
ABSTRACT
Introduction
The oral pathogen Porphyromonas gingivalis is not only associated with periodontitis but also with systemic diseases elsewhere in the body. The mechanisms by which P. gingivalis travels from the oral cavity to other organs in the body are largely unknown. This review describes the four putative mechanisms supported by experimental evidence, which enable translocation of P. gingivalis over the oral mucosa, endothelial barriers and subsequent dissemination into the bloodstream.
Mechanisms
The first mechanism: proteolytic enzymes secreted by P. gingivalis degrade adhesion molecules between tissue cells, and the extracellular matrix. This weakens the structural integrity of the mucosa and allows P. gingivalis to penetrate the tissue. The second is transcytosis: bacteria actively enter tissue cells and transfer to the next layer or the extracellular space. By travelling from cell to cell, P. gingivalis reaches deeper structures. Thirdly, professional phagocytes take up P. gingivalis and travel to the bloodstream where P. gingivalis is released. Lastly, P. gingivalis can adhere to the hyphae forming Candida albicans. These hyphae can penetrate the mucosal tissue, which may allow P. gingivalis to reach deeper structures.
Conclusion
More research could elucidate targets to inhibit P. gingivalis dissemination and prevent the onset of various systemic diseases.
Introduction
The oral cavity can be colonized by more than 700 different species of microbes, including bacteria, fungi, viruses and protozoa. Together they form a complex biological system also known as the oral microbiome. This oral microbiome is the second most diverse microbiome in the human body, after the gut [Citation1]. Several different niches can be distinguished in the oral cavity, each having their own unique microbial composition. The oral microbiome is dynamic and can be influenced by behavioral factors such as diet, smoking and stress; and by physiological changes of the host such as hormonal fluctuations and age [Citation2,Citation3]. When these environmental conditions change the composition and phenotypes of the microbiome also changes. This can lead to disruption of the microbial balance, or dysbiosis of the oral microbiome, characterized by the emergence of pathogenic bacteria. Dysbiosis can result in oral diseases such as caries and gingivitis. Gingivitis, a reversible inflammation of the gingiva, may progress to the more severe disease periodontitis, which is defined by alveolar bone loss and loss of clinical attachment of the periodontal ligament [Citation4].
The major pathogens that are associated with the development of periodontitis are known as the red complex in the pyramid of Socransky. This red complex consists of three bacterial species: Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia [Citation5,Citation6]. The one that is most broadly studied in relation to periodontitis is P. gingivalis, a Gram-negative strictly anaerobic bacterium [Citation7]. This bacterium colonizes the gingival sulcus, predominantly found in patients suffering from periodontitis. P. gingivalis interacts with other oral microorganisms that colonize the gingiva in earlier stages of periodontal disease, such as Streptococcus spp. and Fusobacterium nucleatum. Interactions with these species are important for the colonization of P. gingivalis in the oral cavity [Citation5]. This review is focused on P. gingivalis, which has a variety of virulence factors which aid in invasion or destruction of host tissue and evasion of the host immune response. Major virulence factors of P. gingivalis include its fimbriae and cysteine proteases known as gingipains [Citation8]. Gingipains are essential for the survival of this proteolytic bacterium and play an important role in its pathogenicity. There are two types of gingipains: Arginine-specific gingipains (Rgp) and lysine-specific gingipains (Kgp) can cleave peptide bonds at the C-terminal after arginine and lysine amino acids, respectively. Gingipains can be secreted, expressed on the bacterial surface or released in outer membrane vesicles [Citation9]. They are responsible for various pathogenic activities such as: evasion of host defense systems, activation of kallikrein/kinin system, lysis of fibrin and fibrinogen, and activation of the blood clotting system [Citation10].
In addition to oral disease, periodontal pathogens such as P. gingivalis have been associated with various systemic diseases, including atherosclerosis, diabetes, pneumonia, rheumatoid arthritis, chronic kidney disease and Alzheimer’s disease (AD) [Citation11–14]. P. gingivalis has been found in artery tissues of patients suffering from atherosclerosis and in the brains of patients with Alzheimer’s disease [Citation15,Citation16]. This suggests that the bacterium is able to disseminate into the bloodstream of patients and travel to various parts of the human body. In order to do so, it has to pass the oral mucosa (epithelium) and endothelium of the blood vessel. It is very likely for P. gingivalis to enter the bloodstream easily via bleeding gingiva, a common sign of periodontitis [Citation17], or after tissue trauma due to dental procedures [Citation17,Citation18]. These mechanisms are not specific for P. gingivalis and would result in many different microorganisms from the oral cavity to disseminate into the blood. It was postulated that fewer different bacterial species can be found in the blood than the oral cavity [Citation18]. For example, DNA of oral pathogens was detected in synovial fluid of patients suffering from rheumatoid arthritis which did not correspond to the amount of DNA found in dental plaque. The frequency of Prevotella intermedia and P. gingivalis DNA was almost as high in synovial fluid as in dental plaque, whereas the DNA of other species were not as frequently found in synovial fluid as in dental plaque [Citation19]. This suggests that there is a specific mechanism for these pathogens to enter the blood and/or that these bacteria survive longer in circulation, by evading host defenses [Citation18]. It should be emphasized that a possible passive mechanism such as bleeding should be followed up by an active translocation such as described above, crossing tissue barriers away from the periodontium, such as the brain, where P. gingivalis has been detected [Citation16]. In addition, P. gingivalis can be found in 25% of healthy individuals not suffering from periodontitis [Citation20]. Such a relatively high prevalence in healthy individuals should not be overlooked when considering highly prevalent diseases such as atherosclerosis and AD. Whether these healthy individuals with P. gingivalis also develop these diseases, has not been studied.
The exact mechanism by which P. gingivalis actively crosses tissue barriers is currently unknown. When considering an active role of P. gingivalis in invading the host tissue, it has been reported that gingipains play a crucial role in the breakdown of tissue barriers [Citation21].
P. gingivalis, as well as its secreted gingipains, have been found in the brains of AD patients [Citation16]. In the brain, the gingipains stimulate factors that are associated with AD, such as neuroinflammation, accumulation of amyloid β (Aβ) and phosphorylation of Tau protein [Citation16,Citation22,Citation23]. In one of the more recent studies, mice were orally infected with P. gingivalis for 6 weeks and an increased amount of Aβ plaques was found in their brains. On top of this, gingipain inhibitors had a neuroprotective effect by lowering Aβ accumulation and lowering immune responses, suggesting that gingipains are involved in these processes [Citation16]. For all these effects to occur, P. gingivalis would have to enter the brain and so there should be a mechanism to pass the blood–brain barrier as well as the oral mucosa.
The aim of this review is to describe potential mechanisms allowing translocation of P. gingivalis over the oral mucosa and disseminate into the bloodstream. It is important to understand these mechanisms as it might uncover possible targets for the prevention of these diseases. Based on research into P. gingivalis invasion and/or translocation, four different mechanisms could be identified by which P. gingivalis can translocate the oral mucosa barrier and endothelial linings, based on known mechanisms of either P. gingivalis or other microbes. These four mechanisms are separately described in this review, each with a detailed schematic depiction (). A more simplified overview of all four mechanisms is shown in .
Figure 1. Schematic overview of the first mechanism of translocation. P. gingivalis secretes proteolytic enzymes known as gingipains (Rgp/Kgp) that degrade cell-cell adhesion molecules (yellow) and adhesion molecules (light green) that connect the cell with the extracellular matrix (ECM). In addition, P. gingivalis stimulates the fibroblasts to produce matrix metalloproteases (MMPs) that can degrade ECM molecules. These three effects combined weaken the integrity of the epithelial barrier, which allows P. gingivalis to travel between cells into deeper layers of the tissue.
