located enzyme in the endothelial cells, fibroblasts, keratinocytes, and osteoclasts of the periodontium [Citation197–Citation199]. The ATP stimulation through ligation of P2X7 receptors synergistically increases the production of NADPH oxidase and exaggerate the mitochondrial ROS production in the periodontal tissues [Citation147].
Studies have even confirmed that P. gingivalis infection increases the production of 8 hydroxy 2ʹ deoxyguanosine, superoxide anions, matrix metalloproteinases, caspase-3, catalase enzyme in the host cells [Citation192,Citation197]. P. gingivalis protect itself against the extracellular ATP and oxidative stress by releasing a unique enzyme known as nucleotide-diphosphate-kinase (NDK) () [Citation147]. NDK cleaves the extracellular ATP molecules and down-regulates the P2X7/pannexin 1 receptor that is activated by autocrine action of ATP, within the host [Citation200]. The NDK from P. gingivalis accumulates in the cytoplasm and is subsequently carried along myosin-9 filaments and actin filaments to the host cell periphery [Citation150]. Upon translocation to the extracellular environment through the formation of the P2X7/pannexin 1 channel, NDK hydrolyzes ATP, and decrease the ATP-induced IL-1β release [Citation92,Citation150,Citation152]. Additionally, the NDK inhibits ATP and increase the reactive oxygen species (ROS) production that helps in bacterial persistence [Citation150–Citation152,Citation201-Citation203]
Furthermore, it has been observed that even though P. gingivalis increase the oxidative stress in the host, it remains unaffected by the ROS due to the release of various endogenous antioxidants, like thiol peroxidase, glutathione, superoxide dismutase, rubrerythrin, in and around its cell surface [Citation18,Citation147,Citation180–Citation183,Citation200,Citation204,Citation205]. The endogenously produced antioxidants protect P. gingivalis from the ROS mediated injury and allow it to survive within the host. The increase in the levels of glutathione suppresses the mitochondrial-mediated intrinsic cell death and protects the bacteria against the reactive oxygen species (ROS) that help P. gingivalis to survive within the host [Citation18,Citation147]. P. gingivalis selectively inhibits the macrophage-mediated cytokine production without affecting the production of T cell-mediated chemokines [Citation206] The selective inhibition of the inflammatory process allows it to acquire the essential nutrient derived from the dead bacteria and tissue breakdown products for its growth and provide a competitive advantage to P. gingivalis during the inflammatory process [Citation18,Citation20,Citation119,Citation126,Citation188].
Alteration of T cell function with activation of Th17 cell
Another important mechanism by which P. gingivalis impairs the host immune response is by altering the T cell function and activation of Th17 mediated cytokine production [Citation201–Citation203,Citation205-Citation208]. It has been observed that P. gingivalis can induce TLR2-dependent IL-10 production that ‘leads to inhibition of IFN-γ production by CD4+ and CD8 + T cells [Citation209]. P. gingivalis down-regulates IL12 secretion and increases the release of Interferon-alpha (IFN-alpha) from the CD4 Th1 cells. The decreased levels of IL12 and increase the production of IFN-alpha shifts the immune response from Th1 to Th2 [Citation14]. The outer membrane proteins of P. gingivalis is found to activate the Th1 cells that in turn increase the production of proinflammatory cytokines [Citation210]
Furthermore, P. gingivalis interacts with the dendritic cells of the host and favors the production of Th17 related cytokines such as TNFα, IL8, CXCL8, IL17, IL1β, IL6, IL23, IL12p40, IL21, IL3, granulocyte colony-stimulating factor (GCSF), etc. from both immune and non-immune cells of the host [Citation147–Citation149,Citation202,Citation203,Citation206–Citation210]. The increased production of IL1, IL 6, IL 21, IL23, TGF βeta amplifies and stabilizes the Th17 cell differentiation and favors the onset of dysbiosis and periodontal inflammation. The activation of Th17 cells increases the levels of IL1β, IL6, and TNFα in the periodontal tissues, which is associated with increase bone and soft tissue destruction in the periodontal tissues [Citation210]. IL17 activates the RANKL receptor on the osteoblasts and fibroblasts and induce bone resorption by expressing the ‘osteoclast activating factor’ on the alveolar bone [Citation210]. Furthermore, the arg-gingipain from P. gingivalis is known to modulate and increase the production of proinflammatory cytokines by acting on novel receptors sites like protease-activated receptors [PAR]-2 and soluble triggering receptor expressed on myeloid cells [sTREM-1] [Citation211]. The activation of PAR-1, PAR-2, and PAR-4 induce CD69 and CD25 expression in CD4+ T cells that in turn increase the production of IL17 [Citation211–Citation215]. P. gingivalis also interacts with the NF-κB and RAR-related orphan receptor (ROR) transcription factor on the alveolar bone and increases the production of IL17 mediated bone resorption [Citation213].
2 c. ‘Fratricide and Programmed Cell Death’ of other bacterial species
P. gingivalis facilitates ‘Fratricide and Programmed Cell Death’ of other bacterial species within the biofilm to increase the inflammatory response and promotes its survival [Citation60,Citation216] Fratricide is the pathogenic process by which P. gingivalis kills and obtain DNAs fragments from noncompeting host cells and microbes by inducing their death [Citation216,Citation218–Citation221] During the process of programmed cell death, P. gingivalis promotes a fraction of the microbial population to perform a self-sacrificing ‘suicide’ and discharge its nutrients and extracellular DNA fragments into the environment. The discharged nutrients and extracellular DNA fragments are utilized by P. gingivalis and other members of the microbial community for their growth. The process of ‘Fratricide and Programmed Cell Death’ is tightly regulated by a pattern of genes along with the presence of an intricate toxin and antitoxin system [Citation216]. For example, when P. gingivalis strains are added to the biofilm containing Streptococcus. mitis and Staphylococcus aureus, cell death and altruistic suicide of microbial colonies take place [Citation60] Some of the essential molecules that are linked with Fratricide and Programmed Cell Death include Choline Binding protein D (CbpD), Competence-induced bacteriocins (CibAB), Autolysin-Encoding Gene (LytA, LytC, LrgAB, LytR, and LytSR), CidABC operon, etc [Citation220]. When P. gingivalis is added to biofilms containing S. mitis, apoptosis-like death with release of DNA fragments and production of transposase that marks the onset of programmed cell death have been observed [Citation60]. P. gingivalis is even known to activate the bacterial apoptosis endonuclease (BapE) enzyme that can fragment the chromosomes by sequentially cleaving the supercoiled DNA. The DNA damage and induced chromosome fragmentation activate the process of apoptosis in the entire microbial community and intensify the process of dysbiosis and inflammation in the host [Citation166–Citation168].
