
ABSTRACT
Oral lichen planus (OLP) is a chronic T cell-mediated inflammatory disease that affects the mucus membrane of the oral cavity. We previously proposed a potential role of intracellular bacteria detected within OLP lesions in the pathogenesis of OLP and isolated four Escherichia coli strains from OLP tissues that were phylogenetically close to K-12 MG1655 strain. We sequenced the genomes of the four OLP-isolated E. coli strains and generated 6.71 Gbp of Illumina MiSeq data (166–195x coverage per strain). The size of the assembled draft genomes was 4.69 Mbp, with a GC content of 50.7%, in which 4360 to 4367 protein-coding sequences per strain were annotated. We also identified 368 virulence factors and 53 antibiotic resistance genes. Comparative genomics revealed that the OLP-isolated strains shared more pangenome orthologous groups with pathogenic strains than did the K-12 MG1655 strain, a derivative of K-12 strain isolated from human feces. Although the OLP-isolated strains did not have the major virulence factors (VFs) of the pathogenic strains, a number of VFs involved in adherence/invasion, colonization, or systemic infection were identified. The genomic characteristics of E. coli first isolated from the oral cavity would benefit future investigations on the pathogenic potential of these bacteria.
Introduction
Oral lichen planus (OLP) is a chronic T cell-mediated mucosal disease in which T cells activated by unknown factor(s) infiltrate into the superficial lamina propria, leading to liquefaction degeneration of the epithelial basal layer [Citation1,Citation2]. Although the OLP occurs in 0.5–4% of the global population and is incurable, the etiology and pathogenesis of OLP are not well understood [Citation1,Citation2]. To date, many research groups have suggested strong correlations between the human microbiome and human disease through high-throughput sequencing technology studies [Citation3–5]. Although the oral microbiome is known as the most stable human-associated microbiota, many studies have reported that alterations in the oral microbiome may cause physiological changes, suggesting potential roles of the microbiome in oral disease [Citation6,Citation7].
According to previous studies, OLP patients present dysbiosis in their salivary and buccal mucosal microbiota compared to healthy controls [Citation8–10]. We previously proposed the possibility that bacterial invasion into the lamina propria and the presence of intracellular bacteria detected in OLP tissues trigger T cell activation and infiltration [Citation9]. We recently reported a significant enrichment of Escherichia coli in the intratissue bacterial communities of OLP lesions and isolated four E. coli strains from OLP biopsies. Furthermore, strong signals of E. coli were detected in most OLP tissues [Citation11]. Although E. coli colonizing the human intestine is mostly commensal, there are diverse pathogenic strains that cause enteric diseases, urinary tract infections, and sepsis/meningitis [Citation12].
It is currently unclear whether the E. coli strains isolated from OLP tissues are oral commensals, pathobionts, or pathogens because our attempts to isolate E. coli from buccal swabs of healthy subjects have failed, despite the presence of E. coli in the buccal microbiota of the subjects [Citation10,Citation11]. Pathogenic E. coli strains usually acquire pathogenicity through horizontal gene transfer of virulence factors encoded on transposons, plasmids, bacteriophages, or pathogenicity islands and through deletions or point mutations of regulatory genes [Citation12]. Therefore, the genetic information of newly isolated E. coli strains will guide future investigations on the potential pathogenic role of E. coli in OLP. Here, we describe the genomic characteristics of four E. coli strains isolated from OLP tissues through whole-genome sequencing and comparative genomics to suggest their potential in OLP etiology.
Material and methods
Genomic DNA (gDNA) extraction
Stocks of the OLP-isolated E. coli strains were inoculated into 50 mL of Luria-Bertani liquid culture media in a 250 mL flask and incubated at 37°C on a 200 rpm orbital shaker. For gDNA extraction, 50 mL of culture media with E. coli strains were harvested, and their gDNA was isolated using a commercial QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Through amplification and sequencing of the 16S rRNA gene, contamination of the extracted gDNA by that of other genomes was ruled out.