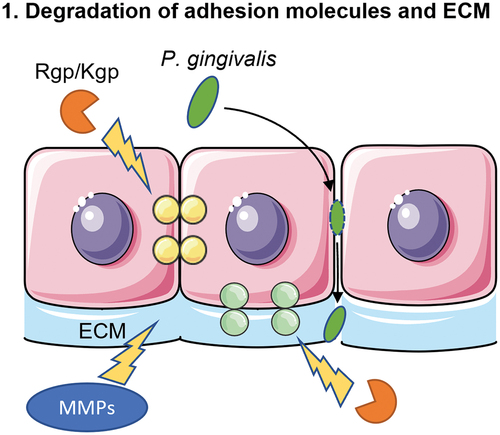
Figure 2. Schematic overview of the second mechanism of translocation. The major fimbriae of P. gingivalis adhere to the β1 integrin receptor of the epithelial cells. This will lead to entry of the bacterium into an early phagosome. In order to survive within the cell, P. gingivalis makes use of the autophagy pathway of the epithelial cell to prevent being transferred to lysosomes, where P. gingivalis would be killed. Lastly, the bacterium exits the cell via the endocytic recycling pathway.
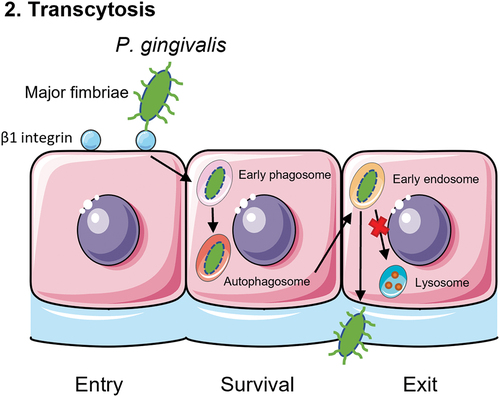
Figure 3. Schematic overview of the third mechanism of translocation. P. gingivalis is recognized by phagocytic cells such as macrophages, monocytes or dendritic cells. Toll-like receptor 4 (TLR4) recognizes the lipopolysaccharide (LPS) of P. gingivalis and subsequently phagocytoses the bacterium. Then, the phagocyte will travel back to the blood stream with P. gingivalis inside, as a ‘Trojan Horse’ mechanism.
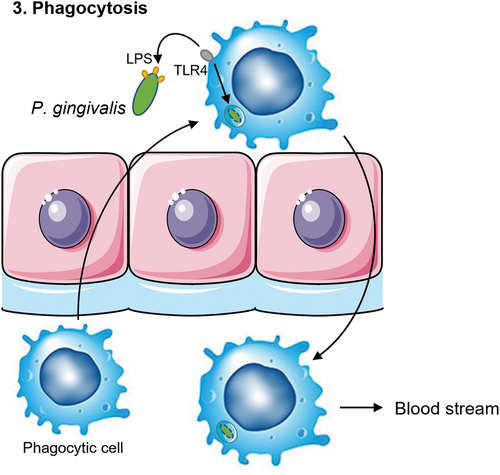
Figure 4. Schematic overview of the fourth mechanism of translocation. C. albicans can form invasive hyphae that insert themselves between the epithelial cells. P. gingivalis can adhere to C. albicans via the Als3 protein on the hyphae of the fungi. Phagocytic cells such as macrophages are attracted to C. albicans. This allows for the phagocytes to come in close contact to the bacterium and take up P. gingivalis to travel back into the blood stream.
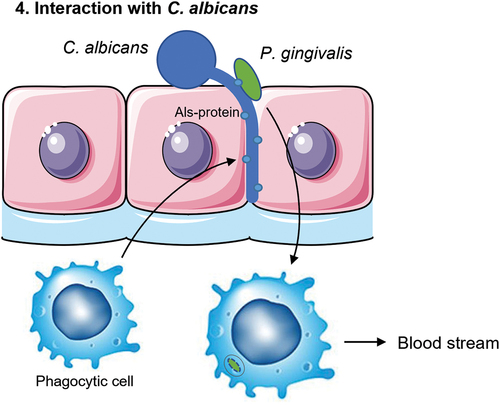
Figure 5. A schematic overview of the four putative mechanisms for P. gingivalis to translocate the oral mucosa or the endothelium of blood vessels. First, proteolytic enzymes known as gingipains (Rgp/Kgp) are secreted by P. gingivalis (green) and degrade cell-cell adherence molecules (yellow circles) and the extracellular matrix (ECM; light blue). The structural integrity of the oral mucosa is weakened so that P. gingivalis is able to pass in between the cells. Second, P. gingivalis can enter the cell after adherence and subsequent endocytosis and exit the cell on the other side of the epithelial layer. P. gingivalis needs to have a mechanism to be able to survive within the cell and not be degraded by phagolysosomes. It can then travel from cell to cell, migrating deeper into the tissue, eventually reaching the basal membrane, and finally the endothelial cells that line the blood vessels. Third, phagocytic cells of the host are able to pick up P. gingivalis within the tissue and transfer it over the endothelial barrier. They can then travel back into the bloodstream taking the bacterium with them. Again, P. gingivalis needs to have a mechanism to survive degradation by the phagocyte. Fourth and lastly, interaction with other microbes such as C. albicans (blue) could allow for travel across the oral mucosa due to the hyphae of C. albicans that can insert themselves between cells. P. gingivalis is able to attach to these hyphae. Macrophages may also play a role in this mechanism as they are attracted to the hyphae of C. albicans and can phagocytose the attached P. gingivalis.
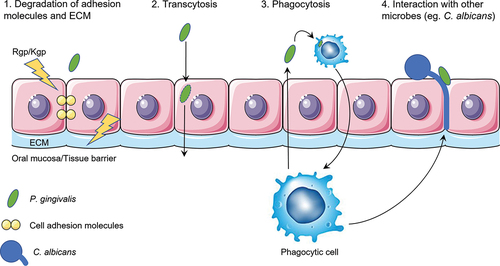
Mechanisms
Degradation of adhesion molecules and extracellular matrix
The oral mucosal barrier can be disrupted by P. gingivalis-mediated degradation of the cellular adhesion molecules and extracellular matrix (ECM) (). The structural integrity of the tissue is compromised after such proteolysis, creating space for the bacteria to penetrate deeper into the tissue. P. gingivalis is able to degrade cell-cell junction complexes Occludin, E-cadherin and β1-integrin between epithelial cells [Citation24]. In addition, using a monolayer of Madin-Darby canine kidney (MDCK) cell line it was found that P. gingivalis decreases the transepithelial resistance (TER). The bacteria were also found to be translocating the monolayer of MDCK cells [Citation24]. This suggests that P. gingivalis is able to invade the connective tissues e.g. by degrading the epithelial tissue structure and travelling between the cells. This allows the spread of the bacteria and may result in dissemination into the blood. It is unsure whether MDCK cells translate well to the oral mucosa but this study does suggest that P. gingivalis is able to invade epithelial barriers.
Following entry of the body by passing through oral mucosa layers, the bacteria would need to pass the endothelium as well to reach the bloodstream. It was found that gingipains are involved in the detachment and apoptosis of endothelial cells [Citation25]. Kinetics of cell adhesion molecule cleavage differed between epithelial and endothelial cells. Gingipains induce cleavage of the adhesion molecules vascular endothelial cadherin (VE-cadherin), neural cadherin (N-cadherin) and integrin β1, as well as E-selectin and platelet endothelial cell adhesion molecule-1 (PECAM-1) [Citation25,Citation26].
