
ABSTRACT
Apical hypertrophic cardiomyopathy is a rare form of hypertrophic cardiomyopathy that involves thickening of the distal portion of the left ventricular wall. Most commonly seen in the Japan, with a prevalence rate of about 15% of all HCM patient, its incidence in the USA is approximately 3% of HCM cases. We report a case of a 46-year-old woman with history of hypertension who presented to emergency department with worsening dyspnea and orthopnea with features of left ventricular hypertrophy (LVH) and diffuse large T-wave inversions in the lateral leads on a 12-lead ECG. Further work up revealed severe concentric LVH, with near obliteration of the LV cavity. Ventriculogram showed severe symmetric hypertrophy of the mid to lower septum, extending to the apex of left ventricle with significant pressure gradient of at least 160 mmHg across the apex to mid septal cavity, with no significant gradient across the left ventricular outflow tract. These findings were consistent with apical hypertrophic cardiomyopathy. She was treated with verapamil and metoprolol and has remained asymptomatic over last 2.5 years of follow-up. Although the clinical presentation of AHCM can be variable and nonspecific; however, hallmark findings on ECG and echo can be extremely important in its diagnosis.
Abbreviations: AHCM: Apical hypertrophic cardiomyopathy; ECG: Electrocardiogram; LVH: Left ventricular hypertrophy; LVOT: Left ventricular outflow tract
1. Background
Hypertrophic cardiomyopathy is a genetic disorder characterized by a different type of asymmetric left ventricular wall thickening [Citation1–Citation3]. Of the four common types, apical hypertrophic cardiomyopathy (AHCM) is a rare form, which was first reported in Japan in 1979 [Citation4]. It is described as non-obstructive hypertrophic cardiomyopathy, where the wall thickening involves the distal portion of the left ventricle wall, i.e., the apex.
2. Case report
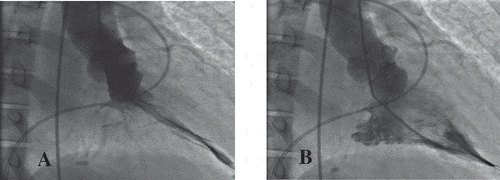
A 46-year-old woman with history of hypertension presented to the emergency department for acute worsening of dyspnea in the past week, which had started about a year ago. Upon initial presentation, she had stable vitals, except for elevated blood pressure (BP) of 181/108 mmHg. She was noted to be in pulmonary edema based on clinical examination, and a 12-lead ECG (electrocardiogram) showed findings of left ventricular hypertrophy (LVH) with diffuse large T wave inversions in the lateral leads (). 2-D echocardiography revealed preserved left ventricular ejection fraction (LVEF) and no significant valvular disease, but noted to have severe concentric LVH and with obliteration of the LV cavity (). Coronary angiography did not reveal any significant epicardial coronary artery disease and left ventriculogram showed severe symmetric myocardial hypertrophy of the mid to lower septum, extending to the apex of the left ventricle. There was over 160 mmHg pressure gradient across the LV apex to mid septum, however there was no significant gradient across the left ventricular outflow tract, consistent with a spade shaped / Japanese variety / apical hypertrophic cardiomyopathy ().
Figure 1. ECG of 46-year-old woman with apical hypertrophic cardiomyopathy, which revealed LVH pattern with diffuse T wave inversions.
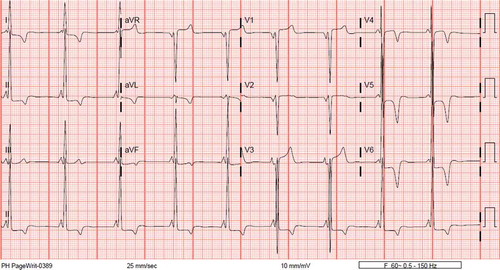
Figure 2. Echocardiogram of 46-year-old woman with apical hypertrophic cardiomyopathy (a and b), which revealed severe symmetric left ventricular hypertrophy with apical hypertrophy with near obliteration of the left ventricular cavity during systole.
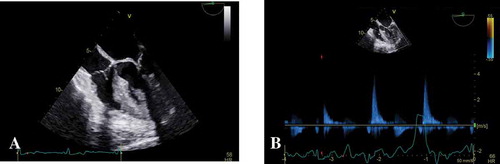
Figure 3. Cardiac catheterization of 46-year-old woman with apical hypertrophic cardiomyopathy during systole (a) and diastole (b). Findings were consistent with a spade shaped / Japanese variety / apical hypertrophic cardiomyopathy.
In view of the above findings, optimization of medical therapy with treatment with beta-blockers and /or calcium channel blockers was advised. She was also monitored for any underlying cardiac arrhythmias and is being closely followed at cardiology (HCM) clinic. She and her family members were offered genetic counseling which the result showed variant of uncertain significance (VUS) and she has been doing well with about two and a half years of follow up.
