
ABSTRACT
Pulmonary alveolar proteinoisis (PAP) is a rare disease characterized by accumulation of proteinaceous material in the alveolar spaces. Here, we report a case of mild dyspnea with incidental ‘crazy-paving’ pattern on chest computed tomography (CT). Further evaluation and bronchoscopy found to have PAP.
1. Case report
A 46-year-old man, former smoker, was admitted to the hospital initially with alcohol intoxication. He reports drinking ‘large amount’ of alcohol at his friend’s house the night before. He consumes one pint of tequila per week. During his admission for alcohol detoxification, he endorsed progressive dyspnea on exertion that started 4 months ago with a non-productive cough. Cough was dry with no hemoptysis or wheezing. He use to run every morning but recently noticed he can not run as far as before. This is the first time he has these symptoms. He reports nocturnal nasal congestion for the past 5 months. He had reactive airway disease as a child exacerbated by the cold. He denies any fever, joint pain, or diarrhea. Medical history was significant for psoriasis and alcoholic liver disease. The psoriasis was diagnosed 6 years ago in El Salvador. He also has alcoholic liver disease with elevated baseline liver enzymes and heterogenous liver on ultrasound. He was born in El Salvador and moved to America many years ago. He works at a dry-cleaning facility for 8 years and is in contact with ammonia-based cleaning agents. His last trip was to El Salvador 6 years ago. He lives with three of his friends and has no pets.
The physical exam revealed an afebrile man in no distress. He was alert, with a blood pressure 141/98 mm Hg, pulse of 90 beats/min, temperature 36.6°C, respiratory rate 16 breaths/min, and oxygen saturation 96% on 3-l nasal cannula. He mild diffuse expiratory wheezes and bilateral basilar crackles. He had a normal cardiac rhythm without murmurs. Skin exam pertinent for psoriatic lesions on his forearms.
The patient’s complete blood count revealed white blood cell count of 4,730 cells/mm3, hemoglobin 17.8 g/dl, and a platelet count of 102 × 104/mm3. The WBC differential were all within the normal range. Procalcitonin was <0.09 ng/mL (1-3)-B-D-Glucan was 34 pg/mL.
Result of the arterial blood gas with 3 L nasal cannula revealed pH 7.44, pCO2 31 mm Hg, pO2 77 mm Hg, HCO3 20.3 with alveolar-arterial oxygen gradient 112 mm Hg. HIV serology was negative. Antinuclear antibody screen and rheumatoid factor were negative.
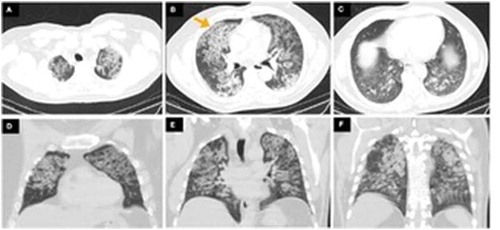
Findings on chest radiograph included extensive reticulonodular changes in both lungs with normal heart size (). Computed tomography (CT) revealed normal lung volumes with interlobular thickening and diffuse ground glass opacities with a ‘crazy paving’ pattern. The ground glass opacities are decreasing in pattern, more prominently in the upper lobes. No pleural effusions or pneumothorax (). There were no previous radiological findings to compare it with.
