
ABSTRACT
Acquired Hemophilia A (AHA) is a bleeding diathesis related to the development of factor VIII inhibitor, which can frequently go undetected. It commonly manifests as spontaneous mucosal bleeds without any known history of a bleeding disorder, but has the potential to cause life threatening bleeding especially in elderly patients with underlying comorbidities. Here we describe a case of AHA in a 78 year old female presenting with spontaneous mucocutaneous bleeding as tongue hematoma and recurrent gastrointestinal (GI) bleeding. Underlying etiology remained unclear in this case. While she did not require any reversal agents to control bleeding, the patient received steroids and rituximab as inhibitor eradication therapy.
1. Background
Acquired Hemophilia A (AHA) is a rare bleeding disorder related to formation of autoantibodies to Factor VIII. AHA is commonly associated with autoimmune disorders, such as rheumatoid arthritis, as well as postpartum state and malignancies, but in approximately half of the cases the etiology remains unclear [Citation1]. It commonly manifests as spontaneous mucosal bleeds without any known history of a bleeding disorder, but has the potential to cause life threatening bleeding especially in elderly patients with underlying comorbidities. The etiopathology remains unclear due to limited access to biological samples from untreated individual, though various speculations have been made regarding involvement of CD4 cells, HLA, and IgG antibodies [Citation2]. While one-third of the cases can be self-limited, steroids and immunosuppressive agents, including rituximab, cyclophosphamide, azathioprine, and others have been used for treatment [Citation3].
2. Case description
A 78 year old African American female presented to the hospital with slurred speech that developed over two days. She arrived at the hospital concerned that she was having a stroke. In addition, the patient described having dysphagia, odynophagia, and that she had noticed purple blotches inside her oral cavity. The patient had developed worsening anemia for past few months and had needed three blood transfusions for symptomatic anemia within past 2 months. She had undergone two endoscopies and was found to have a bleeding gastric ulcer both times, for which she was treated during the endoscopies. She also described having multiple spontaneous bruises and ecchymotic lesions sporadically throughout her body over past few months. In addition to recurrent GI bleeding, past medical history was significant for chronic iron deficiency anemia, type 2 diabetes mellitus and multinodular goiter requiring thyroidectomy. She denied any history of a bleeding diathesis in the family.
On presentation, patient was hemodynamically stable. Physical exam was notable for a large ecchymotic lesion along floor of mouth, as well as on ventral tongue, while the rest of the buccal mucosa was normal (). She also had multiple ecchymoses over her chest and upper extremities.
Figure 1. Demonstrating oral mucosal ecchymoses.
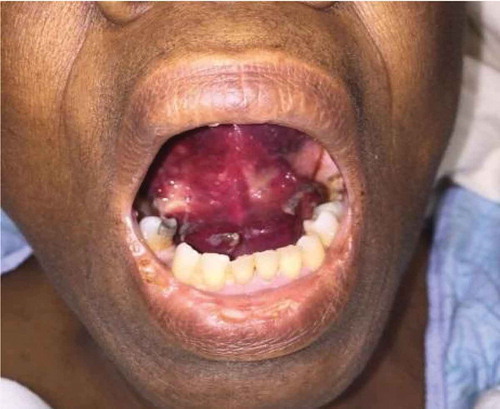
On admission, the differential diagnosis included possible angioedema, oral cavity malignancy, and autoimmune phenomenon. Lab investigations were significant for anemia with hemoglobin of 6.7 g/dL, and prolonged aPTT of 35–48 s (reference range 20–28 s) with normal PT. There were no signs of GI bleeding on admission this time and she was not receiving any heparin products. In this context, tongue swelling and ecchymotic lesion were believed to be limited to the submucosal area and diagnosis of a bleeding diathesis was considered. Due to the prolonged aPTT, a mixing study was performed and did not correct, suggesting presence of a factor inhibitor. Further testing revealed an extremely low factor VIII activity of <1%, high factor VIII inhibitor level of 59.7 bethesda units (reference range </ = 0.50), elevated von Willebrand Factor level of 256% (reference range 52–214%) and elevated Factor IX level of 201% (reference range 78–184%), confirming the diagnosis of AHA. Patient was started on high dose steroids (Methylprednisolone 60 mg IV twice a day) and monitored for any signs of bleeding. She was also transfused 1 unit of packed red blood cells due to symptomatic anemia. Any invasive interventions and procedures were minimized to avoid provoking further bleeding. She continued to do well and did not require any bypassing agents like recombinant factor VIIa and activated prothrombin complex concentrate (aPCC) products; or factor VIII replacement to control bleeding as the submucosal hematoma resolved over next few days. Her hemoglobin remained stable and was 9.7 g/dL She was discharged on high dose Prednisone at 60 mg PO daily with outpatient hematology follow up. The use of prednisone caused side effects, including insomnia, impaired blood glucose control, and fluid retention. Therefore, prednisone was tapered off and she was switched to rituximab. She received four doses of rituximab 375 mg/m2 weekly and tolerated the infusions very well. Factor VIII titers increased from <1% to 11% and inhibitor titer decreased from 59 BU to 2 BU immediately after four doses were given. At 2 months after starting rituximab therapy, labs revealed a normal PT/PTT factor VIII level of 113%, and the bethesda assay was not done due to the normal factor VIII level. The patient did not have any further bleeding episodes or other side effects from treatment. At 6 months, she maintained a normal aPTT, stable hemoglobin (10.7 g/dL) and remained asymptomatic. Patient is being monitored every 3 months through complete blood count and aPTT.
