
ABSTRACT
Rhabdomyosarcoma is an aggressive malignant soft-tissue sarcoma that develop from undifferentiated mesenchymal cells. Less than 1% of all adult solid malignant cancers are sarcomas, and RMSs represent less than 2–5% of adult sarcomas. RMS is divided into three main subtypes: Embryonal, alveolar and pleomorphic RMS (PRMS). Most common subtype in adults is PRMS. Most common primary sites are extremities, trunk wall, and genitourinary organs. Metastasis are often found at diagnosis. 5-year overall survival rates were reported in the Surveillance, Epidemiology, and End Results database (SEER) to be 63% for pediatric patients and 27% for adults. Given the rarity of the adult PRMS, variation in its clinical presentation, characteristics of the tumor itself and the prognosis; there are very limited data available to guide the management of adults with PRMS. Herein we present a case report of pleomorphic rhabdomyosarcoma of the right thigh in a 60-year-old male who achieved a long-term survival (30 months) which was accomplished by multimodality treatment including surgery, radiotherapy, and chemotherapy.
1. Introduction
Rhabdomyosarcoma is an aggressive malignant soft-tissue sarcoma that develop from undifferentiated mesenchymal cells [Citation1]. It has a propensity for advanced disease early in its course [Citation2].
Less than 1% of all adult solid malignant cancers are sarcomas, and RMSs represent less than 2–5% of adult sarcomas [Citation3] RMS is divided into three subtypes: Embryonal, alveolar and pleomorphic RMS (PRMS). The most common subtype of rhabdomyosarcoma to occur in adults, however, is the (PRMS) [Citation3], and its incidence increases with age [Citation4]. The most common primary sites are extremities, trunk wall, and genitourinary organs. Based on recent SEER database of 1071 adults (older than 19 years) with RMS, the most common primary sites included extremities (26%) and trunk (23%) followed by genitourinary tract (17%) and head and neck (9%). With regards to histology, Pleomorphic RMS was present in 19% in adults vs. 1% in children; the estimated 5-year OS rates were 63% for pediatric patients and 27% for adults [Citation5]. Metastasis are often found at diagnosis, commonly to lungs, lymph nodes, and bone marrow [Citation4].
The diagnosis is based on histomorphologic, immunohistochemical, and molecular characteristics. RMS arises predominantly in males between 46 to 62 years of age [Citation2,Citation6,Citation7]. Given the rarity of the PRMS, variation in its clinical presentation, characteristics of the tumor itself and the prognosis; there are very limited data available to guide the management of adults with RMS. Multimodality treatment approach including surgery, Radiation therapy and chemotherapy all has been used in these patients with variable outcomes. Herein we present a case report of pleomorphic rhabdomyosarcoma of the right thigh in a 60-year-old male who achieved a long-term survival (30 months).
2. Case presentation
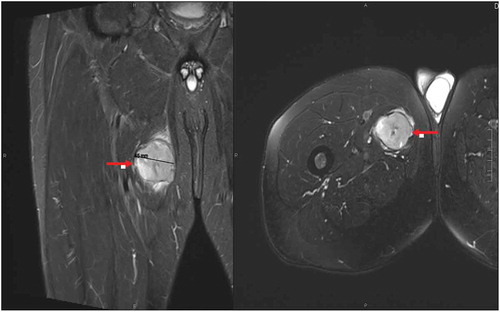
A 60-year-old male who presented to his primary care physician (PCP) with an enlarging painless right thigh mass in December 2016. On examination it was hard but non-tender on palpation, attached to the underlying structures and about the size of a tennis ball, it was located at the upper 1/3rd portion of the thigh medially. Ultrasound of the right groin showed a heterogeneous mass with a concern of soft tissue sarcoma. Subsequent magnetic resonance imaging (MRI) of the area showed hyperintense mass within the adductor brevis muscle measuring 4.5 cm (). Computed tomography (CT) guided biopsy was done and the histological findings were consistent with high-grade pleomorphic rhabdomyosarcoma (PRMS), the cells were strongly reactive with Desmin, and fluorescence in situ hybridization (FISH) for anaplastic lymphoma kinase (ALK) was negative. CT staging examination in January 2017 showed multiple subcentimeter pulmonary nodules in the right lower lobe measuring less than 4 mm concerning for metastasis and the diagnosis of metastatic high-risk PRMS was made. Subsequently, he was treated with three cycles of doxorubicin, ifosfamide, and mesna as neoadjuvant chemotherapy. Follow-up CT scans showed a progression of disease in both primary and lung sites and the patient was treated with local neoadjuvant radiation therapy. In August 2018 he underwent wide excision of the right thigh mass (7 cm), with 80% of it has viable PRMS, and resection margins were negative. He was followed by serial imaging every 3 months. Subsequently, in March 2019 CT chest showed progression of the pulmonary nodules within the right lower lobe increasing in size from 6 to 8 mm and from 4 to 5 mm nodules. MRI of the right lower extremity was repeated at the same time and did not show any evidence of residual or recurrent disease. Patient underwent Right Video-assisted thoracoscopic wedge resection of right lower lobe enlarging nodules. Pathology examination of one of these nodules (8 mm) showed high-grade sarcoma consistent with metastases with negative margins and the 5 mm nodule represented an area of benign fibrosis.