Figure 1. Anterior-posterior chest X-ray showing extensive bilateral nodular densities
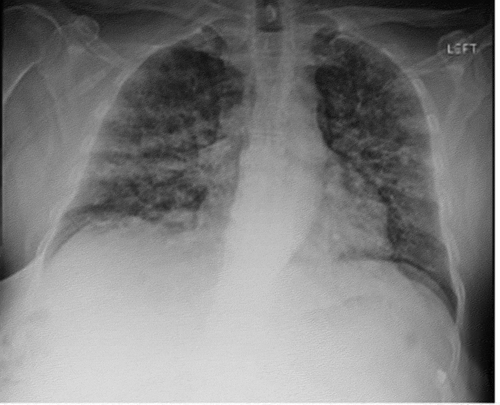
Figure 2. (A-C) Axial computed tomography (CT) showing bilateral ground glass opacities with thickening of the intervening interlobular septa representing a ‘crazy paving’ pattern (yellow arrow). (D–F) Coronal CT showing decreasing pattern of ground glass opacities most prominently affecting in the upper lobes
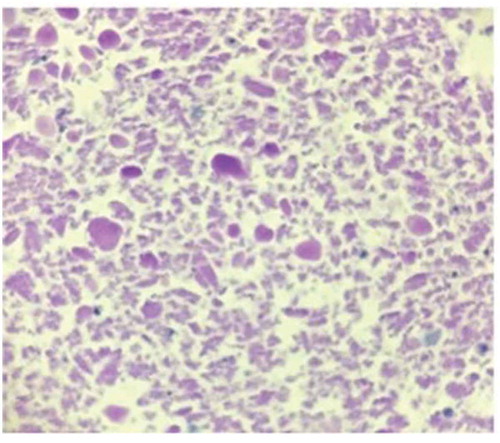
To determine the diagnosis, he underwent a flexible bronchoscopy with bronchoalveolar lavage. BAL from the right middle lobe was hazy with 1,099/cumm WBC and 35,000/cumm. The lavage was negative for pneumocystis jirovecii and acid fast stain. Cytopathology however revealed periodic acid-Schiff (PAS) positive proteinaceous material (). Bronchial cytology was negative for malignancy. Further investigation revealed the presence of serum anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) auto-antibody diagnostic of protein alveolar proteinousis. He had mild symptoms and was managed conservatively. Follow-up in 4 months revealed continuous mild shortness of breath with exertion not meeting the criteria for whole lung lavage.
Figure 3. Periodic acid-Schiff (PAS) stain of the right bronchial middle lobe lavage cell block (100× magnification) showing few pulmonary macrophages in a background of abundant PAS-positive proteinaceous material
2. Discussion
Pulmonary alveolar proteinoisis (PAP) is a rare disease first described by Samuel H. Rosen in 1958 [Citation1]. The overall incidence rate is 0.2 cases per million per year and prevalence of 3.7 to 40 cases per million [Citation2]. It is a disorder characterized by surfactant, lipid and protein accumulation in the alveolar spaces [Citation3]. The etiology of PAP is varied and thought to be due to either autoimmune, secondary/exposure, and/or congenital causes [Citation4,Citation5]. Autoimmune PAP compromises 90% of the cases and it is a result of anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) antibodies that lead to a deficiency of GM-CSF [Citation6]. GM-CSF is an essential cytokine for development of alveolar macrophage metabolic and immune functionCitation5. Secondary PAP is characterized by dysfunctional alveolar macrophages secondary to hematologic or environmental factors [Citation7]. Congenital PAP is a result of genetic mutation in GM-CSF receptor proteins or surfactant production disorders which leads to impaired macrophage maturation [Citation8]. The clinical presentation of PAP is typically insidious with dyspnea as the main complaint in 50% to 90% of the cases followed by a non-productive cough in 23% to 65% of the cases. Up to one-third may be asymptomatic and found incidentally as in our patient[Citation6]. The chest X-ray in PAP usually has a ‘batwing distribution’ with bilateral alveolar opacities in peri-hilar, basilar distribution with peripheral parenchymal sparing. Uncommonly it may present as a unilateral, asymmetric and nodular pattern [Citation7]. The thick lipoproteinaceous material can cause bronchial obstruction presenting as segmental atelectasis [Citation8].
A high-resolution computer tomography often reveals a ‘crazy paving’ appearance secondary to interlobular septal thickening in polygonal shapes superimposed on diffuse ground glass opacities [Citation9].
The diagnosis of PAP is supported by the presence of periodic acid-Schiff (PAS) positive flocculent proteinaceous material on histopathology. Alveolar macrophages are also typically filled with PAS-positive material [Citation11].
Asymptomatic and mild dyspnea are usually treated with supportive care and pulmonary function test monitoring every 6 to 12 months [Citation12]. Moderate to severe symptoms are defined as dyspnea at rest, resting arterial partial pressure of oxygen of less than 65 mmHg, resting alveolar-arterial gradient more than 40 mmHg or oxygen desaturation on 6-min walk test. Those patients often require repeat whole lung lavage treatments to improve their symptoms [Citation12,Citation13].
In refractory PAP, rituximab and plasmapheresis were used separately and shown to be effective in improving oxygenation in a few studies [Citation3,Citation14,Citation15,Citation16]. A recurrence of PAP was observed in lung transplant recipients. Steroids worsen PAP and increase risk for infections [Citation17]. The only proven therapy for secondary PAP is treatment of an associated identified underlying disease [Citation18].