When the adhesion molecules that facilitate the interaction between epithelial cells and the ECM are degraded, the structural integrity of the epithelium is affected. This may facilitate the penetration of the bacteria deeper into the tissue. The gingipains secreted by P. gingivalis are also known to degrade molecules of the ECM, such as fibronectin and tenascin-C. This was found via fragmentation of fibronectin and tenascin-C by purified proteases, observed in a SDS-PAGE gel and via immunofluorescence staining [Citation27,Citation28]. Using a complex three-dimensional models of engineered human oral mucosa (EHOM) it was illustrated that P. gingivalis is able to penetrate into the tissue by degradation of components of the basement membrane [Citation21]. EHOM is composed of epithelial cells and fibroblasts from healthy human biopsy specimens. Using transmission electron microscopy on this model, it was found that wild-type P. gingivalis is able to penetrate into deeper structures of the tissue as compared to gingipain-deficient mutants of P. gingivalis.
Outer membrane protein A-like proteins are known to be involved in ECM binding by bacteria. P. gingivalis can adhere to ECM via outer membrane proteins Pgm6 and Pgm7 they form a heterotrimer (Pgm6/7) [Citation29]. In addition, P. gingivalis is able to adhere to ECM molecule fibronectin via its fimbriae and the affinity of binding is enhanced when fibronectin is degraded by gingipain-like proteases [Citation30]. Thus, gingipains contribute directly and indirectly to adherence and colonization of epithelial surfaces such as the oral mucosa.
P. gingivalis is also indirectly involved in the degradation of the extracellular matrix. Studies have shown that P. gingivalis supernatant stimulates the release of matrix metalloproteases (MMPs) in human gingival fibroblasts on the mRNA level. In addition, the tissue inhibitor of MMPs (TIMP) expression in reduced [Citation31,Citation32]. These MMPs are known to degrade various extracellular matrix proteins such as collagen.
Not only the gingipains are important virulence factors that have an effect on the structural integrity of epithelial and endothelial tissue, this process also occurs indirectly via the immune system. P. gingivalis can dysregulate host immune responses to benefit other pathogens. For example, P. gingivalis can cleave or induce cross-talk in immune cell receptors or suppress the release of cytokines [Citation33,Citation34]. Dysbiosis of the microbial community occurs which causes inflammation in the periodontium [Citation34]. This inflammation could enhance the epithelial cell damage and vascular leakage, which increases the ability of P. gingivalis to translocate.
Invasion of epithelial and endothelial cells (Transcytosis)
Transcytosis is a pathogen invasion process, in principle involving three essential phases: entry, survival and exit [Citation35]. shows a detailed overview of the mechanism. Entry of the pathogen can occur either through active or passive mechanisms. In the active process, the bacteria attach to the surface of a host cell. This can occur either by the interaction of bacterial surface proteins with proteins on the host cell, or the insertion of effectors by the bacterium into the membrane of the host cell [Citation36]. Then, the bacterium is engulfed by the cell and an intracellular vesicle will form which contains the pathogen. This process is dependent on reorganization of the cytoskeleton of the invaded cell [Citation36–38]. The passive mechanism involves phagocytosis by the host cell [Citation35]. Only professional phagocytes such as macrophages can perform this mechanism with high efficiency. They are a group of cells that are specialized to remove microorganisms, whereas non-professional phagocytes such as epithelial cells cannot ingest microorganisms [Citation39].
After entry, the bacterium needs to have a mechanism to survive within the cell. Generally, when an epithelial cell is invaded by a pathogen, the host cell will fuse the pathogen-containing intracellular vesicle with lysosomes in order to kill the invading pathogens [Citation40]. Several mechanisms exist for evasion of this process, each invading pathogen species has its own survival strategy. For example, the pathogen can survive by evading fusion with a lysosome or resisting degradation once inside the phagolysosome [Citation36,Citation38]. Lastly, the bacterium needs to be able to exit the cell on the other side into deeper structures of the tissue. Three main strategies are described by which an intracellular pathogen can exit the cell: Induction of programmed cell death of the host, active destruction of the host cell, and membrane-dependent exit without destruction of the host cell [Citation41]. By travelling from cell to cell, bacteria can travel deeper and deeper into the tissue and can eventually enter the bloodstream.
The first step of transcytosis, cell entry by P. gingivalis, has been researched extensively. Already two decades ago it was discovered that this bacterium is able to rapidly invade primary gingival epithelial cells [Citation42,Citation43]. Adherence and subsequent invasion of P. gingivalis into gingival epithelial cells is mediated by binding of major fimbriae to β1 integrin receptor which induces the phosphorylation of paxillin (integrin-clustering associated protein) [Citation44,Citation45]. While P. gingivalis is generally considered to be a non-motile bacterium, recent research has shown that the bacterium is able to translocate over surfaces when sandwiched between two surfaces [Citation46]. This mechanism might occur when P. gingivalis is located between different epithelial cells. Possibly, this newfound mobility supports the potency of interaction with β1 integrin, in order to adhere and subsequently invade the tissue cell.
After invasion of the epithelial cell layer, the bacterium will enter the extracellular matrix of the connective tissue, and subsequently, it will encounter the endothelial cells that line the blood vessels. These vascular endothelial cells can also be invaded by P. gingivalis. However, they are less susceptible to invasion than oral epithelial keratinocytes [Citation26]. This process was found to be gingipain-dependent. It is suggested that the intracellular P. gingivalis are enclosed in vesicles, which leads to the hypothesis that endocytosis is the mechanism by which P. gingivalis was internalized [Citation26].
For the second step in transcytosis, survival, P. gingivalis is dependent on a mechanism to evade degradation via lysosomes. It is suggested in multiple studies that this is achieved by trafficking via autophagic pathways [Citation37,Citation47,Citation48]. After internalization into gingival epithelial cells, P. gingivalis is incorporated into early phagosomes. Then, P. gingivalis travels to autophagosomes of the host cell to provide a nutrient-rich environment in which the bacterium can survive. Here, P. gingivalis can somehow prevent lysosomal hydrolases from entering the autophagosome and thereby evading degradation. When the autophagic pathway was inhibited, P. gingivalis was transferred to phagolysosomes where they were degraded [Citation47].
The third and final step for P. gingivalis to disseminate into the blood stream by transcytosis is for the bacterium to exit the cells. It was suggested that exit of P. gingivalis from infected cells occurs via the endocytic recycling pathway [Citation49]. The endocytic recycling pathway is a dynamic system inside the cell that is responsible for sorting and exporting membrane components. First, the internalized cargo is transported to the early endosome by membrane fusion. There it is sorted and will either be transported for degradation by lysosomes or returned to the outer membrane of the cell [Citation50].
When P. gingivalis invades a gingival epithelial cell, it remains inside an intracellular vesicle. This vesicle will then fuse with the early endosome. It is suggested that P. gingivalis has a mechanism by which it evades the transfer to the lysosome but instead will enter the recycling pathway back to the cell membrane [Citation51]. Intracellular P. gingivalis was found to be co-localized with transferrin receptor, Rab11 and RaIA, which are components of the recycling pathway. When the recycling pathway was inhibited by knockdown of Rab11 or RaIA, exit of P. gingivalis from gingival epithelial cells was reduced. It was also found that exit of the bacterium from gingival epithelial cells depended on actin polymerization, lipid rafts and microtubule assembly. These processes are also associated with the recycling pathway, suggesting that this pathway and exit of P. gingivalis from gingival epithelial cells might be connected [Citation49].