Conclusion
P. gingivalis is a potential and highly virulent periodontal pathogen that adopts intricate molecular mechanisms to interact with other members of the microbial community and host to pave its path of invasion, survival, and persistence. It adopts unique molecular and intricate mechanisms to survive and persist within the tissues and exaggerate inflammation and dysbiosis. P. gingivalis modulated the entire oral microbiome and creates an environment that not only favors its growth and survival but also facilitates the growth of many other commensal and pathobionts species. The interbacterial interactions, immune evasion, and tissue invasive properties help P. gingivalis to survive even in adverse environmental conditions and reach a distant organ system. However further research on how P. gingivalis invade the epithelial barrier of different organs and induce organ dysfunction is intriguing and needs more research. It is also necessary to explore if the altered immune response by P. gingivalis considerable enough to acquire other opportunistic infections and systemic diseases. More research is necessary at the genomic level to understand and discover the transcriptomes, nucleotide, and metabolites affected in the host by P. gingivalis with the host and other members of the microbial community upon invasion. It is also necessary to explore if these immunologic mechanisms form a link between P. gingivalis and the development of various cancers. Furthermore, it is also critical to explore if any of these cellular and molecular mechanisms are associated with the development of emerging antimicrobial resistance of periodontal pathogens to conventional antibiotics. There is a need to study the strain-specific variation of P. gingivalis on the modulation of host response and systemic inflammation. The paper is of paramount importance to researchers, oral biologists, microbiologists, immunologists, scientists, and pharmacologists across the globe to initiate further research and develop novel therapeutic modalities that can target the specific mechanism and prevent the onset of oral and systemic inflammation in the host
Acknowledgments
We are extremely grateful to Dr. Ashmeet Shah for helping us with the reference section and proofreading of the paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Darveau RP, Tanner A. Page RC The microbial challenge in periodontitis. Periodontol. 2000;4:12–22.
- Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol. 2000;14:9–11.
- Kolenbrander PE, Palmer RJ Jr, Rickard AH, et al. Bacterial interactions and successions during plaque development. Periodontol. 2000;42:47–79.
- Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. 1994;8:263–271.
- Kumar PS, Griffen AL, Moeschberger ML, et al. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J Clin Microbiol. 2005;43:3944–3955.
- Kumar PS, Leys EJ, Bryk JM, et al. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16s cloning and sequencing. J Clin Microbiol. 2006;44:3665–3673.
- Ximenez-fyvie LA, Haffajee AD, Socransky SS. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol. 2000;27:648–657.
- Paster BJ, Boches SK, Galvin JL, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783.
- Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol. 2000;2005(38):72–122.
- Hajishengallis G, Liang S, Payne MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497506.
- Davic RD. Linking keystone species and functional groups: A new operational definition of the keystone species concept - Response. Conserv Ecol. 2003;7:r11.
- Hajishengallis G, Darveau RP, Curtis MA. The Keystone Pathogen Hypothesis. Nat Rev Microbiol. 2012;10:717–725.
- Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490.
- Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol. 2011;11:187–200.
- Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. 2015;21:172–183.
- Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res. 2012;91:816–820.
- Griffen AL, Becker MR, Lyons SR, et al. Prevalence of Porphyromonas gingivalis and periodontal health status. J Clin Microbiol. 1998;36(11):3239–3242.
- Hajishengallis G, Lamont RJ. Breaking bad: manipulation of the host response by Porphyromonas gingivalis. Eur J Immunol. 2014;44:328–338.
- Bostanci N, Belibasakis GN. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett. 2012;333:1–9.
- How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Frontiers Microbiol. 2016;7:53.
- Kuboniwa M, Lamont RJ. (2010) Subgingival biofilm formation. Periodontology. 2000;52:38–52.
- Zenobia C, Hajishengallis G. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence. 2015;6:236–243.
- Ogawa T, Yagi T. Bioactive mechanism of Porphyromonas gingivalis lipid A. Periodontol. 2000;2010:71–77.
- Yoshimura F, Murakami Y, Nishikawa K, et al. Surface components of Porphyromonas gingivalis. J Periodont Res. 2009;44:1–12.
- Frias-Lopez J, Duran-Pinedo A. Effect of periodontal pathogens on the metatranscriptome of a healthy multispecies biofilm model. J Bacteriol. 2012;194:2082–2095.
- Darveau RP, Belton CM, Reife RA, et al. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1999;66:1660–1665.
- Amano A. Disruption of epithelial barrier and impairment of cellular function by Porphyromonas gingivalis. Front Biosci. 2007;12:3965–3974.
- Amano A. Bacterial adhesins to host components in periodontitis. Periodontol. 2000;52:12–37.
- Baek KJ, Ji S, Kim YC, et al. Association of the invasion ability of Porphyromonas gingivalis with the severity of periodontitis. Virulence. 2015;6:274–281.
- Wright CJ, Burns LH, Jack AA, et al. Microbial interactions in building of communities. Mol Oral Microbiol. 2013;28:83–101.
- Jiao Y, Hasegawa M, Inohara N. The role of oral pathobionts in dysbiosis during periodontitis development. J Dent Res. 2014;93:539–546.