3. Discussion
AHA is a rare clinical entity but is associated with significant morbidity and mortality. Incidence is reported to be 1.48/million per year, but is significantly higher in elderly, being 14.7% in patients over 85 years of age [Citation3]. It is likely that incidence is higher due to unreported and undiagnosed cases of the disease. Although most of the cases tend to be idiopathic, underlying disorder exists in about half of the patients. In a survey of 215 non-hemophilic patients who developed inhibitor against factor VIII, most of the patients were over 50 years of age, 8% had rheumatoid arthritis, and 7% were at some peripartum stage. Other associations included asthma, malignancies, allergy to penicillin, and other autoimmune diseases. No cause could be found in 46% of patients [Citation1]. In nonpregnant patients, AHA is equally distributed in men and women. Both lymphoproliferative and solid malignancies are known to be associated with AHA; the most common being chronic lymphocytic leukemia and prostate cancer, followed by lung cancer [Citation4,Citation5]. Recently, CTLA 4 inhibitor Ipilimumab has been reported to lead to the development of factor VIII inhibitor [Citation6].
Typically, patients with AHA present with new onset bleeding symptoms with no previous history of bleeding. The bleeding associated with AHA tends to be in mucocutaneous sites or soft tissue and can lead to recurrent GI, intramuscular, or intracerebral bleeding in elderly patients as compared to hemarthrosis in younger patients with congenital hemophilia [Citation4]. According to EACH2 registry, clinical bleeding occurs in 96.2% of the patients and is clinically severe in 69.5% patients requiring hemostatic therapy. Occasionally, incidental isolated prolongation of aPTT is the only finding [Citation7,Citation8].
Isolated prolongation of aPTT with normal prothrombin time (PT), thrombin time (TT), and platelet count is indicative of either a factor deficiency in intrinsic pathway or a factor inhibitor. Other common causes are heparin therapy or lupus anticoagulant [Citation9,Citation10]. The next step is to perform a mixing study, in which the patient’s plasma is combined with normal plasma in a 1:1 ratio and incubated at 37 °C for at least an hour. If the patient’s sample has a factor deficiency, the mixed sample should correct the aPTT. If an inhibitor is present in the patient’s plasma, the aPTT will continue to be prolonged. Sensitivity of detection of mild inhibitors is increased if ratio of patient to control plasma is increased to 4:1 and incubated for 2 h [Citation11]. To avoid misinterpretation as lupus inhibitor, a confirmatory step is advised to measure Factor VIII activity, which will be low in these cases. After confirming the presence of the inhibitor, the Bethesda assay is used to evaluate inhibitor titer levels. Neither factor VIII activity nor inhibitor titer correlates well with the disease activity or severity of bleeding [Citation10].
Management of AHA consists of management of acute bleeding episodes with hemostatic measures, and inhibitor eradication to prevent recurrent episodes. There are no randomized control data to guide us on management, and most of the data is derived from observational data from the EACH2 registry and from treatment of patients with Congenital Hemophilia with or without inhibitors. International practice guidelines have been derived by Huthe-Kuhne et al., based on analysis of the data available and clinical experience [Citation10].