Phagocytosis
Periodontitis is characterized by systemic inflammation of the gingiva [Citation52]. This attracts immune cells, among which are professional phagocytes such as monocytes and macrophages [Citation53]. Studies have shown that macrophages are found in higher abundance in gingival biopsies of gingivitis and periodontitis patients as compared to healthy gingiva [Citation54,Citation55]. Professional phagocytes such as macrophages are highly specialized to engulf and destroy pathogens. However, some pathogens can evade killing by the macrophage after phagocytosis and use the macrophage as an entry into other tissues of the host. This strategy is known as a ‘Trojan Horse’ mechanism and is used by Bacillus anthracis, for example [Citation56]. The phagocyte takes up the pathogen in the periphery (lungs, intestine or oral cavity) and travels into the bloodstream (). Inside the phagocyte, the pathogens are protected from the host immune system and can multiply. The phagocyte will travel to another organ where the pathogen can exit the phagocyte and causes an infection in that organ. This mechanism can also lead to infection of the brain, when the phagocytes pass the blood–brain barrier [Citation57].
This Trojan Horse mechanism could also be a way for P. gingivalis to disseminate into the blood. One major condition for this to occur is that P. gingivalis has the ability to survive within the phagocyte, and escape effectively. Research has shown that P. gingivalis was able to effectively invade and viably escape macrophages [Citation58]. This was observed with a phagocytosis assay: THP-1 derived macrophages with M0 (naive) or M1 (pro-inflammatory) phenotype and RAW264.7 cells were infected with two different strains of P. gingivalis, Streptococcus gordonii or Escherichia coli and then the extracellular bacteria were killed with antibiotics. After 24 hours (long enough to allow for potential phagolysosome killing of bacteria) the cells were lysed and the number of intracellular bacteria that survived were determined by counting colony forming units (CFU). The efficiency of escape was determined by collecting supernatant after killing the extracellular bacteria, instead of lysing the cells. It appeared that strain W83 in particular was very effective in invading and escaping THP-1 derived macrophages and RAW 264.7 cells. Strain ATCC 33,277 of P. gingivalis also showed invasion and escape, significantly more than other microbes tested (S. gordonii and E. coli) [Citation58]. Active invasion of phagocytic cells was also shown by P. gingivalis. Here, the invasion of macrophages by P. gingivalis is dependent on the major fimbriae. This was found in a study using a fimbriae deficient mutant of P. gingivalis (DPG3) that adherence and invasion of THP-1 cells was compromised, compared to wild type [Citation59]. The mechanism behind this process is not completely clear and more research is needed to fully unravel this process.
Macrophages can polarize into pro-inflammatory M1 or anti-inflammatory M2 phenotypes. The M1 macrophages are responsible for phagocytosis and subsequent killing of bacteria, and production of pro-inflammatory cytokines. On the other hand, M2 macrophages are involved in the reduction of inflammation, wound healing and tissue repair [Citation60,Citation61]. The anti-inflammatory M2 macrophages might be responsible for the survival of bacteria after phagocytosis and thereby act as the Trojan horse to bring bacteria into the blood stream. Studies have shown that bacteria such as Chlamydia pneumoniae and Brucella melitensis are more likely to survive in M2 macrophages compared to M1 macrophages [Citation62,Citation63]. The same was shown for P. gingivalis: they can survive in naïve and M2 macrophages for 24 hours, but not in M1 macrophages [Citation64]. This was found by determining the number of bacteria that are taken up by the macrophages after 1 hour and relating these to the number of bacteria that were still present inside the macrophages after 24 hours using the phagocytosis assay described above.
A capsule-deficient mutant of the P. gingivalis strain W50 was used in order to investigate the effect of the capsule on host response. Phagocytosis was determined by flow cytometry of mouse bone-marrow derived macrophages and dendritic cells after they were exposed to FITC-labeled bacteria. Survival was determined by lysing the cells, plating and counting the CFU. The capsule-deficient mutant of P. gingivalis was phagocytosed much more by the macrophages and dendritic cells than the wild-type strain. In contrast, no surviving capsule-deficient P. gingivalis were found in the phagocytes, whereas the wild-type did survive. In addition, it was found by ELISA that the capsule of P. gingivalis can aid in reducing the inflammatory response of the host [Citation65].
Lipopolysaccharide (LPS) is expressed on Gram-negative bacteria and can activate macrophages via Toll-like receptor 4 (TLR4) to phagocytose the bacteria [Citation66,Citation67]. It was found that P. gingivalis LPS only weakly activates macrophage polarization but does induce a release of cytokines and chemokines that attract other immune cells [Citation68].
In addition to macrophages, dendritic cells (DC) are also phagocytic cells that could be involved in the translocation of P. gingivalis over the mucosal barrier. Already over two decades ago, it was found that P. gingivalis can be internalized into DCs and activate them after penetration into the gingival epithelium [Citation69]. The minor fimbriae of P. gingivalis can target DC-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) and enter the cell. DCs in periodontitis lesions can become activated and will travel to the lamina propria, which is highly vascularized. It is hypothesized that upon infection, P. gingivalis-containing DCs travel through the lamina propria to the bloodstream which will allow the bacterium to reach atherosclerotic plaques [Citation70]. It was found that P. gingivalis is able to survive within monocyte-derived DCs as well, after uptake via DC-SIGN. The bacterium uses its Mfa-1 fimbriae to evade antibacterial autophagy and lysosome fusion. However, the activation of TLR2 can promote killing of the bacterium, which is stimulated by the major fimbriae FimA of P. gingivalis [Citation71].
Interaction with other microbes
The fourth and final mechanism of actively passing penetrating host tissue barriers is via interaction with other (oral) microbes such as Candida albicans (see ). This mechanism is particularly relevant for passing the oral mucosa. C. albicans is a polymorphic fungus, i.e. it can grow as yeast in a spherical appearance or it can adapt the pseudohyphae or hyphae morphology, and it is a common colonizer of the oral cavity. In healthy individuals, C. albicans grows mostly as yeast without causing tissue damage. Formation of hyphae allows for invasion and destruction of epithelial cells. Many oral bacteria interact with C. albicans and some utilize the tissue invading hyphae to cross the oral mucosa barrier [Citation72]. For example, Staphylococcus aureus is able to adhere to hyphae of C. albicans. This adhesion is mediated by the Als3 protein, a member of the agglutinin-like sequence (Als) family of C. albicans. In a murine model of oral candidiasis, interaction with C. albicans allowed co-colonizing S. aureus to invade deeper into the tissue and disseminate into the blood [Citation73,Citation74]. There was no dissemination when the mice were infected with S. aureus alone. However, further investigation showed that adhesion of the bacterium does not progress with the hyphae, meaning S. aureus cannot be actively transported across the oral mucosa in this manner [Citation75]. Another mechanism must be in place in order for C. albicans to increase S. aureus dissemination. This has led to the hypothesis that S. aureus can also disseminate through the previously described Trojan Horse mechanism by surviving inside macrophages after phagocytosis. It has been observed that macrophages are highly attracted to C. albicans hyphae. This attraction is mediated by the interaction between the β-glucan on C. albicans hyphae and dectin-1 on macrophages [Citation76,Citation77]. This attraction leads to an increase in phagocytosis of S. aureus that are adhering to the hyphae of C. albicans and could explain why S. aureus disseminates into the blood much easier in the presence of C. albicans [Citation74,Citation75].