Whole-genome sequencing, assembly, and annotation
Whole-genome sequencing and assembly were performed at ChunLab, Inc. (Seoul, Korea). The concentration and purity of the gDNA samples were determined using a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, USA). To generate libraries, 400 ng of gDNA was fragmented into approximately 550 bp using a M220 Focused-ultrasonicator (Covaris, Brighton, UK), and the fragmented DNA was quantified using a DNA 7500 kit (Agilent, Palo Alto, USA) and a Bioanalyzer 2100 instrument (Agilent). Libraries were then constructed using a TruSeq DNA Library LT kit (Illumina, San Diego, USA) according to the manufacturer’s protocol. Whole-genome sequencing was performed on an Illumina MiSeq platform with 2 × 300 bp paired-end reads in conjunction with an Illumina MiSeq Reagent Kit v3 (600-cycle) (Illumina).
After trimming primer sequences and filtering the low-quality reads, the obtained sequencing data were assembled with SPAdes 3.9.1 [Citation13]. Each assembled genome was then tested for the presence of contaminating genomes by comparing different copies of 16S rRNA in the genome using ContEst16S v1.0 [Citation14], and genome-based species were identified using the TrueBac ID system (v1.92) [Citation15]. Raw sequencing reads obtained from the Illumina MiSeq are available at the NCBI Sequence Read Archive under SRR12284166–SRR12284169 (). The annotated assemblies for 5.1, 7.1, 7.2, and 7.3 have been deposited in GenBank via the NCBI submission portal and can be accessed with the accession numbers listed in .
Table 1. Sequencing and assembly statistics of the four oral lichen planus (OLP)-isolated Escherichia coli strains
Table 2. Genomic characteristics of four oral lichen planus (OLP)-isolated Escherichia coli strains in comparison with other strains
Protein-coding sequences (CDSs) were predicted using Prodigal v2.6.3 [Citation16]. The predicted CDSs were annotated by homology searches using USEARCH v8.1.1861 [Citation17] against the Swiss-Prot [Citation18], KEGG [Citation19], and SEED [Citation20] databases, after which they were assigned to the Clusters of Orthologous Groups of proteins (COG) categories [Citation21] based on their function, with reference to orthologous groups (EggNOG 4.5) [Citation22]. A total of 1293 Pathosystems Resource Integration Center (PATRIC)-curated virulence factor (VF) sequences [Citation23] were downloaded as a FASTA file from the PATRIC site (https://patricbrc.org/view/SpecialtyGeneList/?and(eq(source,PATRIC_VF),eq(evidence,Literature)). Using these sequences as a reference genome, we analyzed the genomes with the PATRIC proteome comparison tool (https://docs.patricbrc.org/user_guides/services/proteome_comparison_service.html) with 70% minimum coverage, 30% minimum identity, and 1e−5 BLAST e-value to identify VFs. Antimicrobial resistance genes within the genome were determined using the Resistance Gene Identifier (RGI) on the Comprehensive Antibiotic Resistance Database (CARD) website (https://card.mcmaster.ca/analyze/rgi) [Citation24]. A total of 4326 proteins of the 7.3 strain were searched against human proteins with 1e−5 BLAST e-value to identify human homologous proteins.
Comparative genomics
For comparative genomics, the genome sequences of related E. coli strains were obtained from the GenBank. For commensal strains, the K-12 MG1655 and Nissle 1917, a strain used as probiotics [Citation25], were selected. For pathogenic strains, the CFT073 that is closely related to Nissle 1917 and uropathogenic E. coli (UPEC) strain [Citation26], another UPEC strain JJ2434, an enterohemorrhagic E. coli (EHEC) strain ATCC BAA-460 (Sakai), and an enteropathogenic Shigella flexneri 2457 T were selected. Although S. flexneri is considered a different entity from E. coli in clinical disease, S. flexneri and E. coli genetically constitute the same species [Citation27]. In a previously reported phylogenetic tree, the OLP-isolated E. coli strains were more closely related to S. flexneri 2457 T than to E. coli Nissle 1917 [Citation11].
After selection of genomes, comparative genomic analyses were performed using ChunLab’s comparative genomics tools (https://www.ezbiocloud.net/genome/view_myCGData). Pangenome orthologous groups (POGs) were determined by the reciprocal best hit (RBH) method [Citation28] using UBLAST [Citation17] with an e-value threshold of 1e−6 and an open reading frame (ORF)-independent method [Citation29] using nucleotide sequences with cut-off values of 70% of minimum gene coverage. Venn diagrams of the calculated POGs were constructed using the jvenn tool [Citation30]. A pairwise ortholog matrix (POM) table was generated by complete calculation of pairwise ortholog detection within the dataset of select strains. The existence of genomic islands (GIs), which provide evidence of horizontal gene transfer, was confirmed only if the genomic region contained five or more consecutive CDSs in a contig, and the same pattern was observed across the four OLP strains but not in the K-12 MG1655 strain, as previously described [Citation31].