- Kolenbrander PE, Andersen RN, Blehert DS, et al. Communication among oral bacteria. Microbiol Mol Biol Rev. 2002;66:486–505.
- Aruni W, Chioma O, Fletcher HM. Filifactor alocis: the newly discovered kid on the block with special talents. J Dent Res. 2014;93:725–732.
- Jiao Y, Darzi Y, Tawaratsumida K, et al. Induction of bone loss by pathobiont-mediated NOD1 signaling in the oral cavity. Cell Host Microbe. 2013;13:595–601.
- Yilmaz O, Verbeke P, Lamont RJ, et al. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect Immun. 2006;74(1):703–710.
- Feuille F, Ebersole JL, Kesavalu L, et al. Mixed infection with Porphyromonas gingivalis and Fusobacterium nucleatum in a murine lesion model: potential synergistic effects on virulence. Infect Immun. 1996;64:2094–2100.
- Ebersole JL, Feuille F, Kesavalu L, et al. Host modulation of tissue destruction caused by periodontopathogens: effects on a mixed microbial infection composed of Porphyromonas gingivalis and Fusobacterium nucleatum. Microb Pathog. 1997;23:23–32.
- Schlafer S, Riep B, Griffen AL, et al. Filifactor alocis–involvement in periodontal biofilms. BMC Microbiol. 2010 Mar 1;10: 66. .
- Hasegawa M, Yang K, Hashimoto M, et al. Differential release and distribution of NOD1 and NOD2 immunostimulatory molecules among bacterial species and environments. J Biol Chem. 2006;281:29054–29063.
- Jorth P, Whiteley M. An evolutionary link between natural transformation and CRISPR adaptive immunity. mBio3. 2012;e00309–12. 10.1128/mBio.00309-12
- Cady KC, O’Toole GA. Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J Bacteriol. 2011;193:3433–3445.
- Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845.
- Watanabe T, Nozawa T, Aikawa C, et al. CRISPR regulation of interspecies diversification by limiting IS transposition and intercellular recombination. Genome Biol Evol. 2013;5:1099–1114.
- Patti JM, Allen BL, McGavin MJ, et al. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol. 1994;48:585–617.
- Makarova KS, Haft DH, Barrangou R, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9(6):467–477.
- Watanabe T, Shibasaki M, Maruyama F, et al. Investigation of potential targets of Porphyromonas CRISPRs among the genomes of Porphyromonas species. PLoS One. 2017 24;12(8):e0183752. eCollection 2017. .
- Phillips P, Progulske-Fox A, Grieshaber S, et al. Expression of Porphyromonas gingivalis small RNA in response to hemin availability identified using microarray and RNA-seq analysis. FEMS Microbiol Lett. 2014;351:202–208.
- Chen T, Olsen I. Porphyromonas gingivalis and its CRISPR-Cas system. Published 2019 J Oral Microbiol. 2019;111:1638196.
- Aruni AW, Zhang K, Dou Y, et al. Proteome analysis of coinfection of epithelial cells with Filifactor alocis and Porphyromonas gingivalis shows modulation of pathogen and host regulatory pathways. Infect Immun. 2014;82(8):3261–3274.
- Nelson KE, Fleischmann RD, DeBoy RT, et al. Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J Bacteriol. 2003;185:5591–5601.
- Slocum C, Coats SR, Hua N, et al. Distinct lipid A moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog. 2014;10:e1004215.
- Soto C, Bugueño I, Hoare A, et al. The Porphyromonas gingivalis O antigen is required for inhibition of apoptosis in gingival epithelial cells following bacterial infection. J Periodontal Res. 2016;51(4):518–528.
- Hajishengallis G, Harokopakis E. Porphyromonas gingivalis interactions with complement receptor 3 [CR3]: innate immunity or immune evasion? Front Biosci. 2007;12:4547–4557.
- Xie H, Cai S, Lamont RJ. Environmental regulation of fimbrial gene expression in Porphyromonas gingivalis. Infect Immun. 1997;65:2265–2271.
- Anaya-Bergman C, Rosato A, Lewis JP. Iron and hemin-dependent gene expression of Porphyromonas gingivalis. Mol Oral Microbiol. 2015;30:39–61.
- Al-Qutub MN, Braham PH, Karimi-Naser LM, et al. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun. 2006;74:4474–4485.
- Moon JH, Lee JH, Lee JY. Microarray analysis of the transcriptional responses of Porphyromonas gingivalis to polyphosphate. BMC Microbiol. 2014;14:218.
- Azelmat J, Larente JF, Grenier D. The anthraquinone rhein exhibits synergistic antibacterial activity in association with metronidazole or natural compounds and attenuates virulence gene expression in Porphyromonas gingivalis. Arch Oral Biol. 2015;60:342–346.
- Curtis MA, Percival RS, Devine D, et al. Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and interaction with the innate host defenses. Infect Immun. 2011a;79:1187–1193.
- James CE, Hasegawa Y, Park Y, et al. LuxS involvement in the regulation of genes coding for hemin and iron acquisition systems in Porphyromonas gingivalis. Infect Immun. 2006;74:3834–3844.
- Duran-Pinedo AE, Baker VD, Frias-Lopez J. The periodontal pathogen Porphyromonas gingivalis Induces expression of transposases and cell death of Streptococcus mitis in a biofilm model. Infect Immun. 2014;82:3374–3382.
- Kuboniwa M, Tribble GD, James CE, et al. Streptococcus gordonii utilizes several distinct gene functions to recruit Porphyromonas gingivalis into a mixed community. Mol Microbiol. 2006;60:121–139.
- Chawla A, Hirano T, Bainbridge BW, et al. Community signaling between Streptococcus gordonii and Porphyromonas gingivalis is controlled by the transcriptional regulator CdhR. Mol Microbiol. 2010;78:1510–1522.
- Park Y, James CE, Yoshimura F, et al. Expression of the short fimbriae of Porphyromonas gingivalis is regulated in oral bacterial consortia. FEMS Microbiol Lett. 2006;262:65–71.