The same mechanism could also occur for P. gingivalis. Studies have shown that P. gingivalis is able to interact with and can adhere to C. albicans [Citation78,Citation79]. This adherence occurs in a similar way as the adherence of S. aureus to C. albicans, namely in an Als3-dependent manner [Citation79–81]. In addition, C. albicans enhances P. gingivalis invasion of human gingival epithelial cells and fibroblasts [Citation82]. Seeing that P. gingivalis is able to survive within macrophages (as described previously), the interaction with C. albicans could be a plausible component of the mechanisms of P. gingivalis to disseminate into the bloodstream [Citation58]. However, a more recent study found that immune responses in THP-1 derived macrophages were reduced when exposed to a mixed biofilm of P. gingivalis and C. albicans, compared to supernatant of bacterial-only biofilms. This was measured by ELISA of cytokines and chemokines IL-1β, IL-8 and TNFα. While IL-1β production was increased, production of IL-8 and TNFα was significantly reduced when exposed to the mixed biofilm. These results were supported with an in vivo model using C57BL/6 mice, where it was found that the mice survived longer when infected with both species compared to infection with P. gingivalis alone. In addition, the kidney and spleen of the mice were found to be infected with P. gingivalis to a higher extent when the mice were infected with P. gingivalis alone, as compared to mixed species infection. It is suggested that this is due to a lower inflammatory response, as it was also found that dual-species infection leads to a reduced elastase and myeloperoxidase production, which are both neutrophil granular enzymes associated with inflammation [Citation83]. The suggestion that C. albicans protects P. gingivalis from host immune responses would support the theory that C. albicans is involved in P. gingivalis survival, but the study does not strengthen the hypotheses that C. albicans is involved in P. gingivalis dissemination, and the role of macrophages in this process.
Next to C. albicans, there is also evidence that the bacterium Fusobacterium nucleatum is involved in the ability of P. gingivalis to invade the tissue. F. nucleatum is also commonly found in subgingival plaque and is considered to be part of the ‘orange complex’ in the pyramid of Socransky [Citation6]. The influence of F. nucleatum on cell invasion by P. gingivalis was observed using antibiotic protection assays, as described previously in this review. In this study, the cell line Ca9–22 was used as a model for the gingival epithelium and Human aorta endothelial cells (HAEC) were used as a model for the endothelium. Invasion into both cell types by P. gingivalis was significantly enhanced in the presence of F. nucleatum. It is suggested that coaggregation of the two bacterial species may influence the expression of virulence factors. However, the exact molecular mechanism of how F. nucleatum enhances P. gingivalis invasion into gingival epithelial and endothelial cells remains to be elucidated.
Discussion
P. gingivalis has been associated with multiple systemic diseases, in addition to oral disease. For this to occur, the bacterium needs a mechanism to travel from the oral cavity into the bloodstream so that it can reach other organs. Easy access into the blood stream would be via passive entry by bleeding gums, which is common in periodontitis. Considering that P. gingivalis can be found in other organs unlike many other oral microorganisms, it is plausible that P. gingivalis also has an active mechanism to enter the blood stream through epithelial layers. In addition, active translocation from the blood circulation is needed to reach other organs in the body such as the brain. Four active mechanisms have been proposed by which P. gingivalis can cross tissue barriers such as the oral mucosa.
Most components of the described active mechanisms have been tested in in vitro experiments such as in monolayer tissue cultures, with some studies using primary gingival epithelial cells, but also studies using cells that are not from the oral epithelial origin, such as kidney cells [Citation24]. Apparently, within the limitations of these systems which are not always representative for the oral mucosa; there could be interplay between the different cell types found in the tissue and extracellular matrix surrounding the cells. Therefore, such studies should first of all be repeated on genuine oral epithelial cells, in order to establish in a pure culture system whether for instance P. gingivalis disrupts cell-cell adhesion structures here as well. To study the effects of P. gingivalis infection on cell-cell adhesion or the release of certain factors such as cytokines, these monolayer cultures are sufficient. However, in order to study penetration into the deeper tissue layers and the interaction between the different cells of the tissue layers, these monocultures would not be sufficient. Use of a more sophisticated three-dimensional model of the oral mucosa could give more insight in the effects of P. gingivalis on the integrity of the tissue structure. There are already some studies where such a model is being developed, which can be used to observe bacterial penetration into host tissue [Citation84,Citation85].
In addition, the studies using phagocytotic cells such as macrophages generally make use of THP-1 derived macrophages. This is a monocytic cell line derived from human leukemic cells. Likely, under culturing conditions these cells will phenotypically deviate from primary monocytes and macrophages. However, studies on the phagocytosis of P. gingivalis using macrophages that are derived from primary monocytes are currently lacking in literature.
In one of the studies where the degradation of adhesion molecules was studied, it was also found that the trans-epithelial resistance decreases in the presence of P. gingivalis. This shows that the barrier functionality of the epithelium is affected and might therefore allow P. gingivalis to penetrate [Citation24]. This suggests that the mechanism for P. gingivalis to invade the deeper epithelium occurs via the intercellular pathway rather than intracellular, to eventually invade the blood capillaries. This was previously also suggested as the most likely entry by P. gingivalis in a review covering various transepithelial models to study host–pathogen interactions [Citation86]. Cell invasion might then preferably be used by P. gingivalis as a mechanism for evasion of the host immune system and resistance from antibiotic treatment by hiding inside the epithelial and endothelial cells, rather than a way to transport itself further into the tissue [Citation26]. A combination of pathways might also occur in order for P. gingivalis to more effectively cross the oral mucosa barrier. For instance, the involvement of C. albicans might enhance this mechanism of degrading adhesion molecules, as the hyphae are known to cause tissue damage and penetrate into the oral epithelium. On the other hand, macrophages provide both protection from other host immune responses and antibiotic treatment as well as a way to cross the oral mucosa, which would suggest it would be easiest mechanism for P. gingivalis to travel to the bloodstream. It is also a possibility that P. gingivalis actively infiltrates the macrophages, instead of them being phagocytosed, combining transcytosis with the ‘Trojan Horse’ mechanism. However, only a small number of bacteria would be able to travel this way as it was observed in vitro that 2–3% of bacteria invade macrophages and 10–20% of these survive [Citation64]. Of course, the attraction of macrophages to C. albicans might increase the amount of phagocytosis of P. gingivalis that are adhered to C. albicans as opposed to P. gingivalis alone. Very little research has been done for the third and fourth mechanism of translocation described in this review however, and more research might elucidate further how P. gingivalis penetrates the oral mucosa (and possibly other tissue barriers) and whether a combination of these mechanisms occur (see ).
It has to be noted that not all of the discussed mechanisms are experimentally supported, especially those on phagocytosis by macrophages and the involvement of C. albicans. These are still hypothetical and research needs to be done to confirm whether these mechanisms can actually occur. However, in the case of C. albicans, a lot of research has been done on the interaction between S. aureus and C. albicans [Citation73–75,Citation80]. Unpublished results in our group indicate that some aspects of this interaction are also apparent for P. gingivalis. There are some studies on the interaction between P. gingivalis and C. albicans, however this is more limited [Citation78,Citation81,Citation83]. In this review it was hypothesized that P. gingivalis might have a similar interaction with C. albicans as S. aureus. That P. gingivalis can adhere to C. albicans in a similar way as S. aureus has been observed before, but the interaction with macrophages still has to be proven.
In the case of being a risk factor for Alzheimer’s disease, in addition to the oral mucosa the bacterium would also need to pass the blood–brain barrier (BBB). Being composed of many cell types, the BBB is another challenging structural and functional barrier for microorganisms. The vessels in the brain do not contain any pores and its cells are tightly adhered together [Citation87]. Transport across the barrier is regulated by specific transport proteins. This makes the blood–brain barrier highly selective and it is specialized to protect the brain against pathogens and toxins [Citation88,Citation89]. However, infection of the brain has been known to occur for various microorganisms. Multiple reviews about bacterial translocation of the BBB describe three of the four mechanisms described in the current review, including: disruption of adherence molecules, transcytosis and the ‘Trojan Horse’ mechanism via macrophages [Citation90–92]. Research into the blood–brain barrier is challenging as it is difficult to represent this barrier in vitro.
