
ABSTRACT
Background: Thrombotic thrombocytopenic purpura (TTP) is a hematological emergency requiring prompt plasmapheresis. Conversely, vitamin B12 deficiency is a relatively benign diagnosis that can mimic microangiopathic hemolytic anemia, characterized by the presence of anemia, thrombocytopenia, indirect hyperbilirubinemia, markers of hemolysis, and schistocytes. This case series highlights the association of vitamin B12 deficiency and its TTP-like presentations.
Cases: The first case describes a 72-year-old man with shortness of breath and weakness. Diagnostics were notable for pancytopenia, schistocytes, and a low reticulocyte index. Intriguingly, total bilirubin was only mildly elevated however LDH and Haptoglobin were elevated and low, respectively. Additional diagnostic workup demonstrated an undetectable B12, elevated methylmalonic acid and elevated homocysteine. Initiation of B12 supplementation resolved his pancytopenia.
The second case describes a 57-year-old man with chest tightness, dyspnea on exertion, and night sweats. Diagnostic evaluation demonstrated pancytopenia, schistocytes, a low reticulocyte index, and a remarkably low B12. He had associated high methylmalonic acid and homocysteine levels, confirming the diagnosis. B12 supplementation resolved his pancytopenia.
Conclusion: The polysymptomatic presentation of vitamin B12 deficiency-induced pseudothrombotic microangiopathy highlights the vitamin’s role in essential physiological cellular functions. Rapid recognition of the underlying etiology of microangiopathic hemolytic anemia is necessary as treatment approaches diverge greatly.
1. Background
Thrombotic thrombocytopenic purpura (TTP) is a microangiopathic hemolytic anemia caused by deficiency of ADAMTS13, a disintegrin and von Willebrand factor metalloproteinase with a thrombospondin type 1 motif, member 13. Whether the deficiency is due to congenital, genetic mutations or acquired through the development of auto-antibodies, the resultant enzyme insufficiency impedes the breakdown of von Willebrand factor (vWF) multimers and causes progressive multimer accumulation on endothelial surfaces. This incites intravascular platelet adhesion, microthrombi formation, and subsequent erythrocyte shearing and fragmentation. Together, this pathophysiology is responsible for the classic clinical pentad of fever, thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage (i.e., renal failure, neurological manifestations). Management requires prompt initiation of plasmapheresis, designed to provide normal ADAMTS13 or remove ADAMTS13 inhibitory autoantibodies in cases of congenital or acquired TTP, respectively.
Vitamin B12 (cobalamin) deficiency, in contrast, is a relatively benign nutritional deficiency that may result in a macrocytic, megaloblastic anemia, pancytopenia, and neurological sequelae such as subacute combined degeneration of the myelinated dorsal columns of the spinal cord. The etiology may be related to poor nutritional intake or poor absorption in the setting of a variety of causes – alcohol abuse, atrophic gastritis, pernicious anemia, and inflammatory bowel disease. Laboratory diagnostics demonstrate elevated levels of methylmalonic acid (MMA) and homocysteine, precursors that accumulate secondary to reduced function of cobalamin-requiring enzymes, methylmalonyl coenzyme A mutase and methionine synthase, respectively. In rare cases, cobalamin deficiency may mimic a microangiopathic hemolytic anemia such as TTP, with laboratory diagnostics demonstrating anemia, thrombocytopenia, elevated lactate dehydrogenase (LDH), and low haptoglobin, with a critically distinguishing feature, reticulocyte hypoproliferation. In the case of cobalamin deficiency, macrocytic, immature, rigid red blood cells undergo intramedullary hemolysis resulting in a hemolytic anemia. Additionally, there is a hypothesized role for elevated homocysteine levels causing intravascular hemolysis due to activation of the clotting cascade and associated endothelial cell dysfunction with resultant clot formation. This case series will highlight the association of vitamin B12 deficiency, nuclear-cytoplasmic desynchrony, intramedullary hemolysis, thrombocytopenia, and venous thrombosis, with specific focus on thrombotic thrombocytopenic purpura (TTP)-like presentations.
2. Case report 1
A 72-year-old gentleman with a history of hypertension, hyperlipidemia, uncontrolled diabetes mellitus, prior deep venous thrombosis/pulmonary embolism, alcohol use disorder, and benign prostatic hyperplasia presented to the emergency department with shortness of breath and generalized fatigue. He reported usual health until 1 week prior to presentation at which time he noted exertional shortness of breath with minimal ambulation as well as a productive cough, nasal congestion, and sore throat, without chest discomfort, edema, fever, or chills. He furthermore denied hematochezia, melena, easy bruising, paresthesia, or gait disturbance, but did report chronic alcohol use, approximately 1–2 beers every other day, and taking a baby aspirin daily.