- Park Y, Simionato MR, Sekiya K, et al. Short fimbriae of Porphyromonas gingivalis and their role in coadhesion with Streptococcus gordonii. Infect Immun. 2005;73:3983–3989.
- Simionato MR, Tucker CM, Kuboniwa M, et al. Porphyromonas gingivalis genes involved in community development with Streptococcus gordonii. Infect Immun. 2006;74:6419–6428.
- Zhang W, Ju J, Rigney T, et al. Fimbriae of Porphyromonas gingivalis are important for initial invasion of osteoblasts, but not for inhibition of their differentiation and mineralization. J Periodontol. 2011;82:909–916.
- Lamont RJ, El-Sabaeny A, Park Y, et al. Role of the Streptococcus gordonii SspB protein in the development of Porphyromonas gingivalis biofilms on streptococcal substrates. Microbiology. 2002 Jun;148(Pt 6):1627–1636.
- Daep CA, Novak EA, Lamont RJ, et al. Structural dissection and in vivo effectiveness of a peptide inhibitor of Porphyromonas gingivalis adherence to Streptococcus gordonii. Infect Immun. 2011;79:67–74.
- Maeda K, Nagata H, Yamamoto Y, et al. Glyceraldehyde-3-phosphate dehydrogenase of Streptococcus oralis functions as a coadhesin for Porphyromonas gingivalis major fimbriae. Infect Immun. 2004;72:1341–1348.
- Chung WO, Demuth DR, Lamont RJ. Identification of a Porphyromonas gingivalis receptor for the Streptococcus gordonii SspB protein. Infect Immun. 2000;68:6758–6762.
- Lamont RJ, Bevan CA, Gil S, et al. Involvement of Porphyromonas gingivalis fimbriae in adherence to Streptococcus gordonii. Oral Microbiol Immunol. 1993;8:272e276.
- Hamada N, Sojar HT, Cho MI, et al. Isolation and characterization of a minor fimbria from Porphyromonas gingivalis. Infect Immun. 1996;64:4788–4794.
- Brooks W, Demuth DR, Gil S, et al. Identification of a Streptococcus gordonii SspB domain that mediates adhesion to Porphyromonas gingivalis. Infect Immun. 1997;65:3753–3758.
- Demuth DR, Irvine DC, Costerton JW, et al. Discrete protein determinant directs the species-specific adherence of Porphyromonas gingivalis to oral streptococci. Infect Immun. 2001;69:5736–5741.
- Kuboniwa M, Lamont RJ. (2010) Subgingival biofilm formation. Periodontol. 2000;52:38–52.
- Cook GS, Costerton JW, Lamont RJ. Biofilm formation by Porphyromonas gingivalis and Streptococcus gordonii. J Periodontol Res. 1998;33:323–327.
- Kuboniwa M, Hendrickson EL, Xia Q, et al. of Porphyromonas gingivalis within a model oral microbial community. BMC Microbiol. 2009;9:98.
- Hendrickson EL, Wang T, Dickinson BC, et al. Proteomics of Streptococcus gordonii within a model developing oral microbial community. BMC Microbiol. 2012;12:211.
- Ohshima J, Wang Q, Fitzsimonds ZR, et al. Streptococcus gordonii programs epithelial cells to resist ZEB2 induction by Porphyromonas gingivalis. Proc Natl Acad Sci U S A. 2019;116(17):8544–8553.
- Wright CJ, Xue P, Hirano T, et al. Characterization of a bacterial tyrosine kinase in Porphyromonas gingivalis involved in polymicrobial synergy. Microbiol Open. 2014;3:383–394.
- Shizukuishi S, Lewis JP, Demuth DR, et al. A Porphyromonas gingivalis tyrosine phosphatase is a multifunctional regulator of virulence attributes. Mol Microbiol. 2008;69:1153–1164.
- Maeda K, Tribble GD, Tucker CM, et al. A Porphyromonas gingivalis tyrosine phosphatase is a multifunctional regulator of virulence attributes. Mol Microbiol. 2008;69:1153‐1164.
- Kishi M, Hasegawa Y, Nagano K, et al. Identification and characterization of novel glycoproteins involved in growth and biofilm formation by Porphyromonas gingivalis. Mol Oral Microbiol. 2012;27:458‐470.
- Park Y, Xie H, Lamont RJ. Transcriptional organization of the Porphyromonas gingivalis fimA locus. FEMS Microbiol Lett. 2007;273:103–108.
- Patankar AV, Gonzalez JE. Orphan LuxR regulators of quorum sensing. FEMS Microbiol Rev. 2009;33:739–756.
- James CE, Hasegawa Y, Park Y, et al. LuxS involvement in the regulation of genes coding for hemin and iron acquisition systems in Porphyromonas gingivalis. Infect Immun. 2006;74:3834–3844.
- Burgess NA, Kirke DF, Williams P, et al. LuxS-dependent quorum sensing in Porphyromonas gingivalis modulates protease and haemagglutinin activities but is not essential for virulence. Microbiol. 2002;148:763–772.
- Byrne DP, Potempa J, Olczak T, et al. Evidence of mutualism between two periodontal pathogens: co-operative haem acquisition by the HmuY haemophore of Porphyromonas gingivalis and the cysteine protease interpain A [InpA] of Prevotella intermedia. Mol Oral Microbiol. 2013;28:219–229.
- Mcnab R, Ford SK, El-Sabaeny A, et al. LuxS-based signaling in Streptococcus gordonii: autoinducer 2 controls carbohydrate metabolism and biofilm formation with Porphyromonas gingivalis. J Bacteriol. 2003;185:274–284.
- Sroka A, Sztukowska M, Potempa J, et al. Degradation of host heme proteins by lysine- and arginine-specific cysteine proteinases [Gingipains] of Porphyromonas gingivalis. J Bacteriol. 2001;183:5609–5616.