Kanamycin resistance test
Kanamycin resistance of the four OLP-isolated and two commensal strains was determined according to the measurements of growth rates in the presence of kanamycin. Growth rates were measured in Luria-Bertani broth at 37°C with or without kanamycin using a Tecan Spark 10 M 96-well plate reader (Tecan, Zurich, Switzerland). Each well was inoculated with a 1000-fold dilution of overnight culture in triplicate per kanamycin concentration. The cultures were allowed to grow for 10 hours under shaking at 200 rpm, and the OD600 was measured every 10 minutes. The calculations of exponential growth rates were based on OD600 values between 0.02 and 0.1 [Citation32].
Results
General genomic features of the OLP-isolated E. coli strains
As previously reported, E. coli 5.1 and 7.1–7.3 strains were isolated from the biopsy tissues obtained from OLP patient 5 and 7, respectively. The patient 5 was a 74-year-old male and had reticular and erythematous lesions at two and one areas, respectively. The patient 7 was a 60-year-old female and had reticular, erythematous, and ulcerative lesions at one, three, and one areas, respectively. While the biopsy of the patient 5 presented acanthosis and band-shaped infiltrate only in some areas, that of the patient 7 showed atrophy and heavy infiltrate in all areas, in addition to the liquefaction degeneration of the epithelial basal layer which was a common feature [Citation11].
We generated 6.71 Gbp of Illumina MiSeq short-read data, which resulted in 166–195x coverage per strain (). The short reads were assembled into 123–134 contigs, with an N50 of 95–106 Kbp per strain. The size of genomes was 4.69 Mbp (with a GC content of 50.7%), which was 0.05 Mbp larger than that of the K-12 MG1655 and 0.09 Mbp larger than that of a pathogen S. flexneri 2457 T; however, it was smaller than that of another commensal strain (Nissle 1917) and three selected pathogenic E. coli strains ().
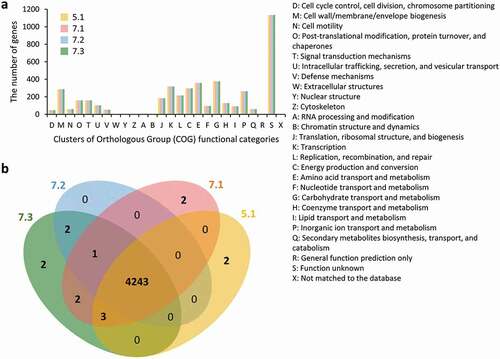
Genome annotation revealed 4360 to 4367 CDSs. The numbers of CDSs were larger than that of MG1655 but smaller than those of Nissle 1917 and four pathogenic strains (.) The CDSs were assigned to 20 of the 26 COG categories, and the largest portion of CDSs belonged to the S category with unknown function. Although 5.1 and the other three strains were isolated from two different OLP patients, the four strains shared most of the CDSs. Only the M, K, and L categories differed among these strains by just one or two CDSs ( and b)). Each of the 5.1, 7.1, and 7.3 strains had two unique CDSs, which encode four hypothetical proteins, a putative prophage Qin DNA-packaging protein NU1-like protein in the 5.1 strain and a DNA-directed RNA polymerase in the 7.1 strain.
Figure 1. Distribution of annotated genes in four oral lichen planus (OLP)-isolated Escherichia coli strains. (a) Distribution based on the Clusters of Orthologous Groups of proteins (COG) functional categories, (b) A Venn diagram showing the distribution of shared and unique pangenome orthologous groups (POGs) among the OLP-isolated strains
Comparison with commensal strains
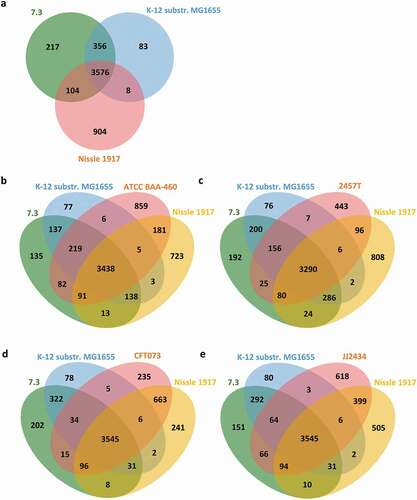
We previously reported that the OLP-isolated strains were closely related to the K-12 MG1655 strain [Citation11]. Gene content-based comparative genomic analysis revealed nine GIs between the OLP-isolated strains and the K-12 MG1655 strain, suggesting multiple horizontal gene transfer events (Supplementary Table 1). Comparative genomic analysis of 7.3, which contained the largest number of CDSs among the OLP strains, and two commensal strains also revealed that the 7.3 strain shared more POGs with the K-12 MG1655 strain than with Nissle 1917 and that 217 POGs were unique to the 7.3 strain ()). The 217 POGs included multiple genes that encode machinery for conjugation found in GI4, toxin-antitoxin systems for plasmid maintenance, and phage-related proteins (Supplementary Table 2).