For acute bleeding episodes, hemostatic therapy should be initiated as soon as possible. Bypassing agents are currently first line treatment, which include recombinant factor VIIa (rFVIIa) and aPCCs like factor VIII inhibitor bypassing agent (FEIBA). rFVIIa works by initiating formation of complex between tissue factor and FVIIa, leading to increased thrombin production and accelerated fibrin clot formation at sites of vascular injury bypassing the intrinsic pathway. rFVIIa has been noted to be effective/partially effective in 90% nonsurgical and 86% surgical cases [Citation12]. Some studies have reported it to have an effective response in 100% cases when used as first line therapy, 75% good or partial response as second line therapy, and 17% response when used as salvage therapy [Citation10,Citation13]. aPCC is an alternative to rVIIa as a bypassing agent. aPCC products contain four anticoagulation factors (II, VII, IX, and X) in activated and inactivated forms. This is different from other PCC products, which contains three factor (II, IX, and X) or four factor as above but contain inactive forms only. Factor eight inhibitor bypassing activity (FEIBA) is the only aPCC available in USA, containing activated factor VII along with non-activated factors II, IX, and X [Citation14]. FEIBA has been known to have a response rate of 76% in severe bleeding and 100% in moderate bleeding as observed in a retrospective study [Citation10,Citation15]. Based on the data in the EACH2 registry, bypassing agents were superior when compared with desmopressin or factor VIII concentrates, however there is no head-to-head comparison of the efficacy of aPCC and rVIIa. Major adverse effects are thromboembolic events including myocardial infarction, disseminated intravascular coagulation (DIC), pulmonary embolism, and stroke, which are noted to be higher in elderly population due to underlying comorbidities. Rates of thrombotic events were noted to be 2.9% with rVIIa and 4.8% with aPCC [Citation7]. Therefore, caution is advised when using them in elderly patients.
If bypassing agents are unavailable, alternative treatment, such as recombinant factor VIII concentrates or desmopressin should be used. These agents can also be used in patients with low inhibitor titer (<5 Bethesda Units) or insignificant bleeding. Treatment failure should be considered if there is no change in rate of blood loss, if the hemoglobin fails to increase despite transfusions, or if there is any new bleed or increasing pain at the hematoma site despite receiving first-line hemostatic treatment, and an alternative agent should be used. Combination therapy of rfVIIa and aPCC, although effective, is associated with higher rate of life- or limb-threatening thromboembolic complications [Citation10].
AHA carries a mortality rate of up to 22% and major bleeding episodes have occurred in about 87% cases [Citation1]. The rate of inhibitor related complications is high. Therefore, all patients with AHA should undergo inhibitor eradication therapy. As bleeding risk is unrelated to inhibitor level, the levels should not be used to decide whether or not to start immunosuppressive therapy. A variety of regimens have been tested for efficacy and are available. Initial treatment should consist of a corticosteroid with or without cyclophosphamide. The recommended dose of prednisone is 1 mg/kg/day for 4–6 weeks while the recommended dose for cyclophosphamide is 1.5–2 mg/kg/day for 6 weeks. The rate of inhibitor eradication has been found to be 60–70% with the use of prednisone alone and 70–80% when used in combination with oral cyclophosphamide (given at doses of 50–150 mg/day). There was observed no difference in overall survival and disease-free survival between prednisone alone and the combination [Citation3].
If first-line therapy fails or is contraindicated, rituximab can be used as second line therapy as was the case with our patient. Rituximab is a chimeric human/murine monoclonal antibody to the CD20 antigen on surface of B-cells. It is currently used extensively to treat CD20-positive hematologic malignancies and has been used to treat many autoimmune diseases, and hence can also be used in AHA [Citation16]. In a systematic review by Franchini et al., rituximab was found to have an efficacy rate of over 90% without any significant adverse events or opportunistic infections at a standard dose of 375 mg/m2 given weekly for 4 weeks. Data from small, uncontrolled studies, and case reports indicate that a standard course of rituximab is more effective in treating low titer inhibitors, while higher titers may require longer duration of treatment or the addition of cyclophosphamide [Citation17]. As of today, there are no randomized, placebo-controlled trials to assess the efficacy of rituximab, but it can be inferred from the available data that it is a good option for inhibitor eradication in patients with AHA. However, if there is no response in 6–8 weeks, alternative combination therapies should be considered. IVIg has been used in the treatment of a variety of autoimmune conditions, and although it seems logical to use IVIg in patients with AHA, it has been found to have low efficacy. Even in patients with low inhibitor titers, IVIg had a reported inhibitor eradication rate of 10–12% [Citation18,Citation19]. Currently, IVIg is not a recommended therapy for inhibitor eradication [Citation9]. Other alternatives that have been used include azathioprine, vincristine, mycophenolate, and cyclosporine. All immunosuppression regimens have potential side effects, including cytopenias, alopecia, sepsis, and opportunistic infections. Treatment should be individualized based on potential risks and benefits for each patient.