- Gao J, Kwan AH, Yammine A, et al. Structural properties of a haemophore facilitate targeted elimination of the pathogen Porphyromonas gingivalis. Nat Commun. 2018;9(4097). DOI:10.1038/s41467-018-06470-0
- Patankar AV, Gonzalez JE. Orphan LuxR regulators of quorum sensing. FEMS Microbiol Rev. 2009;33:739–756.
- Subramoni S, Venturi V. LuxR‐family ‘solos’: bachelor sensors/regulators of signalling molecules. Microbiology. 2009;155:1377–1385.
- Brown JL, Yates E, Bielecki M, et al. A potential role for Streptococcus gordonii-derived hydrogen peroxide in haem acquisition by Porphyromonas gingivalis. Mol Oral Microbiol. 2018;33(4):322–335.
- Yilmaz O, Jungas T, Verbeke P, et al. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to the survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751.
- Wu J, Lin X, Xie H. Regulation of hemin binding proteins by a novel transcriptional activator in Porphyromonas gingivalis. J Bacteriol. 2009;191:115–122.
- Capestany CA, Kuboniwa M, Jung IY, et al. Role of the Porphyromonas gingivalis InlJ protein in homotypic and heterotypic biofilm development. Infect Immun. 2006;74:3002–3005.
- Holt SC, Kesavalu L, Walker S, et al. Virulence factors of Porphyromonas gingivalis. Periodontol. 2000;1999:168–238.
- Chen W, Honma K, Sharma A, et al. A universal stress protein of Porphyromonas gingivalis is involved in stress responses and biofilm formation. FEMS Microbiol Lett. 2006;264:15–21.
- Lin X, Wu J, Xie H. Porphyromonas gingivalis minor fimbriae are required for cell–cell interactions. Infect Immun. 2006;74:6011–6015.
- Nakao R, Senpuku H, Watanabe H. Porphyromonas gingivalis galE is involved in lipopolysaccharide O‐antigen synthesis and biofilm formation. Infect Immun. 2006;74:6145–6153.
- Capestany CA, Tribble GD, Maeda K, et al. Role of the Clp system in stress tolerance, biofilm formation, and intracellular invasion in Porphyromonas gingivalis. J Bacteriol. 2008;190:1436–1446.
- Li Y, Krishnan K, Duncan MJ. Post‐translational regulation of a Porphyromonas gingivalis regulator. J Oral Microbiol. 2018;10:1487743.
- Krishnan K, Duncan MJ. Role of sodium in the RprY‐dependent stress response in Porphyromonas gingivalis. PLoS ONE. 2013;8:e63180.
- Potempa M, Potempa J, Kantyka T, et al. a cysteine proteinase from Prevotella intermedia, inhibits complement by degrading complement factor C3. PLoS Pathog. 2009;5:e1000316.
- Stanley NR, Lazazzera BA. Environmental signals and regulatory pathways that influence biofilm formation. Mol Microbiol. 2004;52:917–924.
- Grenier D. Porphyromonas gingivalis outer membrane vesicles mediate coaggregation and piggybacking of Treponema denticola and Lachnoanaerobaculum saburreum. Int J Dent. 2013;2013:305476.
- Grenier D. Nutritional interactions between two suspected periodontopathogens, Treponema denticola and Porphyromonas gingivalis. Infect Immun. 1992;60:5298–5301.
- Tan KH, Seers CA, Dashper SG, et al. Porphyromonas gingivalis and Treponema denticola exhibit metabolic symbioses. PLoS Pathog. 2014;10:e1003955.
- Meuric V, Martin B, Guyodo H, et al. Treponema denticola improves adhesive capacities of Porphyromonas gingivalis. Mol Oral Microbiol. 2013;28:40–53.
- Potempa J, Sroka A, Imamura T, et al. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis structure, function, and assembly of multidomain protein complexes. Curr Protein Pept Sci. 2003;4:397–407.
- Xie H, Hong J, Sharma A, et al. Streptococcus cristatus ArcA interferes with Porphyromonas gingivalis pathogenicity in mice. J Perio Res. 2012;47:578–583.
- Wang BY, Wu J, Lamont RJ, et al. Negative correlation of distributions of Streptococcus cristatus and Porphyromonas gingivalis in subgingival plaque. J Clin Microbiol. 2009;47:3902–3906.
- Lin X, Lamont RJ, Wu J, et al. Role of differential expression of streptococcal arginine deiminase in inhibition of fimA expression in Porphyromonas gingivalis. J Bacteriol. 2008;190:4367–4371.
- Butler CA1, Veith PD1, Nieto MF1, et al. Lysine acetylation is a common post-translational modification of key metabolic pathway enzymes of the anaerobe Porphyromonas gingivalis. J Proteomics. 2015 Oct 14;128:352–364.
- Sakanaka A, Kuboniwa M, Takeuchi H, et al. Arginine-Ornithine Antiporter ArcD Controls Arginine Metabolism and Interspecies Biofilm Development of Streptococcus gordonii. J Biol Chem. 2015;290(35):21185–21198.
- Dong Y, Chen YY, Burne RA. Control of expression of the arginine deiminase operon of Streptococcus gordonii by CcpA and Flp. J Bacteriol. 2004;186(8):2511–2514.
- Kaplan A, Kaplan CW, He X, et al. Characterization of aid1, a novel gene involved in Fusobacterium nucleatum interspecies interactions. Microb Ecol. 2014;68(2):379.
- Nascimento MM, Browngardt C, Xiaohui X, et al. The effect of arginine on oral biofilm communities. Mol Oral Microbiol. 2014;29:45–87.
- Saito A, Inagaki S, Kimizuka R, et al. Fusobacterium nucleatum enhances invasion of human gingival epithelial and aortic endothelial cells by Porphyromonas gingivalis. FEMS Immunol Med Microbiol. 2008;54(3):349–355.
- Kuboniwa M, Hasegawa Y, Mao S, et al. P. Microbes Infect. 2008;10:122–128.