Figure 2. Venn diagrams of shared and unique pangenome orthologous groups (POGs)
The 217 POGs may also include genes that confer the ability to colonize the oral cavity. For example, ECC84_03920 encodes a protein 99% identical to TraT, an outer membrane protein involved in surface exclusion of conjugating E. coli [Citation33]. TraT also confers complement resistance [Citation34], a competitive trait in the oral cavity where functionally active complement systems are present [Citation35]. ECC84_04059 encodes OmpT which is 70% identical to another copy of OmpT encoded by ECC84_04087, which is shared in two commensal strains. OmpT cleaves urinary antimicrobial peptides and increases bacterial adhesion/invasion of bladder epithelial cells, which are also important in colonizing the oral mucosa [Citation36,Citation37].
Fimbriae determine the ability of E. coli to bind to biotic/abiotic surfaces and thus to colonize specific environments. A total of 13 fimbrial gene clusters were found, which are identical to those present in the K-12 MG1655 strain [Citation38]. Among them, the ydeQRST, yfcOPQRSTUV, and gltF-yhcF clusters had a deletion, stop codon, and insertional element, respectively, as in the K-12 MG1655 strain. However, the ygiL-yqiGHI cluster contained no insertional element as is found in the K-12 MG1655 strain, thus it was expected to be functional.
Comparison with pathogenic strains and virulence factors
To predict the pathogenic potential of the OLP-isolated strains, the genomes of the 7.3, K-12 MG1655, and Nissle 1917 strains were compared with that of each of the pathogenic strains. The 7.3 strain shared more POGs with all the pathogenic strains than did the K-12 MG1655 strain. With respect to the CFT073 and JJ2434 strains, however, the Nissle 1917 strain shared more POGs than did the 7.3 strain () and Supplementary Tables 3–6). However, the major virulence factors of the selected pathogenic strains, such as Shiga toxin, type III secretion system, locus of enterocyte effacement, mannose-resistant hemagglutinins, and pap pili [Citation39,Citation40], were not found in the OLP-isolated strains.
Using a tool and database provided by the PATRIC, we further analyzed the genome of 7.3 for the presence of VFs and identified 368 VFs (Supplementary Table 7). Among them, only 10 were missing in the K-12 MG1655 strain, which included the aforementioned TraT- and OmpT homolog-encoding genes. Interestingly, five of the 10 VFs absent in the K-12 MG1655 strain are reportedly involved in colonization of EPEC or UTEC. ECC84_01765 encodes an ABC transporter, 99% identical to that encoded by ycjV; ECC84_03063 encodes an uncharacterized lipoprotein, 97% identical to YdeK; ECC84_04058 encodes a hypothetical protein, and ECC84_04181 encodes a phage tail tip assembly protein L, both of which are 76% identical to those identified in the EHEC O157:H7 str. EDL933. These four VFs have been shown to be involved in colonization of the bovine intestinal tract by EHEC strains [Citation41,Citation42]. ECC84_04284 encodes a putative transferase, 99% identical to that identified in the UPEC CFT073 strain. This gene is required to maintain the hypercolonization phenotype of the dsdA-deleted CFT073 strain [Citation43]. The remaining three VFs absent in the K-12 MG1655 strain are reportedly involved in systemic infection. ECC84_04204 encodes TraJ, which is 100% identical to that identified in avian pathogenic E. coli (APEC). The importance of TraJ in early systemic dissemination of infection from the mesenteric lymph nodes to the liver, spleen, bloods, and central nervous system has been shown in a neonatal rat model [Citation44]. ECC84_04206 encodes an X polypeptide that is 76% identical to a putative transglycosylase encoded by the Salmonella enterica serovar Typhimurium gene finP. This gene is required for long-term systemic Salmonella infection in mice [Citation45]. ECC84_04369 encodes a protein that is 79% identical to OmpD identified in S. enterica and 100% identical to OmpC of E. coli, as determined by Blast search. OmpC-deleted APEC presents reduced virulence in duck and mouse models accompanied with reduced invasion into the brains, lungs, and bloods [Citation46].