Beyond immunosuppressive agents, immunoadsorption protocols are newer techniques that have been successfully used in severe cases with life-threatening bleed to eradicate inhibitors. At this time, these can only be done in centers with necessary expertise and experience. The modified Bonn-Malmo protocol involves administration of high dose Factor VIII, IVIg, and immunosuppression, and has a reported success rate of 91% in all patients and 97% in patients without cancer. No recurrence was noted at a median follow-up of 48 months [Citation20,Citation21]. Other protocols are also available with similar results.
In all patients receiving inhibitor eradication therapy, CBC, aPTT, FVIII activity, and inhibitor titer should be measured on a weekly or biweekly basis during the treatment in order to closely monitor their response. In patients who achieve complete remission (defined by undetectable inhibitor and normal FVIII activity), aPTT or Factor VIII:C assays can be used to monitor for relapse. Median relapse time appears to be 7–9 months after cessation of therapy; therefore labs should be checked 2–3 months for first year [Citation9].
4. Conclusion
AHA is a rare hematologic disorder and can often go undetected. It is associated with significant morbidity and mortality due to the occurrence of severe and life threatening bleeding. Early detection is vital to improving outcomes. Due to the rarity of the disorder, it is hard to conduct randomized clinical trials to guide management of the bleeding and inhibitor eradication. Management is mostly based on case reports, small cohorts, uncontrolled trials, and retrospective studies. More research is warranted given the severity of the disease.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost. 1981;45(3):200.
- Mahendra A, Padiolleau-Lefevre S, Kaveri SV, et al. Do proteolytic antibodies complete the panoply of the autoimmune response in acquired haemophilia A? Br J Haematol. 2012;156:3.
- Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the UK: a 2-year national surveillance study by the UK Haemophilia Centre Doctors’ Organisation. Blood. 2007;109:1870.
- Reeves BN1, Key NS. Acquired hemophilia in malignancy. Thromb Res. 2012 Apr;129(Suppl 1):S66–8.
- Franchini M, Gandini G, Di Paolantonio T, et al. Acquired hemophilia A: a concise review. Am J Hematol. 2005;80:55.
- Delyon J, Mateus C, Lambert T. Hemophilia A induced by ipilimumab. N Engl J Med. 2011;365:1747.
- Baudo F, Collins P, Huth-Kühne A, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) registry. Blood. 2012;120:39.
- Knoebl P, Marco P, Baudo F, et al. EACH2 registry contributors. demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012 Apr;10(4):622–631.
- Sborov DW1, Rodgers GM. How I manage patients with acquired haemophilia A. Br J Haematol. 2013 Apr;161(2):157–165. Epub 2013 Feb 4.
- Huth-Kühne A, Baudo F, Collins P, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica. 2009;94(4):566–575.
- Lossing TS, Kasper CK, Feinstein DI. Detection of factor VIII inhibitors with the partial thromboplastin time. Blood. 1977;49:793.
- Sumner MJ, Geldziler BD, Pedersen M, et al. Treatment of acquired haemophilia with recombinant activated FVII: a critical appraisal. Haemophilia. 2007;13:451–461.
- Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost. 1997;78:1463–1467.
- Awad NI, Cocchio C. Activated prothrombin complex concentrates for the reversal of Anticoagulant-Associated Coagulopathy. Pharm Ther. 2013;38(11):696–701.
- Sallah S. Treatment of acquired haemophilia with factor eight inhibitor bypassing activity. Haemophilia. 2004;10:169–173.
- Boye J, Elter T, Engert A. An overview of the current clinical use of the anti-CD20 monoclonal antibody rituximab. Ann Oncol. 2003;14(4):520–535.
- Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systemic review. Crit Rev Oncol Hematol. 2007;63(1):47–52.
- Crenier L, Ducobu J, Des Grottes J-M, et al. Low response to high-dose intravenous immunoglobulin in the treatment of acquired factor VIII inhibitor. Br J Haematol. 1996;95:750–753.
- Schwartz R, Gabriel D, Aledort L, et al. A prospective study of treatment of acquired (autoimmune) factor VIII inhibitors with high-dose intravenous gammaglobulin. Blood. 1995;86(2):797–804. [Accessed 2017 Jan 25]
- Nemes L, Pitlik E. New protocol for immune tolerance induction in acquired hemophilia. Haematologica. 2000;85(10 Suppl):64–68.
- Zeitler H, Ulrich-Merzenich G, Hess L, et al. Treatment of acquired hemophilia by the Bonn-Malmo protocol: documentation of an in vivo immunomodulating concept. Blood. 2005;105:2287–2293.