- Mao S, Park Y, Hasegawa Y, et al. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 2007;9:1997–2007.
- Tribble GD, Lamont RJ. Bacterial invasion of epithelial cells and spreading in periodontal tissue. Periodontol. 2000;52:68–83.
- Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4:305–314.
- Jia L, Han N, Du J, et al. Pathogenesis of Important Virulence Factors of Porphyromonas gingivalis via Toll-Like Receptors. Front Cell Infect Microbiol. 2019;9:262. Published 2019 Jul 18.
- Belton CM, Izutsu KT, Goodwin PC, et al. Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol. 1999;1:215–223.
- Yilmaz O, Young PA, Lamont RJ, et al. Gingival epithelial cell signaling and cytoskeletal responses to Porphyromonas gingivalis invasion. Microbiol. 2003;149:2417–2426.
- Takeuchi H, Furuta N, Morisaki I, et al. Exit of intracellular Porphyromonas gingivalis from gingival epithelial cells is mediated by endocytic recycling pathway. Cell Microbiol. 2011;13:677–691.
- Lamont RJ, Chan A, Belton CM, et al. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63:3878–3885.
- Hasegawa Y, Tribble GD, Baker HV, et al. Role of Porphyromonas gingivalis SerB in gingival epithelial cell cytoskeletal remodeling and cytokine production. Infect Immun. 2008;76:2420–2427.
- Olsen I, Hajishengallis G. Major neutrophil functions subverted by Porphyromonas gingivalis. J Oral Microbiol. 2016;8. DOI:10.3402/jom.v8.30936.
- Zaric S, Shelburne C, Darveau R, et al. Impaired immune tolerance to Porphyromonas gingivalis lipopolysaccharide promotes neutrophil migration and decreased apoptosis. Infect Immun. 2010;78:4151–4156.
- Gibson FC, Ukai T, Genco C. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Front Biosci. 2008;13:2041–2059.
- Hayashi C, Gudino CY, Gibson FC, et al. Review: pathogen-induced inflammation at sites distant from oral infection: bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Mol Oral Microbiol. 2010;25:305–316.
- Hajishengallis G, Harokopakis E. Porphyromonas gingivalis interactions with complement receptor 3 [CR3]: innate immunity or immune evasion? Front Biosci. 2007;12:4547–4557.
- Moffatt CE, Inaba H, Hirano T, et al. Porphyromonas gingivalis SerB-mediated dephosphorylation of host cell cofilin modulates invasion efficiency. Cell Microbiol. 2012;14:577–588.
- Tribble GD, Mao S, James CE, et al. A Porphyromonas gingivalis haloacid dehalogenase family phosphatase interacts with human phosphoproteins and is important for invasion. Proc Natl Acad Sci U S A. 2006;103:11027–11032.
- Bainbridge B, Verma RK, Eastman C, et al. Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect Immun. 2010;78:4560–4569.
- Nishita M, Aizawa H, Mizuno K. Stromal cell-derived factor 1α activates LIM kinase 1 and induces cofilin phosphorylation for T-cell chemotaxis. Mol Cell Biol. 2002;22:774–783.
- Amano T, Kaji N, Ohashi K, et al. Mitosis-specific activation of LIM motif-containing protein kinase and roles of cofilin phosphorylation and dephosphorylation in mitosis. J Cell Biol. 2002;277:22093–22102.
- Okreglak V, Drubin DG. Cofilin recruitment and function during actin-mediated endocytosis dictated by actin nucleotide state. J Cell Biol. 2007;178:1251–1264.
- Arber S, Barbayannis FA, Hanser H, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809.
- Huang TY, Dermardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol. 2006;18:26–31.
- Yilmaz O, Yao L, Maeda K, et al. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cell Microbiol. 2008;10:863–875.
- Yao L, Jermanus C, Barbetta B, et al. Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol Oral Microbiol. 2010;25:89–101.
- Choi CH, Spooner R, Deguzman J, et al. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol. 2013;15:961–976.
- Benakanakere MR, Li Q, Eskan MA, et al. Modulation of TLR2 protein expression by miR-105 in human oral keratinocytes. J Biol Chem. 2009;284:23107–23115.
- Moffatt CE, Lamont RJ. Porphyromonas gingivalis induction of microRNA-203 expression controls suppressor of cytokine signaling 3 in gingival epithelial cells. Infect Immun. 2011;79:2632–2637.
- Reife RA, Coats SR, Al-Qutub M, et al. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell Microbiol. 2006;8:857–868.
- Abe T, Hosur KB, Hajishengallis E, et al. Local complement-targeted intervention in periodontitis: proof of- concept using a C5a receptor [CD88] antagonist. J Immunol. 2012;189:5442–5448.
- Huang GT, Kim D, Lee JK, et al. Interleukin-8 and intercellular adhesion molecule 1 regulation in oral epithelial cells by selected periodontal bacteria: multiple effects of Porphyromonas gingivalis via antagonistic mechanisms. Infect Immun. 2001;69:1364–1372.
- Hajishengallis G, Wang M, Liang S, et al. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci USA. 2008;105:13532–13537.
- Takeuchi H, Hirano T, Whitmore SE, et al. The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-κB RelA/p65. PLoS Pathog. 2013;9:e1003326.
- Wang M, Krauss JL, Domon H, et al. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal. 2010;3:ra11.
- Maekawa T, Krauss JL, Abe T, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. 2014;15:768–778.
- Bainbridge BW, Coats SR, Darveau RP. Porphyromonas gingivalis lipopolysaccharide displays functionally diverse interactions with the innate host defense system. Ann Periodontol. 2002;7:1–9.
- Herath TD, Wang Y, Seneviratne CJ, et al. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity differentially modulates the expression of IL-6 and IL-8 in human gingival fibroblasts. J Clin Periodontol. 2011;38:694–701.
- Coats SR, Jones JW, Do CT, et al. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4ʹ-phosphatase activities. Cell Microbiol. 2009;11:1587–1599.