3. Discussion
Apical HCM / spade shaped /Japanese variety / yamaguchi syndrome is a rare morphologic variant of HCM in which the hypertrophy of the myocardium predominantly involves the apex of the left ventricle [Citation5]. These patients do not have LVOT obstruction but have midventricular obstruction. It has been noted to be more common in the Japanese population, where it accounts for about 15% of the HCM cases, versus 3% in the non-east asian population [Citation6]. Given the rarity and low incidence, little is known about the prevalence of mutations and genotype–phenotype correlations in apical hypertrophic cardiomyopathy.
3.1. Clinical presentation
Most patients present with symptoms in their 4th decade of life and this is more commonly seen in men. More than half of them are asymptomatic, and the ones that have clinical manifestations, can present with chest pain, palpitations, heart failure, dyspnea, atrial fibrillation or syncope [Citation7]. Unlike other variants of HCM, the prognosis of AHCM is relatively benign [Citation8]. The overall mortality rate of AHCM patients was 10.5% [Citation9]. However, one third of them may experience potential life-threatening complications such as myocardial infarction, apical infarction with aneurysm, ventricular tachycardia, sudden cardiac death or stroke [Citation10]. Klarich et al., followed up 193 patients over a median follow up of 78 months, and found that women had higher incidence of heart failure, atrial fibrillation, or death than men. Survival was also noted to be worse than expected, the observed versus expected 20-year survival was 47% versus 60% [Citation11]. Given limited data available due to the low prevalence, periodic lifelong follow-up is indicated for even asymptomatic patients and family screening should be considered.
3.2. Classical findings and diagnosis
The most frequent ECG findings are classic deep T-wave inversions in the precordial leads found in 93% of patients and about 65% of them have LVH [Citation12]. Negative T-waves with a depth >10 mm are found in 47% of patients with AHCM [Citation13].
The most useful non-invasive diagnostic studies are 2D echocardiography and cardiac MRI [Citation14,Citation15]. Two-dimensional echocardiography is the initial diagnostic study of apical hypertrophic cardiomyopathy [Citation16]. The diagnostic criteria for AHCM includes asymmetric LV hypertrophy, confined to the LV apex, with an apical wall thickness ≥15 mm and a ratio of maximal apical to posterior wall thickness ≥1.5 mm [Citation13,Citation17]. Detecting apical cardiomyopathy may be difficult, especially when the quality of transthoracic echocardiogram imaging is suboptimal; use of contrast echocardiography increases the yield of the study [Citation18].
Morphologically, AHCM can be divided into three types: pure focal, pure diffuse and mixed, of which pure focal is most common [Citation19]. However in clinical practice this sub-classification is not widely used and its clinical relevance is unknown. Choi et al. subtyped 182 patients with AHCM and found that mixed-type had a higher risk for development of atrial fibrillation, increased left atrial volume index and left ventricular longitudinal dysfunction [Citation19].
On contrast ventriculography AHCM shows a distinctive LV ‘spade-like’ configuration with significant gradient across the LV cavity. Unlike septal HCM, there is usually no significant gradient across the LVOT in AHCM [Citation20,Citation21].
3.3. Genetics
HCM is a genetic disorder of the myocardium, inherited in an autosomal dominant pattern with variable expressivity and age-related penetrance. So far, over 1500 mutations in 15 or more genes encoding the thick or thin myofilament proteins of the sarcomere have been reported in about 65% of the patients. Mutations in the cardiac myosin binding protein C gene (MYBPC3) are most common, accounting for up to 50% of the mutations identified. Mutations in the cardiac beta-myosin heavy chain gene (MYH7 gene) are second in frequency, followed by mutations in the troponin I (TNNI3), troponin T (TNNT2), and alpha-tropomyosin genes (TPM1). Up to 5% of patients have multiple mutations and about 30% have genetically unexplained disease.
Apical HCM has also been recognized as familial disease, implicating a role for genetics in the development of this morphological pattern of hypertrophy [Citation22]; however this has been poorly explained. Genetic analyses based on studies of small sized population with apical HCM identified most common mutation in sarcomere gene ACTC 1 (Actin, Alpha, Cardiac Muscle 1) - Glu101Lys missense mutation which was inherited in an autosomal dominant nature. It was also noted that few of them had family history of HCM. Based on a cohort study by Towe et al., which has been so far the largest cohort study for this population, (25%) had a positive genetic test, with the majority of mutations found in MYBPC3 (six; 35%) and MYH7 (six; 35%) [Citation23].
3.4. Treatment and follow up
Initial medical management with cautious use of beta-blockers and calcium channel blockers are recommended to decrease heart rate and increase diastolic filling, which decreases myocardial oxygen requirement [Citation24]. Surgical myotomy or alcohol septal ablation has been used in relieving LVOT obstruction in septal HCM, but there is very limited data and experience about its use in AHCM. a few cases have been reported where alcohol injection into the small vessels supplying a limited segment of mid-LV obstructive muscle or surgical apical myectomy helped alleviate the gradient and improve symptoms [Citation9].
Given the risk for sudden death, expert consensus supports the use of implantable defibrillator for primary prevention in those with one of the five following risk factors: a family history of sudden cardiac death, syncope, asymptomatic non-sustained ventricular tachycardia, an abnormal blood pressure response to exercise and a left ventricular wall thickness > 30 mm [Citation24].