Antibiotic resistance
Disease-associated E. coli has a higher prevalence of antibiotic resistance than commensals [Citation47]. A search against the CARD revealed 53 antibiotic resistance genes, among which 43 encode efflux pumps for multiple antibiotics, including fluoroquinolones, aminocoumarin, penams, cephamycins, carbapenems, rifamycins, tetracyclines, aminoglycosides, macrolides, and phenicols. Five genes encode proteins that alter targets for peptide antibiotics or penams and cephamycins. Five genes encode proteins that inactivate penams, cephamycins, macrolides, or aminoglycosides. Most of the antibiotic resistance genes were ubiquitously present in the strains included in the current study, but the APH(3ʹ)-IIa gene that inactivates aminoglycosides was present only in the OLP-isolated strains (Supplementary Table 8).
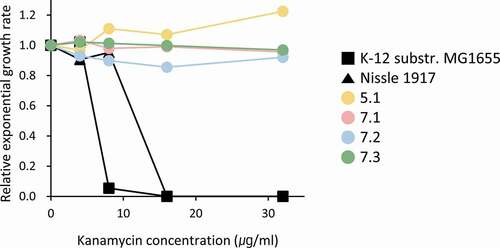
To validate the annotation and comparative genomic data, the four OLP-isolated and two commensal strains were tested for resistance to kanamycin. While K-12 MG1655 and Nissle 1917 were sensitive to kanamycin, the four OLP strains were resistant, which coincided with the genotype of each strain ().
Figure 3. Kanamycin resistance test. Kanamycin resistance of the four OLP-isolated and two commensal strains was determined by the measurements of growth rates in the presence of various concentrations of kanamycin
Discussion
E. coli was identified as one of pathogenic species in the oral microbiome (buccal mucosa, supragingival plaque, and tongue dorsum) of healthy subjects in the Human Microbiome Project through metagenomics shot-gun sequencing [Citation48]. However, the presence of E. coli in the oral cavity had been questioned by many researchers. Interestingly, the relative abundance of E. coli in buccal mucosal microbiota is reduced in OLP lesions compared with healthy subjects but substantially enriched within the tissues compared with the surface of OLP lesions [Citation10,Citation11]. Here, we report the genomic characteristics of four E. coli strains isolated from OLP tissues.
In contrast to human intestinal commensal and pathogenic isolates that are distributed among diverse phylogenetic groups [Citation47,Citation49,Citation50], all four strains isolated from two OLP patients belonged to the A phylogenetic group. Furthermore, the inter-strain differences in gene contents were all less than 10 regardless of the patient source. Thus, E. coli detected in other OLP biopsies by in situ hybridization must be genetically similar to those presented here. However, the OLP-isolated strains were clearly different from the K-12 MG1655 strain isolated from human feces, as evidenced by nine GIs and unique POGs in each strain. The POGs present in the OLP-isolated strains but not in MG1655 included multiple genes encoding machinery for conjugation and plasmid maintenance. This coincides with the fact that MG1655 is derived from K-12 W1485 strain by curing the F plasmid [Citation51]. The F pilus forms not only the mating channels but also the presumptively static structures without the channel, the latter of which promotes nonspecific aggregation [Citation52]. Indeed, in contrast to the MG1655 strain, all the OLP-isolated strains severely aggregated during liquid culture (data not shown). Since the genomes reported in the current study are draft assemblies, further investigation is required to confirm the presence of plasmid(s) in the OLP-isolated strains.