The patient is still alive and in complete remission from his PRMS. His long-term survival has reached more than 30 months at the last follow-up in May 2019.
3. Discussion
There is limited data to guide management of adults with RMS given its rarity. Multiple studies have used a variety of multidrug chemotherapy regimens, including cyclophosphamide or ifosfamide, doxorubicin, and/or dactinomycin with or without vincristine or other drugs such as cisplatin, carboplatin, and etoposide.
A retrospective study at MD Anderson for 82 adult patients with RMS treated with chemotherapy regimens containing vincristine and cyclophosphamide with dactinomycin or doxorubicin, the 10-year overall, disease-free, and metastasis-free survival rates were 47%, 45%, and 59%, respectively [Citation8].
Another large retrospective study evaluated 171 RMS patients, out of them 37 patients had PRMS. Surgery was the main treatment in patients with PRMS (74%) followed by radiotherapy and chemotherapy. Median follow up was 28 months. Five-year Event Free Survival rate and OS were 29.9% and 53.4%, respectively [Citation3]. Another small retrospective study investigated the effect of chemotherapy in 11 adult patients with PRMS treated with vincristine, doxorubicin, and ifosfamide. Overall response rate of 86%; the 2-year Overall Survival and disease-free survival rates were 55% and 64%, respectively [Citation9]. Generally, there are not much recommendations available regarding the chemotherapy sensitivity in adults with RMS [Citation2,Citation3,Citation7,Citation8]. However, in the absence of data from prospective clinical trials, there are no definitive, optimal regimens for the management of adult RMS. We strongly recommend that all adult patients with RMS should be referred to institutions with expertise in treating soft tissue sarcomas. Evaluation by a multidisciplinary team involving medical, surgical, and radiation oncologists is strongly encouraged.
4. Conclusion
This case report demonstrates an exceptionally rare malignancy in an adult patient with an impressive long-term survival. His survival exceeding 30 months was achieved by multimodality treatment including surgery, radiotherapy, and chemotherapy.
Figure 1. T2 MRI scan of the right thigh showing 4.5 cm mass (red arrows) within the adductor brevis muscle with mild surrounding edema.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Hawkins WG, Hoos A, Antonescu CR, et al. Clinicopathologic analysis of patients with adult rhabdomyosarcoma. Cancer. 2001;91(4):794–803.
- Esnaola NF, Rubin BP, Baldini EH, et al. Response to chemotherapy and predictors of survival in adult rhabdomyosarcoma. Ann Surg. 2001;234(2):215–223.
- Ferrari A, Dileo P, Casanova M, et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer. 2003;98(3):571–580.
- Simon JH, Paulino AC, Ritchie JM, et al. Presentation, prognostic factors and patterns of failure in adult rhabdomyosarcoma. Sarcoma. 2003;7(1):1–7.
- Sultan I, Qaddoumi I, Yaser S, et al. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27(20):3391–3397.
- Stock N, Chibon F, Nguyen Binh MB, et al. Adult-type rhabdomyosarcoma: analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am J Surg Pathol. 2009;33(12):1850–1859.
- Furlong MA, Mentzel T, Fanburg-Smith JC. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14(6):595–603.
- Little DJ, Ballo MT, Zagars GK, et al. Adult rhabdomyosarcoma: outcome following multimodality treatment. Cancer. 2002;95(2):377–388.
- Ogilvie CM, Crawford EA, Slotcavage RL, et al. Treatment of adult rhabdomyosarcoma. Am J Clin Oncol. 2010;33(2):128–131.