Unraveling the mechanism by which P. gingivalis may cross rigid, protective structures such as the oral epithelium, will provide insight for eventual dissemination into the blood, but equally important: similar mechanisms could occur for P. gingivalis to travel from blood into tissue, as similar barriers have to be crossed. It is essential to find possible targets to prevent the bacterium to affect other organs in the body leading to systemic diseases such as atherosclerosis and Alzheimer’s disease. For example, a gingipain inhibitor could be used for periodontitis patients in order to reduce the dissemination of P. gingivalis into the bloodstream and its subsequent entry into other organs. Not only P. gingivalis but other periodontal pathogens are associated with systemic diseases [Citation11–13]. Unraveling the dissemination mechanisms for P. gingivalis might open some doors for research into other oral pathogens that might have similar mechanisms. In addition, these oral pathogens might strengthen each other’s dissemination mechanisms, such as the interaction between P. gingivalis and C. albicans. This emphasizes the importance of a balanced oral microbiome and how disbalance can have an impact on systemic health.
Author contributions
CdJ, BK, TdV, FB and SG conceived the plan for the manuscript. CdJ wrote the initial draft of the manuscript. BK, TdV, FB and SG critically commented and suggested revisions on all versions of the manuscript.
Acknowledgments
Parts of the image in were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
References
- Deo PN, Deshmukh R. Oral microbiome: unveiling the fundamentals. J Oral Maxillofac Pathol. 2019;23(1):122–13. Epub 2019/05/22. doi: 10.4103/jomfp.JOMFP_304_18
- Kilian M, Chapple IL, Hannig M, et al. The oral microbiome - an update for oral healthcare professionals. Br Dent J. 2016;221(10):657–666. Epub 2016/11/20. DOI:10.1038/sj.bdj.2016.865.
- Cornejo Ulloa P, van der Veen MH, Krom BP. Review: modulation of the oral microbiome by the host to promote ecological balance. Odontology. 2019;107(4):437–448. Epub 2019/02/06. DOI:10.1007/s10266-019-00413-x.
- Scannapieco FA, Dongari-Bagtzoglou A. Dysbiosis revisited: understanding the role of the oral microbiome in the pathogenesis of gingivitis and periodontitis: a critical assessment. J Periodontol. 2021;92(8):1071–1078. Epub 2021/04/27. DOI:10.1002/JPER.21-0120
- Mohanty R, Asopa SJ, Joseph MD, et al. Red complex: polymicrobial conglomerate in oral flora: a review. J Family Med Prim Care. 2019;8(11):3480–3486. Epub 2019/12/06. DOI:10.4103/jfmpc.jfmpc_759_19.
- Socransky SS, Haffajee AD, Cugini MA, et al. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25(2):134–144. Epub 1998/03/12. DOI:10.1111/j.1600-051x.1998.tb02419.x
- Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a potential community activist for disease. Epub 2012/07/10 J Dent Res. 2012;91(9):816–820. DOI:10.1177/0022034512453589
- How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol. 2016;7:53. Epub 2016/02/24. DOI:10.3389/fmicb.2016.00053.
- Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol 2000. 2010;54(1):15–44. Epub 2010/08/18. DOI:10.1111/j.1600-0757.2010.00377.x
- Imamura T. The role of gingipains in the pathogenesis of periodontal disease. J Periodontol. 2003;74(1):111–118. Epub 2003/02/21. DOI:10.1902/jop.2003.74.1.111
- Bui FQ, Almeida-da-Silva CLC, Huynh B, et al. Association between periodontal pathogens and systemic disease. Biomed J. 2019;42(1):27–35. Epub 2019/04/17. DOI:10.1016/j.bj.2018.12.001.
- Linden GJ, Lyons A, Scannapieco FA. Periodontal systemic associations: review of the evidence. J Clin Periodontol. 2013;40(Suppl 14):S8–19. Epub 2013/05/03. DOI:10.1111/jcpe.12064
- Wang RP, Ho YS, Leung WK, et al. Systemic inflammation linking chronic periodontitis to cognitive decline. Brain Behav Immun. 2019;81:63–73. Epub 2019/07/08. DOI:10.1016/j.bbi.2019.07.002.
- Kriauciunas A, Gleiznys A, Gleiznys D, et al. The influence of Porphyromonas gingivalis bacterium causing periodontal disease on the pathogenesis of rheumatoid arthritis: systematic review of literature. Cureus. 2019;11(5):e4775. Epub 2019/08/01. DOI:10.7759/cureus.4775.
- Mougeot JC, Stevens CB, Paster BJ, et al. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J Oral Microbiol. 2017;9(1):1281562. Epub 2017/03/23. DOI:10.1080/20002297.2017.1281562
- Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1). DOI:10.1126/sciadv.aau3333
- Olsen I, Singhrao SK. Can oral infection be a risk factor for Alzheimer’s disease? J Oral Microbiol. 2015;7(1):7.
- Parahitiyawa NB, Jin LJ, Leung WK, et al. Microbiology of odontogenic bacteremia: beyond endocarditis. Clin Microbiol Rev. 2009;22(1):46–64. Table of Contents. Epub 2009/01/13. DOI:10.1128/CMR.00028-08
- Martinez-Martinez RE, Abud-Mendoza C, Patino-Marin N, et al. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J Clin Periodontol. 2009;36(12):1004–1010. Epub 2009/11/26. DOI:10.1111/j.1600-051X.2009.01496.x
- Griffen AL, Becker MR, Lyons SR, et al. Prevalence of Porphyromonas gingivalis and periodontal health status. J Clin Microbiol. 1998;36(11):3239–3242. Epub 1998/10/17. DOI:10.1128/JCM.36.11.3239-3242.1998
- Andrian E, Grenier D, Rouabhia M. In Vitro models of tissue penetration and destruction by Porphyromonas gingivalis. Infect Immun. 2004;72(8):4689–4698. Epub 2004/07/24. DOI:10.1128/IAI.72.8.4689-4698.2004
- Ilievski V, Zuchowska PK, Green SJ, et al. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE. 2018;13(10):e0204941. Epub 2018/10/04. DOI:10.1371/journal.pone.0204941
- Kanagasingam S, Chukkapalli SS, Welbury R, et al. Porphyromonas gingivalis is a strong risk factor for Alzheimer’s disease. J Alzheimers Dis Rep. 2020;4(1):501–511. Epub 2021/02/04. DOI:10.3233/ADR-200250
- Katz J, Sambandam V, Wu JH, et al. Characterization of Porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect Immun. 2000;68(3):1441–1449. Epub 2000/02/26. DOI:10.1128/IAI.68.3.1441-1449.2000
- Sheets SM, Potempa J, Travis J, et al. Gingipains fromPorphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect Immun. 2005;73(3):1543–1552. Epub 2005/02/26. DOI:10.1128/IAI.73.3.1543-1552.2005
- Farrugia C, Stafford GP, Potempa J, et al. Mechanisms of vascular damage by systemic dissemination of the oral pathogen Porphyromonas gingivalis. FEBS J. 2021;288(5):1479–1495. Epub 2020/07/19. DOI:10.1111/febs.15486.