Examination revealed a fatigued-appearing African-American male with conjunctival pallor who was afebrile, mildly tachycardic (100–110 beats per minute), and hypertensive (SBP 160–180 mmHg), with a preserved ambient-air oxygen saturation. Cardiopulmonary examination was notable only for a flow murmur best appreciated at the upper sternal border with delayed capillary refill. There was no evidence of hepatosplenomegaly, peripheral edema, nor rash. Neurological examination was unremarkable with preserved strength, gait, proprioception, sensation (fine touch and pain), and reflexes. Diagnostic evaluation revealed pancytopenia (white blood cell count of 3.6 k/uL, a hemoglobin of 4.6 g/dL, and a platelet count of 74 k/uL) (). His marked anemia was notably macrocytic (MCV 128 fL) with an elevated distribution width (RDW 19.5% [normal range, 11.5 to 15.5]) and reticulocyte hypo-proliferation (1.8%; Reticulocyte Index 0.23). Peripheral blood smear demonstrated no atypical lymphocytes, nor any evidence of abnormal platelets; however, red blood cell morphology included ovalocytes, target cells, moderate schistocytes, and teardrop cells. Basophilic stippling and hyper-segmented neutrophils were also seen (). Metabolic panel demonstrated a mildly elevated AST (65 units/L) with a normal ALT, a mildly elevated total and indirect bilirubin (1.2 mg/dL [0.1–1.0 mg/dL] and 0.86 mg/dL, respectively), and a normal alkaline phosphatase (76 units/L). Lactate dehydrogenase was markedly elevated (3000 units/L) with a low serum haptoglobin (<8 mg/dL). Prothrombin time, partial thromboplastin time, INR, and D-Dimer were mildly elevated (20.3 sec, 40.2 sec, 1.7, and 1.15 mcg/mL, respectively), with a normal fibrinogen. There was no evidence of a gamma gap. Diagnostic imaging included a CT of the chest, abdomen, and pelvis that showed no evidence of pulmonary embolism, nor organomegaly/adenopathy, but the presence of a shrunken, nodular appearing liver and left lingular pneumonia.
Table 1. Significant values on pre-hospitalization, presentation, hospital course and follow-up after vitamin B12 repletion for Case 1
Figure 1. Light Microscopy of a Peripheral Blood Smear in a patient with megaloblastic anemic secondary to severe vitamin B12 deficiency. Light Microscopy demonstrates the presence of numerous red blood cell morphologies including ovalocytes (arrow), target cells (arrowhead), teardrop cells (curved arrow), and schistocytes (asterisk); Panel A. Numerous Howell-Jolly bodies (arrows), basophilic stippling (arrowhead), and a nucleated red blood cell (asterisk) are also seen; Panel B. A solitary hyper-segmented neutrophil is appreciated on a background of macro-ovalocytes; Panel C
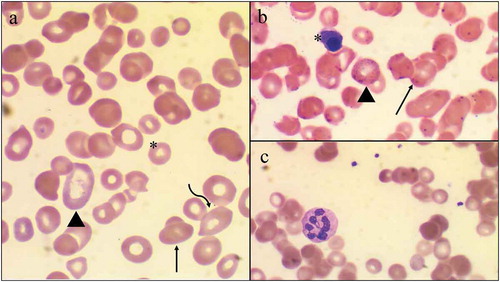
In the setting of the noted anemia, schistocytes on peripheral smear, thrombocytopenia, hyperbilirubinemia, markedly elevated LDH, low haptoglobin, and coagulopathy, there was concern that the patient’s presentation could represent a microangiopathic hemolytic anemia, specifically thrombotic thrombocytopenic purpura; however, the lack of concomitant renal disease, neurological symptoms, skin lesions, reticulocyte hyperproliferation, and the presence of marked macrocytosis seemed to make this etiology less likely. Longitudinal review of his laboratory diagnostics demonstrated a chronic macrocytosis (MCV 128 fL) with borderline anemia (10–12 g/dL) and borderline thrombocytopenia (150 k/uL) (). Furthermore, the patient’s vitamin B12 was noted to be low (65 ng/mL) 2 years prior to presentation, but he was not initiated on B12 supplementation at that time. On the current presentation, his vitamin B12, MMA, and homocysteine were subsequently checked and confirmed the diagnosis of marked vitamin B12 deficiency (B12 < 60 pg/mL, MMA 8.81 umol/L, homocysteine 33.0 umol/L). Intrinsic factor antibody, anti-parietal cell antibody, and H. pylori testing were notably negative, as were additional viral etiologies.