Although there are some exceptions such as Nissle 1917, pathogenic E. coli usually have a larger genome size with more genes than commensals [Citation53]. The genome sizes of the OLP-isolated strains were smaller than most sequenced pathogenic strains [Citation39]. The OLP-isolated strains shared more POGs with the selected pathogenic strains than did the K-12 MG1655 strain but lacked the major VFs of the pathogenic strains. Pathogenic E. coli strains are not the members of commensals colonizing the disease sites. For example, pathogenic strains causing enteric diseases are obtained via foodborne transmission, and UPEC strains causing urinary tract infection are derived from the gut [Citation54]. We previously reported detection of E. coli in the epithelium, but not in the lamina propria, of control tissues with a histologically normal appearance, suggesting the importance of the invasion of E. coli beyond the barrier tissue in OLP development [Citation11]. Furthermore, E. coli was detected in the buccal mucosal microbiota of healthy individuals, and all other species detected in the intratissue bacterial communities of OLP lesions belonged to human oral microbiome [Citation10,Citation11,Citation48]. Based on these findings, we propose that the OLP-isolated strains are the member of oral microbiome and pathobionts rather than overt pathogens.
Although there is a possibility that E. coli colonizing the oral mucosa of healthy subjects are genetically different from the OLP-isolated strains, the OLP strains are more likely to be commensals but increase virulence in the altered environment of OLP patients. Isolation of E. coli strains from the oral mucosa of healthy individuals will clarify this issue. Despite the presence of lacZ, lacY, and lacA genes, the OLP-isolated strains did not form purple-black colonies on a selective and differential medium, such as eosin methylene blue agar. Because E. coli was substantially enriched within the OLP tissues (up to 60%), it was possible to isolate E. coli without colour differentiation. In the buccal swabs of healthy subjects where E. coli accounts for 0.03–7% of total bacteria, however, other Gram-negative species such as Haemophilus spp., Neisseria spp., and Lautropia mirabilis, but not E. coli, have been isolated from cultures on the eosin methylene blue agar.
It is well known that commensal strains have pathogenic potential. Actually, 358 out of 368 VFs identified in the OLP-isolated strains were shared in the genome of the K-12 MG1655 strain. Interestingly, six of the 10 VFs absent in the K-12 MG1655 strain are involved in bacterial adhesion/invasion and colonization, while the other four VFs are involved in complement resistance and systemic dissemination of infection. Together with the fimbriae, these VFs may contribute to invasion of E. coli into tissues, leading to T cell recruitment and OLP development. However, all these potential roles of E. coli and its VFs in the pathogenesis of OLP should be experimentally confirmed in future studies.
OLP has been regarded as a chronic inflammatory disease with autoimmune features. Although the presence of serum autoantibodies against desmoglein ΙΙΙ or antinuclear antibodies has been reported in some OLP patients [Citation55], no autoantigens targeted by the infiltrated T cells have been identified. Microbial infection can promote autoimmunity via molecular mimicry or bystander activation of self-reactive T cells [Citation56]. We previously reported the existence of bacterial orthologs for autoantigens in human-associated bacteria [Citation57]. Through homology searches, 962 proteins encoded by the E. coli 7.3 genome were found to have homologous human proteins (Supplementary Table 9). For example, E. coli Fe-S cluster assembly scaffold IscU had 77% identity and 89% similarity with the human mitochondrial iron-sulfur cluster assembly enzyme ISCU chain D, and an E. coli acyltransferase had 21% identity and 41% similarity with the human 1-acyl-sn-glycerol-3-phosphate acyltransferase epsilon (Supplementary Figure 1). Because only one or two anchor residues are critically involved in the binding of peptide antigens to MHC molecules, the T cells specific to E. coli acyltransferase may be able to cross-react with human acyltransferase. When host cells are infected with microbes, antigens presented by the MHC molecules are rapidly changed from self- to microbial peptides. Therefore, the epithelial cells infected with bacteria in the OLP lesions are unlikely to present self-antigens. However, the cross-reactivity of bacteria-specific T cells with self-antigens would allow the T cells to target not only the infected but also non-infected epithelial cells. Carrying human homologs is universal to all bacteria. Notably, the OLP-isolated E. coli strains have almost twice as many CDSs as those of most oral bacterial species, which have approximately 2000–2500 CDSs, thus, are expected to have increased numbers of human homologs.
The genomic characteristics of the OLP-isolated E. coli strains would benefit future investigations to clarify their potential role in OLP etiology.
Supplemental Material
Download Zip (1,012.9 KB)Acknowledgments
This work was supported by the National Research Foundation of Korea under grants 2016R1E1A1A01942402 and 2019R1A2B5B02100662.