Unlike other types of HCM, genetic association of AHCM has not been clearly established. A limited number of sarcomere gene defects, in particular, cardiac actin Glu101Lys, has shown to be associated with AHCM, however no particular defect has been found in many of the patients. Genetic testing should be offered to the patient and family members to help uncover patients at risk and to uncover unknown mutations.
4. Conclusion
The diagnosis of apical hypertrophic cardiomyopathy is important because most of these patients are relatively young. Classic findings include giant T inversions on ECG and characteristic spade shaped apical hypertrophy of the myocardium sparing the proximal septum. Even though, long standing prognosis is better that other types of HCM, this data is limited and these patient need aggressive surveillance to prevent sudden cardiac death. Genetic testing should be offered to the patient and family members, which can also help uncover unknown mutations.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Wigle ED, Sasson Z, Henderson MA, et al. Hypertrophic cardiomyopathy. The importance of the site and the extent ofhypertrophy. A review. Prog Cardiovasc Dis. 1985 Jul-Aug;28(1):1–83.
- Maron BJ. Hypertrophic cardiomyopathy. Lancet. 1997 Jul 12;350(9071):127–133.
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002 Mar 13;287(10):1308–1320.
- Sakamoto T, Tei C, Murayama M, et al. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasono-cardio tomographic study. Jpn Heart J. 1976 Sep;17(5):611–629.
- Arad M, Penas-Lado M, Monserrat L, et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation. 2005 Nov 1;112(18):2805–2811.
- Kitaoka H, Doi Y, Casey SA, et al. Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol. 2003 Nov 15;92(10):1183–1186.
- Yusuf SW, Bathina JD, Banchs J, et al. Apical hypertrophic cardiomyopathy. World J Cardiol. 2011;3(7):256–259.
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002 Feb 20;39(4):638–645.
- Soler R, Rodríguez E, Rodríguez JA, et al. Magnetic resonance imaging of apical hypertrophic cardiomyopathy. J Thorac Imaging. 1997 Jul;12(3):221–225.
- Kereiakes DJ, Anderson DJ, Crouse L, et al. Apical hypertrophic cardiomyopathy. Am Heart J. 1983 May;105(5):855–856.
- Sakamoto T. Apical hypertrophic cardiomyopathy (apical hypertrophy): an overview. J Cardiol. 2001;37(Suppl 1):161–178. Review.
- Choi EY, Rim SJ, Ha JW, et al. Phenotypic spectrum and clinical characteristics of apical hypertrophic cardiomyopathy: multicenter echo-Doppler study. Cardiology. 2008;110(1):53–61.
- Sakamoto T, Amano K, Hada Y, et al. Asymmetric apical hypertrophy: ten years experience. Postgrad Med J. 1986 Jun;62(728):567–570.
- Webb JG, Sasson Z, Rakowski H, et al. Apical hypertrophic cardiomyopathy: clinical follow-up and diagnostic correlates. J Am Coll Cardiol. 1990 Jan;15(1):83–90.
- Abinader EG, Sharif D, Shefer A, et al. Novel insights into the natural history of apical hypertrophic cardiomyopathy during long-term follow-up. Isr Med Assoc J. 2002 Mar;4(3):166–169.
- Ward RP, Pokharna HK, Lang RM, et al. Resting “Solar Polar” map pattern and reduced apical flow reserve: characteristics of apical hypertrophic cardiomyopathy on SPECT myocardial perfusion imaging. J Nucl Cardiol. 2003 Sep-Oct;10(5):506–512.
- Stainback RF. Apical hypertrophic cardiomyopathy. Tex Heart Inst J. 2012;39(5):747–749.
- Klarich KW, Attenhofer Jost CH, Binder J, et al. Risk of death in long-term follow-up of patients with apical hypertrophic cardiomyopathy. Am J Cardiol. 2013 Jun 15;111(12):1784–1791.
- Thanigaraj S, Pérez JE. Apical hypertrophic cardiomyopathy: echocardiographic diagnosis with the use of intravenous contrast image enhancement. J Am Soc Echocardiogr. 2000 Feb;13(2):146–149.
- Louie EK, Maron BJ. Apical hypertrophic cardiomyopathy: clinical and two-dimensional echocardiographic assessment. Ann Intern Med. 1987 May;106(5):663–670.
- Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol. 1979 Sep;44(3):401–412.
- Moro E, D’Angelo G, Nicolosi GL, et al. Long-term evaluation of patients with apical hypertrophic cardiomyopathy. Correlation between quantitative echocardiographic assessment of apical hypertrophy and clinical-electrocardiographic findings. Eur Heart J. 1995 Feb;16(2):210–217.
- Towe EC, Bos JM, Ommen SR, et al. Genotype–phenotype correlations in apical variant hypertrophic cardiomyopathy. Congenit Heart Dis. 2015;10:E139–E145.
- Gersh BJ, Maron BJ, Bonow RO, et al. ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary. Circulation. 2011;124:2761–2796.