- Ruggiero S, Cosgarea R, Potempa J, et al. Cleavage of extracellular matrix in periodontitis: gingipains differentially affect cell adhesion activities of fibronectin and tenascin-C. Biochim Biophys Acta. 2013;1832(4):517–526. Epub 2013/01/15. DOI:10.1016/j.bbadis.2013.01.003
- Scragg MA, Cannon SJ, Rangarajan M, et al. Targeted disruption of fibronectin-integrin interactions in human gingival fibroblasts by the ri protease of Porphyromonas gingivalis W50. Infect Immun. 1999;67(4):1837–1843. Epub 1999/03/20. DOI:10.1128/IAI.67.4.1837-1843.1999
- Murakami Y, Hasegawa Y, Nagano K, et al. Characterization of wheat germ agglutinin lectin-reactive glycosylated ompa-like proteins derived from Porphyromonas gingivalis. Infect Immun. 2014;82(11):4563–4571. Epub 2014/08/20. DOI:10.1128/IAI.02069-14
- Kontani M, Kimura S, Nakagawa I, et al. Adherence of Porphyromonas gingivalis to matrix proteins via a fimbrial cryptic receptor exposed by its own arginine-specific protease. Mol Microbiol. 1997;24(6):1179–1187. Epub 1997/06/01. DOI:10.1046/j.1365-2958.1997.4321788.x
- Zhou J, Windsor LJ. Porphyromonas gingivalis affects host collagen degradation by affecting expression, activation, and inhibition of matrix metalloproteinases. J Periodontal Res. 2006;41(1):47–54. Epub 2006/01/18. DOI:10.1111/j.1600-0765.2005.00835.x
- Pattamapun K, Tiranathanagul S, Yongchaitrakul T, et al. Activation of mmp-2 by Porphyromonas gingivalis in human periodontal ligament cells. J Periodontal Res. 2003;38(2):115–121. Epub 2003/03/01. DOI:10.1034/j.1600-0765.2003.01650.x
- Olsen I, Taubman MA, Singhrao SK. Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and Alzheimer’s disease. J Oral Microbiol. 2016;8:33029. Epub 2016/11/25. DOI:10.3402/jom.v8.33029.
- Hajishengallis G, Diaz PI. Porphyromonas gingivalis: immune subversion activities and role in periodontal dysbiosis. Curr Oral Health Rep. 2020;7(1):12–21. Epub 2020/12/22. DOI:10.1007/s40496-020-00249-3
- Casadevall A. Evolution of intracellular pathogens. Annu Rev Microbiol. 2008;62:19–33. Epub 2008/09/13. DOI:10.1146/annurev.micro.61.080706.093305.
- Ribet D, Cossart P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015;17(3):173–183. Epub 2015/02/02. DOI:10.1016/j.micinf.2015.01.004
- Lamont RJ, Yilmaz O. In or out: the invasiveness of oral bacteria. Periodontol 2000. 2002;30:61–69. Epub 2002/09/19. DOI:10.1034/j.1600-0757.2002.03006.x.
- Cossart P, Sansonetti PJ. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 2004;304(5668):242–248. Epub 2004/04/10. DOI:10.1126/science.1090124.
- Uribe-Querol E, Rosales C. Phagocytosis: our current understanding of a universal biological process. Front Immunol. 2020;11:1066. Epub 2020/06/26. DOI:10.3389/fimmu.2020.01066.
- Mostowy S. Autophagy and bacterial clearance: a not so clear picture. Cell Microbiol. 2013;15(3):395–402. Epub 2012/11/06. DOI:10.1111/cmi.12063
- Flieger A, Frischknecht F, Hacker G, et al. Pathways of host cell exit by intracellular pathogens. Microb Cell. 2018;5(12):525–544. Epub 2018/12/12. DOI:10.15698/mic2018.12.659
- Lamont RJ, Chan A, Belton CM, et al. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63(10):3878–3885. Epub 1995/10/01. DOI:10.1128/iai.63.10.3878-3885.1995
- Belton CM, Izutsu KT, Goodwin PC, et al. Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol. 1999;1(3):215–223. Epub 2001/02/24. DOI:10.1046/j.1462-5822.1999.00022.x
- Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4(5):305–314. Epub 2002/05/25. DOI:10.1046/j.1462-5822.2002.00192.x
- Yilmaz O, Young PA, Lamont RJ, et al. Gingival epithelial cell signalling and cytoskeletal responses to Porphyromonas gingivalis invasion. Microbiology (Reading). 2003;149(Pt 9):2417–2426. Epub 2003/09/02. DOI:10.1099/mic.0.26483-0
- Moradali MF, Ghods S, Angelini TE, et al. Amino acids as wetting agents: surface translocation by Porphyromonas gingivalis. Isme J. 2019;13(6):1560–1574. Epub 2019/02/21. DOI:10.1038/s41396-019-0360-9
- Belanger M, Rodrigues PH, Dunn WA Jr., et al. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy. 2006;2(3):165–170. Epub 2006/07/29. DOI:10.4161/auto.2828
- Lee K, Roberts JS, Choi CH, et al. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence. 2018;9(1):845–859. Epub 2018/04/05. DOI:10.1080/21505594.2018.1454171
- Takeuchi H, Furuta N, Morisaki I, et al. Exit of intracellular Porphyromonas gingivalis from gingival epithelial cells is mediated by endocytic recycling pathway. Cell Microbiol. 2011;13(5):677–691. Epub 2010/12/16. DOI:10.1111/j.1462-5822.2010.01564.x
- O’Sullivan MJ, Lindsay AJ. The endosomal recycling pathway—At the crossroads of the cell. Int J Mol Sci. 2020;21(17):6074. Epub 2020/08/28. DOI:10.3390/ijms21176074.
- Takeuchi H, Takada A, Kuboniwa M, et al. Intracellular periodontal pathogen exploits recycling pathway to exit from infected cells. Cell Microbiol. 2016;18(7):928–948. Epub 2015/12/01. DOI:10.1111/cmi.12551
- Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 2021;21(7):426–440. Epub 2021/01/30. DOI:10.1038/s41577-020-00488-6
- Silva N, Abusleme L, Bravo D, et al. Host response mechanisms in periodontal diseases. J Appl Oral Sci. 2015;23(3):329–355. Epub 2015/07/30. DOI:10.1590/1678-775720140259.
- Garaicoa-Pazmino C, Fretwurst T, Squarize CH, et al. Characterization of macrophage polarization in periodontal disease. J Clin Periodontol. 2019;46(8):830–839. Epub 2019/06/04. DOI:10.1111/jcpe.13156.
- Zhou LN, Bi CS, Gao LN, et al. Macrophage polarization in human gingival tissue in response to periodontal disease. Oral Dis. 2019;25(1):265–273. Epub 2018/10/05. doi: 10.1111/odi.12983
- Guidi-Rontani C. The alveolar macrophage: the Trojan horse of Bacillus anthracis. Trends Microbiol. 2002;10(9):405–409. Epub 2002/09/10. DOI:10.1016/s0966-842x(02)02422-8
- Santiago-Tirado FH, Doering TL. False friends: phagocytes as Trojan horses in microbial brain infections. PLOS Pathog. 2017;13(12):e1006680. Epub 2017/12/15. DOI:10.1371/journal.ppat.1006680
- Werheim ER, Senior KG, Shaffer CA, et al. Oral pathogen Porphyromonas gingivalis can escape phagocytosis of mammalian macrophages. Microorganisms. 2020;8(9). Epub 2020/09/24. DOI:10.3390/microorganisms8091432.
- Giacona MB, Papapanou PN, Lamster IB, et al. Porphyromonas gingivalis induces its uptake by human macrophages and promotes foam cell formation in vitro. FEMS Microbiol Lett. 2004;241(1):95–101. Epub 2004/11/24. DOI:10.1016/j.femsle.2004.10.009.
- Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 2014;5:514. Epub 2014/11/05. DOI:10.3389/fimmu.2014.00514.
- Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015;2015:816460. Epub 2015/06/20. DOI:10.1155/2015/816460.
- Buchacher T, Ohradanova-Repic A, Stockinger H, et al. M2 polarization of human macrophages favors survival of the intracellular pathogen Chlamydia pneumoniae. PLoS ONE. 2015;10(11):e0143593. Epub 2015/11/26. DOI:10.1371/journal.pone.0143593.
- Wang Y, Xi J, Yi J, et al. Brucella induces M1 to M2 polarization of macrophages through stat6 signaling pathway to promote bacterial intracellular survival. Res Vet Sci. 2022;145:91–101. Epub 2022/02/19. DOI:10.1016/j.rvsc.2022.02.006.