Disclosure statement
The authors report no conflict of interests.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Farhi D, Dupin N. Pathophysiology, etiologic factors, and clinical management of oral lichen planus, part I: facts and controversies. Clin Dermatol. 2010;28(1):100–10.
- Paul M, Shetty DC. Analysis of the changes in the basal cell region of oral lichen planus: an ultrastructural study. J Oral Maxillofac Pathol. 2013;17:10–16.
- Ruff WE, Greiling TM, Kriegel MA. Host-microbiota interactions in immune-mediated diseases. Nat Rev Microbiol. 2020;18(9):521–538.
- Durack J, Lynch SV. The gut microbiome: relationships with disease and opportunities for therapy. J Exp Med. 2019;216(1):20–40.
- Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30:492–506.
- Deo PN, Deshmukh R. Oral microbiome: unveiling the fundamentals. J Oral Maxillofac Pathol. 2019;23(1):122–128.
- Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, et al. The oral metagenome in health and disease. Isme J. 2012;6(1):46–56.
- Wang K, Lu W, Tu Q, et al. Preliminary analysis of salivary microbiome and their potential roles in oral lichen planus. Sci Rep. 2016;6:22943.
- Choi YS, Kim Y, Yoon HJ, et al. The presence of bacteria within tissue provides insights into the pathogenesis of oral lichen planus. Sci Rep. 2016;6:29186.
- Baek K, Choi Y. The microbiology of oral lichen planus: is microbial infection the cause of oral lichen planus? Mol Oral Microbiol. 2018;33(1):22–28.
- Baek K, Lee J, Lee A, et al. Characterization of intratissue bacterial communities and isolation of Escherichia coli from oral lichen planus lesions. Sci Rep. 2020;10:3495.
- Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–140.
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477.
- Lee I, Chalita M, Ha SM, et al. ContEst16S: an algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int J Syst Evol Microbiol. 2017;67:2053–2057.
- Ha SM, Kim CK, Roh J, et al. Application of the whole genome-based bacterial identification system, truebac ID, using clinical isolates that were not identified with three matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) Systems. Ann Lab Med. 2019;39:530–536.
- Hyatt D, Chen GL, Locascio PF, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010;11:119.
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26:2460–2461.
- Pundir S, Marti MJ, Donovan C. UniProt protein knowledgebase. Methods Mol Biol. 2017;1558:41–55.
- Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:353–361.
- Overbeek R, Begley T, Butler RM, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:691–702.
- Galperin MY, Makarova KS, Wolf YI, et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:261–269.
- Huerta-Cepas J, Szklarczyk D, Forslund K, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:286–293.
- Mao C, Abraham D, Wattam AR, et al. Curation, integration and visualization of bacterial virulence factors in PATRIC. Bioinformatics. 2015;31:252–258.
- Alcock BP, Raphenya AR, Lau TTY, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48:517–525
- Sonnenborn U. Escherichia coli strain Nissle 1917-from bench to bedside and back: history of a special Escherichia coli strain with probiotic properties. FEMS Microbiol Lett. 2016;363:212.
- Vejborg RM, Friis C, Hancock V, et al. A virulent parent with probiotic progeny: comparative genomics of Escherichia coli strains CFT073, Nissle 1917 and ABU 83972. Mol Genet Genomics. 2010;283:469–484.
- Chattaway MA, Schaefer U, Tewolde R, et al. Identification of Escherichia coli and Shigella Species from Whole-Genome Sequences. J Clin Microbiol. 2017;55:616–623.
- Ward N, Moreno-Hagelsieb G. Quickly finding orthologs as reciprocal best hits with BLAT, LAST, and UBLAST: how much do we miss? PLoS ONE. 2014;9:e101850.
- Chun J, Grim CJ, Hasan NA, et al. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc Natl Acad Sci USA. 2009;106:15442–15447.
- Bardou P, Mariette J, Escudie F, et al. An interactive Venn diagram viewer. BMC Bioinformatics. 2014;15:293.
- Ha SM, Chalita M, Yang SJ, et al. Comparative genomic analysis of the 2016 vibrio cholerae outbreak in South Korea. Front Public Health. 2019;7:228.
- Gullberg E, Cao S, Berg OG, et al. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011;7:e1002158.
- Achtman M, Kennedy N, Skurray R. Cell–cell interactions in conjugating Escherichia coli: role of traT protein in surface exclusion. Proc Natl Acad Sci USA. 1977;74(11):5104–5108.