The patient initially received three units of packed red blood cells for symptomatic anemia and was initiated on intramuscular 1000 mcg vitamin B12 injection as well as folate and iron pills in anticipation of folate and iron depletion upon resumption of erythropoiesis. At the time of discharge, the patient’s liver function tests had completely resolved (AST 21 units/L, ALT 24 units/L), and he was discharged on a high-dose vitamin B12 supplementation (1000 mcg IM weekly for 4 weeks) and instructed to follow-up with outpatient hematology 1 month later. At this follow-up visit, the patient’s laboratory data revealed significant improvement in the vitamin B12 level (above 6000 pg/mL) with resolution of his pancytopenia (white blood cell count of 4.9 k/uL, hemoglobin of 13.7 gm/dL [MCV 107.3 fL] and platelet count of 134 k/uL). At that point, he was instructed to continue vitamin B12 shots once a month with serial vitamin B12 levels and CBC monitoring. The plan was to transition back to oral vitamin B12 supplementation, but unfortunately the patient stopped vitamin B12 supplementation. Six months after the first presentation he was once again noted to have vitamin B12 deficiency (190 pg/mL [normal range 200–900 pg/mL]), with associated return of adjunctive symptoms including shortness of breath and dyspnea on exertion. CBC nor MMA or homocysteine were collected.
3. Case report 2
A 57-year-old African-American gentleman with a history of obesity and prostate cancer status-post prostatectomy presented with a one-week history of chest tightness, dyspnea on exertion, and night sweats. He reported usual health until 1 month prior at which time he noted a decreasing exercise capacity, attributing his symptoms to work-related stress as a university professor. His symptoms culminated in the week prior to presentation as an inability to complete his standard 35-minute treadmill regimen and marked dyspnea on climbing a flight of steps. Review of systems was positive for intermittent night sweats of six months duration and notably negative for palpitations, hemoptysis, hematochezia/melena, adenopathy, and easy bruising. His family history was remarkable for acute lymphocytic leukemia and chronic myeloid leukemia in second-degree relatives.
Physical examination revealed a well-appearing African-American male who was hemodynamically stable. Sclera were anicteric and no evidence of adenopathy (occipital, cervical, axillary, or inguinal) was appreciated. Cardiopulmonary as well as abdominal examination was unremarkable. Diagnostic evaluation demonstrated pancytopenia (white blood cell count of 3.9k/uL, a normocytic anemia with a hemoglobin of 6.8 gm/dL [MCV 96.1 fL], and a platelet count of 122 k/uL) (). Reticulocyte count, reticulocyte index and immature platelet fraction were 0.4%, 0.07 and 4.3%, respectively, indicative of ineffective erythropoiesis. Smear demonstrated moderate anisocytes, occasional teardrop cells and schistocytes, and a few enlarged platelets. Metabolic panel demonstrated a normal creatinine, an elevated AST (363 units/L) and ALT (121 units/L), a normal alkaline phosphatase, a normal total bilirubin, and normal gamma gap (3.2 gm/dL). Additional diagnostic workup for evaluation of the noted pancytopenia and transaminitis revealed negative serologies for HIV, Hepatitis A/B/C, but a history of EBV infection (EBV VCA IgG >750.0 u/mL, EBV nuclear antigen IgG 518 u/mL, EBV IgG/early antigen 34.1 u/mL, negative IgM). Serum protein electrophoresis, serum immunoglobulins, and kappa/lambda light chains were normal. ESR and CRP were notably elevated at 51 mm/hr and 6.49 mg/L, respectively. D-Dimer was notably elevated to 9.83 mcg/mL, with a PT 15.8 sec (normal 11–15 sec), PTT 36.6 sec (normal 25–40 sec), and INR 1.3. LDH was markedly elevated to >4000 units/L with an undetectable haptoglobin (<8 mg/dL), seemingly concerning for hemolysis, in light of the noted anemia. Diagnostic imaging including chest radiograph and CT of the chest, abdomen, and pelvis remained unremarkable. In the setting of the noted anemia, schistocytes, thrombocytopenia, low haptoglobin, elevated LDH, and elevated D-Dimer, there was minor concern that this could represent an atypical presentation of microangiopathic hemolytic anemia. Given the atypical presentation, iron studies and vitamin B12 levels were sent which demonstrated an iron level 167 μg/dL (normal 60–170 μg/dL), TIBC 187 μg/dL (normal 240–450 μg/dL), iron saturation 89% (normal 20–50%), consistent with anemia of chronic disease, and a remarkably low vitamin B12 (64 pg/mL), likely contributing to the normal MCV. He had an associated high methylmalonic acid (16.87 umol/L, normal 0.0–0.4 umol/L) and homocysteine (140.0 umol/L), confirming the diagnosis. Intrinsic factor antibodies were negative and anti-parietal cell antibodies were within normal limits (12.2 units, normal <24.9 units).