- Darveau RP, Cunningham MD, Bailey T, et al. Ability of bacteria associated with chronic inflammatory disease to stimulate E-selectin expression and promote neutrophil adhesion. Infect Immun. 1995;63:1311–1317.
- Burns E, Bachrach G, Shapira L, et al. TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300.
- Yoshioka H, Yoshimura A, Kaneko T, et al. Analysis of the activity to induce Toll-like receptor TLR 2 and TLR4-mediated stimulation of supragingival plaque. J Periodontol. 2008;79:920–928.
- Krauss JL, Potempa J, Lambris JD, et al. Complementary Tolls in the periodontium: how periodontal bacteria modify complement and Toll-like receptor responses to prevail in the host. Periodontol. 2000;52:141–162.
- Ding PH, Wang CY, Darveau RP, Jin LJ. Porphyromonas gingivalis LPS stimulates the expression of LPS-binding protein in human oral keratinocytes in vitro. Innate Immun. 2013;19:66–75.
- Ding PH, Wang CY, Darveau RP, Jin LJ. Nuclear factor-κB and p38 mitogen-activated protein kinase signaling pathways are critically involved in Porphyromonas gingivalis lipopolysaccharide induction of lipopolysaccharide-binding protein expression in human oral keratinocytes. Mol Oral Microbiol. 2013;28:129–141.
- Ding PH, Jin LJ. The role of lipopolysaccharide-binding protein in innate immunity: a revisit and its relevance to oral/periodontal health. J Periodont Res. 2014;49:1–9.
- Coats SR, Pham TT, Bainbridge BW, et al. MD-2 mediates the ability of tetraacylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol. 2005;175:4490–4498.
- Popadiak K, Potempa J, Riesbeck K, et al. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol. 2007;178:7242–7250.
- Nociti FH Jr, Foster BL, Barros SP, et al. Cementoblast gene expression is regulated by Porphyromonas gingivalis lipopolysaccharide partially via toll-like receptor-4/MD-2. J Dent Res. 2004;83[8]: 602-607.
- Liang S, Krauss JL, Domon H, et al. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. 2011;186:869–877.
- Chen YY, Peng B, Yang Q, et al. The outer membrane protein LptO is essential for the O-deacylation of LPS and the co-ordinated secretion and attachment of A-LPS and CTD proteins in Porphyromonas gingivalis. Mol Microbiol. 2011;79(5):1380–1401.
- Jusko M, Potempa J, Karim AY, et al. A metalloproteinase karilysin present in the majority of Tannerella forsythia isolates inhibits all pathways of the complement system. J Immunol. 2012;188:2338–2349.
- Gokyu M, Kobayashi H, Nanbara H, et al. Thrombospondin-1 production is enhanced by Porphyromonas gingivalis lipopolysaccharide in THP-1 cells. PLoS ONE. 2014;10:e0139759.
- Na H, Lim E, Jeong S, et al. Plasminogen activator inhibitor type 1 expression induced by lipopolysaccharide of Porphyromonas gingivalis in human gingival fibroblast. J Microbiol. 2014;52:154–160.
- Vinolo MA, Rodrigues HG, Hatanaka E, et al. Short-chain fatty acids stimulate the migration of neutrophils to inflammatory sites. Clin Sci (Lond). 2009;117(9):331–338.
- Nathan C. Role of iNOS in human host defense. Science. 2006;312:1874–1875.
- Bao K, Belibasakis GN, Thurnheer T, et al. Role of Porphyromonas gingivalis gingipains in multi-species biofilm formation. BMC Microbiol. 2014;14:258.
- Yoneda M, Hirofuji T, Anan H, et al. Mixed infection of Porphyromonas gingivalis and Bacteroides forsythus in a murine abscess model: involvement of gingipains in a synergistic effect. J Periodontal Res. 2001;36:237–243.
- Jusko M, Potempa J, Karim AY, et al. A metalloproteinase karilysin present in the majority of Tannerella forsythia isolates inhibits all pathways of the complement system. J Immunol. 2012;188:2338–2349.
- Bostanci N, Thurnheer T, Aduse-Opoku J, et al. Porphyromonas gingivalis regulates TREM-1 in human polymorphonuclear neutrophils via its gingipains. PLoS ONE. 2013;8:e75784.
- Baruah S, Keck K, Vrenios M, et al. Identification of a novel splice variant isoform of TREM-1 in human neutrophil granules. J Immunol. 2015;195(12):5725–5731.
- Tammaro A, Derive M, Gibot S, et al. TREM-1 and its potential ligands in non-infectious diseases: from biology to clinical perspectives. Pharmacol Ther. 2017;177:81–95.
- Nylund KM, Ruokonen H, Sorsa T, et al. Association of the salivary triggering receptor expressed on myeloid cells/its ligand peptidoglycan recognition protein 1 axis with oral inflammation in kidney disease. J Periodontol. 2017;28:1–17.
- Bugueno IM, Batool F1, Korah L, et al. Porphyromonas gingivalis Differentially Modulates Apoptosome Apoptotic Peptidase Activating Factor 1 in Epithelial Cells and Fibroblasts. Am J Pathol. 2018 Feb;188(2):404–416. .
- Meghil MM, Tawfik OK, Elashiry M, et al. Disruption of Immune Homeostasis in Human Dendritic Cells via Regulation of Autophagy and Apoptosis by Porphyromonas gingivalis. Front Immunol. 2019;10:2286. Published 2019 Sep 24.
- Watanabe K, Yilmaz O, Nakhjiri SF, et al. Association of mitogen-activated protein kinase pathways with gingival epithelial cell responses to Porphyromonas gingivalis infection. Infect Immun. 2001;69(11):6731–6737.
- Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40.
- Taxman TDJ, Swanson KV, Broglie PM, Wen H, Holley-Guthrie E, Huang MTH, Callaway JB, Eitas TK, Duncan JA, Ting JPY. Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis. J Biol Chem. 2012;287:32791–32799.