- Lam RS, O’Brien-Simpson NM, Holden JA, et al. Unprimed, M1 and M2 macrophages differentially interact with Porphyromonas gingivalis. PLoS ONE. 2016;11(7):e0158629. Epub 2016/07/08. DOI:10.1371/journal.pone.0158629.
- Singh A, Wyant T, Anaya-Bergman C, et al. The capsule of Porphyromonas gingivalis leads to a reduction in the host inflammatory response, evasion of phagocytosis, and increase in virulence. Infect Immun. 2011;79(11):4533–4542. Epub 2011/09/14. DOI:10.1128/IAI.05016-11.
- Fujihara M, Muroi M, Tanamoto K, et al. Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: roles of the receptor complex. Pharmacol Ther. 2003;100(2):171–194. Epub 2003/11/12. DOI:10.1016/j.pharmthera.2003.08.003
- Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304(5673):1014–1018. Epub 2004/05/15. DOI:10.1126/science.1096158.
- Holden JA, Attard TJ, Laughton KM, et al. Porphyromonas gingivalis lipopolysaccharide weakly activates M1 and M2 polarized mouse macrophages but induces inflammatory cytokines. Infect Immun. 2014;82(10):4190–4203. Epub 2014/07/23. DOI:10.1128/IAI.02325-14
- Cutler CW, Jotwani R, Palucka KA, et al. Evidence and a novel hypothesis for the role of dendritic cells and Porphyromonas gingivalis in adult periodontitis. J Periodontal Res. 1999;34(7):406–412. Epub 2000/02/24. DOI:10.1111/j.1600-0765.1999.tb02274.x
- Zeituni AE, Carrion J, Cutler CW. Porphyromonas gingivalis –dendritic cell interactions: consequences for coronary artery disease. J Oral Microbiol. 2010;2:2. Epub 2010/01/01. DOI:10.3402/jom.v2i0.5782.
- El-Awady AR, Miles B, Scisci E, et al. Porphyromonas gingivalis evasion of autophagy and intracellular killing by human myeloid dendritic cells involves dc-sign-tlr2 crosstalk. PLOS Pathog. 2015;10(2):e1004647. Epub 2015/02/14. DOI:10.1371/journal.ppat.1004647.
- Krom BP, Kidwai S, Ten Cate JM. Candida and other fungal species: forgotten players of healthy oral microbiota. J Dent Res. 2014;93(5):445–451. Epub 2014/02/04. DOI:10.1177/0022034514521814
- Schlecht LM, Peters BM, Krom BP, et al. Systemic Staphylococcus aureus infection mediated by Candida albicans hyphal invasion of mucosal tissue. Microbiology (Reading). 2015;161(Pt 1):168–181. Epub 2014/10/22. DOI:10.1099/mic.0.083485-0.
- Van Dyck K, Viela F, Mathelie-Guinlet M, et al. Adhesion of Staphylococcus aureus to Candida albicans during co-infection promotes bacterial dissemination through the host immune response. Front Cell Infect Microbiol. 2020;10:624839. Epub 2021/02/20. DOI:10.3389/fcimb.2020.624839.
- Allison DL, Scheres N, Willems HME, et al. The host immune system facilitates disseminated Staphylococcus aureus disease due to phagocytic attraction to Candida albicans during coinfection: a case of bait and switch. Infect Immun. 2019;87(11). Epub 2019/08/28. DOI:10.1128/IAI.00137-19.
- Lewis LE, Bain JM, Lowes C, et al. Stage specific assessment of Candida albicans phagocytosis by macrophages identifies cell wall composition and morphogenesis as key determinants. PLOS Pathog. 2012;8(3):e1002578. Epub 2012/03/23. DOI:10.1371/journal.ppat.1002578.
- Ballou ER, Avelar GM, Childers DS, et al. Lactate signalling regulates fungal beta-glucan masking and immune evasion. Nat Microbiol. 2016;2:16238. Epub 2016/12/13. DOI:10.1038/nmicrobiol.2016.238.
- Karkowska-Kuleta J, Bartnicka D, Zawrotniak M, et al. The activity of bacterial peptidylarginine deiminase is important during formation of dual-species biofilm by periodontal pathogen Porphyromonas gingivalis and opportunistic fungus Candida albicans. Pathog Dis. 2018;76(4). Epub 2018/04/19. DOI:10.1093/femspd/fty033
- Bartnicka D, Karkowska-Kuleta J, Zawrotniak M, et al. Adhesive protein-mediated cross-talk between Candida albicans and Porphyromonas gingivalis in dual species biofilm protects the anaerobic bacterium in unfavorable oxic environment. Sci Rep. 2019;9(1):4376. Epub 2019/03/15. DOI:10.1038/s41598-019-40771-8.
- Peters BM, Ovchinnikova ES, Krom BP, et al. Staphylococcus aureus adherence to Candida albicans hyphae is mediated by the hyphal adhesin als3p. Microbiology (Reading). 2012;158(Pt 12):2975–2986. Epub 2012/08/25. DOI:10.1099/mic.0.062109-0.
- Sztukowska MN, Dutton LC, Delaney C, et al. Community development between Porphyromonas gingivalis and Candida albicans mediated by inlj and als3. MBio. 2018;9(2): Epub 2018/04/25. DOI:10.1128/mBio.00202-18.
- Tamai R, Sugamata M, Kiyoura Y. Candida albicans enhances invasion of human gingival epithelial cells and gingival fibroblasts by Porphyromonas gingivalis. Microb Pathog. 2011;51(4):250–254. Epub 2011/07/12. DOI:10.1016/j.micpath.2011.06.009
- Bartnicka D, Gonzalez-Gonzalez M, Sykut J, et al. Candida albicans shields the periodontal killer Porphyromonas gingivalis from recognition by the host immune system and supports the bacterial infection of gingival tissue. Int J Mol Sci. 2020;21(6):1984. DOI:10.3390/ijms21061984
- Shang L, Deng D, Buskermolen JK, et al. Multi-species oral biofilm promotes reconstructed human gingiva epithelial barrier function. Sci Rep. 2018;8(1):16061. Epub 2018/10/31. DOI:10.1038/s41598-018-34390-y.
- Shang L, Deng D, Buskermolen JK, et al. Commensal and pathogenic biofilms alter toll-like receptor signaling in reconstructed human gingiva. Front Cell Infect Microbiol. 2019;9:282. Epub 2019/08/27. DOI:10.3389/fcimb.2019.00282.
- McCormick BA. The use of transepithelial models to examine host-pathogen interactions. Curr Opin Microbiol. 2003;6(1):77–81. Epub 2003/03/05. DOI:10.1016/s1369-5274(02)00003-6
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16(1):1–13. Epub 2004/06/23. DOI:10.1016/j.nbd.2003.12.016
- Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–1596. Epub 2013/12/07. DOI:10.1038/nm.3407
- Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1):a020412. Epub 2015/01/07. DOI:10.1101/cshperspect.a020412
- Dando SJ, Mackay-Sim A, Norton R, et al. Pathogens penetrating the central nervous system: infection pathways and the cellular and molecular mechanisms of invasion. Clin Microbiol Rev. 2014;27(4):691–726. Epub 2014/10/04. DOI:10.1128/CMR.00118-13.
- Pulzova L, Bhide MR, Andrej K. Pathogen translocation across the blood-brain barrier. FEMS Immunol Med Microbiol. 2009;57(3):203–213. Epub 2009/09/08. DOI:10.1111/j.1574-695X.2009.00594.x
- Kim KS. Mechanisms of microbial traversal of the blood-brain barrier. Nat Rev Microbiol. 2008;6(8):625–634. Epub 2008/07/08. DOI:10.1038/nrmicro1952