- Binns MM, Mayden J, Levin RP. Further characterization of complement resistance conferred on Escherichia coli by the plasmid genes traT of R100 and iss of CoIV,I-K94. Infect Immun. 1982;35:654–659.
- Andoh A, Fujiyama Y, Kimura T, et al. Molecular characterization of complement components (C3, C4, and Factor B) in Human Saliva. J Clin Immunol. 1997;17:404–407.
- He XL, Wang Q, Peng L, et al. Role of uropathogenic Escherichia coli outer membrane protein T in pathogenesis of urinary tract infection. Pathog Dis. 2015;73:3.
- Hui CY, Guo Y, He QS, et al. Escherichia coli outer membrane protease OmpT confers resistance to urinary cationic peptides. Microbiol Immunol. 2010;54(8):452–459.
- Archer CT, Kim JF, Jeong H, et al. The genome sequence of E. coli W (ATCC 9637): comparative genome analysis and an improved genome-scale reconstruction of E. coli. BMC Genomics. 2011;12:9.
- Rasko DA, Rosovitz MJ, Myers GS, et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 2008;190(20):6881–6893.
- Mattock E, Blocker AJ. How do the virulence factors of shigella work together to cause disease? Front Cell Infect Microbiol. 2017;7:64.
- Van Diemen PM, Dziva F, Stevens MP, et al. Identification of enterohemorrhagic Escherichia coli O26: h−genes required for intestinal colonization in calves. Infect Immun. 2005;73(3):1735–1743.
- Dziva F, Van Diemen PM, Stevens MP, et al. Identification of Escherichia coli O157 : H7 genes influencing colonization of the bovine gastrointestinal tract using signature-tagged mutagenesis. Microbiology. 2004;150:3631–3645.
- Haugen BJ, Pellett S, Redford P, et al. In vivo gene expression analysis identifies genes required for enhanced colonization of the mouse urinary tract by uropathogenic Escherichia coli strain CFT073 dsdA. Infect Immun. 2007;75(1):278–289.
- Hill VT, Townsend SM, Arias RS, et al. TraJ-dependent Escherichia coli K1 interactions with professional phagocytes are important for early systemic dissemination of infection in the neonatal rat. Infect Immun. 2004;72(1):478–488.
- Lawley TD, Chan K, Thompson LJ, et al. Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLos Pathog. 2006;2(2):e11.
- Hejair H, Zhu Y, Ma J, et al. Functional role of ompF and ompC porins in pathogenesis of avian pathogenic Escherichia coli. Microb Pathog. 2017;107:29–37.
- Salipante SJ, Roach DJ, Kitzman JO, et al. Large-scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains. Genome Res. 2015;25(1):119–128.
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214.
- Tenaillon O, Skurnik D, Picard B, et al. The population genetics of commensal Escherichia coli. Nat Rev Microbiol. 2010;8(3):207–217.
- Johnson JR, Delavari P, Kuskowski M, et al. Phylogenetic distribution of extraintestinal virulence-associated traits in Escherichia coli. J Infect Dis. 2001;183(1):78–88.
- Jensen KF. The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J Bacteriol. 1993;175(11):3401–3407.
- Hu B, Khara P, Christie PJ. Structural bases for F plasmid conjugation and F pilus biogenesis in Escherichia coli. Proc Natl Acad Sci U S A. 2019;116(28):14222–14227.
- Sahl JW, Morris CR, Rasko DA. Comparative genomics of pathogenic Escherichia coli. p21-43. In: Donnenberg MS, editor. Escherichia coli pathotypes and principles of pathogenesis. 2nd ed. Academic Press; New York, NY: 2013.
- Chen SL, Wu M, Henderson JP, et al. Genomic diversity and fitness of E. coli strains recovered from the intestinal and urinary tracts of women with recurrent urinary tract infection. Sci Transl Med. 2013;5(184):184ra60.
- Sun S, Zhong B, Li W, et al. Immunological methods for the diagnosis of oral mucosal diseases. Br J Dermatol. 2019;181(1):23–36.
- Chervonsky AV. Microbiota and autoimmunity. Cold Spring Harb Perspect Biol. 2013;5(3):a007294.
- Alam J, Kim YC, Choi Y. Potential role of bacterial infection in autoimmune diseases: a new aspect of molecular mimicry. Immune Netw. 2014;14(1):7–13.