- Huck O, Elkaim R, Davideau J-L, et al. Porphyromonas gingivalis-impaired innate immune response via NLRP3 proteolysis in endothelial cells. Innate Immun. 2015;21:65-72.
- Meghil MM, Tawfik OK, Elashiry M, et al. Disruption of Immune Homeostasis in Human Dendritic Cells via Regulation of Autophagy and Apoptosis by Porphyromonas gingivalis. Front Immunol. 2019;10:2286. Published 2019 Sep 24.
- Carvalho-Filho PC, Moura-Costa LF, Ana CMP, et al. Apoptosis Transcriptional Profile Induced by Porphyromonas gingivalis HmuY. Mediators Inflamm. 2019. Article ID 6758159, 8, 2019. DOI:10.1155/2019/6758159.
- O’Brien-Simpson NM, Pathirana RD, Walker GD, et al. Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect Immun. 2009;77:1246–1261.
- Yang X, Pan Y, Xu X, et al. Sialidase Deficiency in Porphyromonas gingivalis Increases IL-12 Secretion in Stimulated Macrophages Through Regulation of CR3, IncRNA GAS5 and miR-21. Front Cell Infect Microbiol. 2018;8:100. Published 2018 Apr 5.
- Hung SC, Choi CH, Said-Sadier N, et al. P2X4 assembles with P2X7 and Pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS ONE. 2013;8:e70210.
- Wang H, Zhou H, Duan X, et al. Porphyromonas gingivalis-Induced Reactive Oxygen Species activate JAK2 and regulate production of inflammatory cytokines through c-JunBäumler AJ. Infect Immun. 2014;82:4118–4126.
- Gölz L, Memmert S, Rath-Deschner B, et al. LPS from Pgingivalis and hypoxia increases oxidative stress in periodontal ligament fibroblasts and contributes to periodontitis. Mediators Inflammation. 2014;2014:986264.
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Rev. 2007;87:245–313.
- KH K. Tissue distribution and putative physiological function of NOX family NADPH oxidases. Japanese J Infectious Dis. 2004;57:S28–S29.
- Yang S, Zhang Y, Ries W, et al. Expression of Nox4 in osteoclasts. J Cellul Biochem. 2004;92:238–248.
- Mydel P, Takahashi Y, Yumoto H, et al. Roles of the host oxidative immune response and bacterial antioxidant rubrerythrin during Porphyromonas gingivalis infection. PLoS Pathog. 2006;2:e76.
- Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–619.
- Sztukowska M, Bugno M, Potempa J, et al. Role of rubrerythrin in the oxidative stress response of Porphyromonas gingivalis. Mol Microbiol. 2002;44:479–488.
- Almeida-da-Silva CLC, Ramos-Junior ES, Morandini AC, et al. P2X7 receptor-mediated leukocyte recruitment and Porphyromonas gingivalis clearance requires IL-1β production and autocrine IL-1 receptor activation. Immunobiology. 2019;224(1):50–59.
- Sztukowska M, Bugno M, Potempa J, et al. Role of rubrerythrin in the oxidative stress response of Porphyromonas gingivalis. Mol Microbiol. 2002;44:479–488.
- Kikuchi Y, Ohara N, Sato K, et al. Novel stationary-phase-upregulated protein of Porphyromonas gingivalis influences production of superoxide dismutase, thiol peroxidase and thioredoxin. Microbiol. 2005;151:841–853.
- Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol. 2000;43:14–40.
- Moutsopoulos NM, Kling HM, Angelov N, et al. Porphyromonas gingivalis promotes Th-17 inducing pathways in chronic periodontitis. J Autoimmun. 2012;39:294–303.
- Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th-17 cells and IL-17. J Dent Res. 2008;87:817–828.
- Sato K, Suematsu A, Okamoto K, et al. Th-17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–2682.
- Ji JD, Park-Min KH, Shen Z, et al. Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-gamma in human osteoclast precursors. J Immunol. 2009;183:7223–7233.
- Cardoso CR, Garlet GP, Crippa GE, et al. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol. 2009;24:1–6.
- Wilson NJ, Boniface K, Chan JR, et al. De Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957.
- Gaddis DE, Maynard CL, Weaver CT, et al. Role of TLR2-dependent IL-10 production in the inhibition of the initial IFN-γ T cell response to Porphyromonas gingivalis. J Leukoc Biol. 2013;93(1):21–31.
- Gonzales JR1, Groeger S, Johansson A, et al. T helper cells from aggressive periodontitis patients produce higher levels of interleukin-1 beta and interleukin-6 in interaction with Porphyromonas gingivalis. Clin Oral Investig. 2014;18(7):1835–1843.
- Yun LW, Decarlo AA, Hunter N. Blockade of protease-activated receptors on T cells correlates with altered proteolysis of CD27 by gingipains of Porphyromonas gingivalis. Clin Exp Immunol. 2007;150(2):217–229.
- Bayles KW. The biological role of death and lysis in biofilm development. Nat Rev Microbiol. 2007;5:721–726.
- Belibasakis GN, Bostanci N, Reddi D. Regulation of protease-activated receptor-2 expression in gingival fibroblasts and Jurkat T cells by Porphyromonas gingivalis. Cell Biol Int. 2010;34:287–292.
- Rice KC, Bayles KW. Molecular control of bacterial death and lysis. Microbiol Mol Biol Rev. 2008;72:85–109.
- Bayles KW. Bacterial programmed cell death: making sense of a paradox. Nat Rev Microbiol. 2014;12:63–69.
- Eldholm V, Johnsborg O, Haugen K, et al. Fratricide in Streptococcus pneumoniae: contributions and role of the cell wall hydrolases CbpD, LytA, and LytC. Microbiol. 2009;155:2223–2234.
- Claverys JP, Martin B, Håvarstein LS. Competence-induced fratricide in streptococci. Mol Microbiol. 2007;64:1423–1433.