Disease course varies and prognosis is unpredictable. Autoimmune PAP treated with whole lung lavage showed a 5-year survival rate of 95% [Citation14]. The most common causes of death were respiratory failure and superimposed infection. Spontaneous remission occurred in less than 10% of the patients [Citation18]. Secondary PAP median survival is less than 15 months and the most common cause of death is an associated underlying hematologic disease such as acute myeloid leukemia [Citation7].
3. Conclusion
In our patient, autoimmune pulmonary alveolar proteinosis was suspected based on his initial CT findings and confirmed by subsequent pathologic and serologic testing. The presence of IgG anti-GM-CSF antibodies is highly sensitive and specific for autoimmune PAP. Given our patient’s mild symptoms, he was managed conservatively. Follow-up in 4 months revealed continuous mild shortness of breath with exertion still not meeting the criteria for whole lung lavage. Familiarity with the key clinical and diagnostic features of pulmonary alveolar proteinosis will help raise awareness and potential new treatment options.
Disclosure statement
The authors note that they have no conflicts of interest to disclose.
References
- Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958 Jun 5;258(23):1123–1142.
- Borie R, Debray MP, Laine C, et al. Rituximab therapy in autoimmune pulmonary alveolar proteinosis. Eur Respir J. 2009;33:1503.
- Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349:2527–2539.
- Kamboj A, Lause M, Duggirala V. Severe pulmonary alveolar proteinosis in a young adult. Am J Med. 2018 May;131(5):e199–e200.
- Shah PL, Hansell D, Lawson PR, et al. Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis. Thorax. 2000;55:67. Borie R, Danel C, Debray MP, Taille C, Dombret MC, Aubier M, Epaud R, Crestani B. Pulmonary alveolar proteinosis. Eur Respir Rev. 2011 Jun;20(120):98-107
- Inoue Y, Trapnell BC, Tazawa R, et al. Characteristics of a large cohort of autoimmune pulmonary alveolar proteinosis patients in Japan. Am J Respir Crit Care Med. 2008;177:752–762.
- Claypool WD, Rogers RM, Matuschak GM. Update on the clinical diagnosis, management, and pathogenesis of pulmonary alveolar proteinosis (phospholipidosis). Chest. 1984;85:550.
- Miller PA, Ravin CE, Smith GJ, et al. Pulmonary alveolar proteinosis with interstitial involvement. AJR Am J Roentgenol. 1981;137:1069.
- Rossi SE, Erasmus JJ, Volpacchio M, et al. “Crazy-Paving” pattern at thin section CT of the lungs: radiologic-pathologic overview. Radiographics. 2003;23:1509.
- De Wever W, Meersschaert J, Coolen J, et al. The crazy-paving pattern: a radiological-pathological correlation. Insights Imaging. 2011;2(2):117–132.
- Bonella F, Bauer PC, Griese M, et al. Pulmonary alveolar proteinosis: new insights from a single-center cohort of 70 patients. Respir Med. 2011;105(12):1908.
- Kumar A, Abdelmalak B, Inoue Y, et al. Pulmonary alveolar proteinosis in adults: pathophysiology and clinical approach. Lancet Respir Med. 2018;6:554.
- Sui X, Du Q, Xu KF, et al. Quantitative assessment of Pulmonary Alveolar Proteinosis (PAP) with ultra-dose CT and correlation with Pulmonary Function Tests (PFTs). PLoS One. 2017;12:e0172958.
- Beccaria M, Luisetti M, Rodi G, et al. Long-term durable benefit after whole lung lavage in pulmonary alveolar proteinosis. Eur Respir J. 2004;23:526. Ishii H, Tazawa R, Kaneko C, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. Eur Respir J 2011; 37: 465–468
- Soyez B, Borie R, Menard C, et al. Rituximab for auto-immune alveolar proteinosis, a real life cohort study. Respir Res. 2018;19(1):74.
- Garber B, Albores J, Wang T, et al. A plasmapheresis protocol for refractory pulmonary alveolar proteinosis. Lung. 2015;193:209.
- Akasaka K, Tanaka T, Kitamura N, et al. Outcome of corticosteroid administration in autoimmune pulmonary alveolar proteinosis: a retrospective cohort study. BMC Pulm Med. 2015;15:88.
- Chaulagain CP, Pilichowska M, Brinckerhoff L, et al. Secondary pulmonary alveolar proteinosis in hematologic malignancies. Hematol Oncol Stem Cell Ther. 2014;7:127.