Table 2. Significant values on presentation, hospital course and follow-up after vitamin B12 repletion for Case 2
The patient initially received a transfusion of packed red blood cells for his symptomatic anemia and was initiated on high-dose parenteral cobalamin as well as folate and iron in anticipation of depletion following erythropoiesis. At the time of discharge, liver function tests had begun to resolve (AST 113 units/L, ALT 133 units/L), and he was discharged on high-dose vitamin B12 supplementation regimen (1000 mcg IM weekly for 4 weeks) and was instructed to follow-up with outpatient hematology. Follow-up laboratory diagnostics one-week after discharge showed resolution of the pancytopenia (white blood cell count of 5.1 k/uL, hemoglobin of 9.3 gm/dL [MCV 99.0 fL], and platelet count of 221 k/uL). He was instructed to continue taking weekly vitamin B12 shots for a total of four weeks followed by monthly treatment. Five months after his first follow-up, his CBC continued to demonstrate complete resolution of pancytopenia (WBC count of 5.4 k/uL, hemoglobin of 13.1 gm/dL, hematocrit of 41.2% (MCV 86.9 fL), and a platelet count of 205 k/uL).
4. Discussion
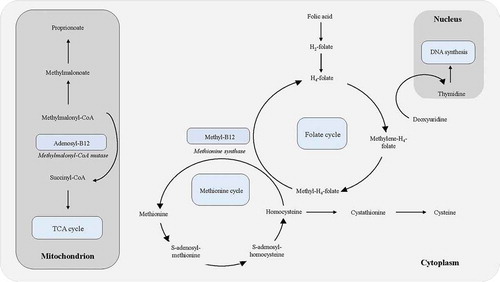
Vitamin B12 is a water-soluble vitamin with an essential role in DNA synthesis, hematopoiesis, and myelination. Vitamin B12 is a necessary cofactor for the conversion of methylmalonyl coenzyme A to succinyl coenzyme A via methylmalonyl coenzyme A mutase (), enabling the breakdown of odd-chained fatty acids and some amino acids. It is also a required cofactor for methionine synthase, which catalyzes the conversion of homocysteine to methionine, ultimately allowing for the generation of tetrahydrofolate, a biologically active form of folate needed in DNA synthesis (). Insufficient levels of vitamin B12 lead to reduced function of methylmalonyl coenzyme A mutase and methionine synthase, with a resultant accumulation of the precursors methylmalonyl coenzyme A and homocysteine, respectively. Classically, vitamin B12 deficiency manifests as a macrocytic, megaloblastic anemia and in severe cases, subacute combined degeneration of the posterior columns of the spinal cord. In rare cases, severe vitamin B12 deficiency can present as a pseudo-microangiopathic hemolytic anemia/TTP, characterized by thrombocytopenia and hemolytic anemia (i.e., elevated LDH, low haptoglobin, hyperbilirubinemia, and schistocytes). In contrast to classic microangiopathic hemolytic anemia/TTP, vitamin B12-related pseudo-TTP presents as a macrocytic, megaloblastic anemia with reticulocyte hypoproliferation, elevated levels of homocysteine and MMA, and a low vitamin B12. Management of this masked deficiency requires only vitamin B12 supplementation in contrast to initiation of plasmapheresis required for true TTP.
Figure 2. Vitamin B12 (Cobalamin) in cellular metabolism
4.1. Megaloblastic anemia and hypersegmented neutrophils
As previously noted, vitamin B12 is critically involved in DNA synthesis, catalyzing the conversion of homocysteine to methionine, while at the same time acting as a methyl-group acceptor to allow for the regeneration of tetrahydrofolate, a biologically active form of folate required for DNA synthesis. Defective DNA synthesis leads to nucleo-cytoplasmic asynchrony [Citation1] wherein the cell is unable to progress from the G2 growth stage to mitosis, leading to progressive cell growth without division. On blood smear, this presents as erythrocytic megaloblastosis (i.e., large, dysfunctional red blood cells), and neutrophil hypersegmentation (greater than five nuclear lobes) [Citation2]. With chronic deficiency, further dyspoiesis may occur, with resultant leukopenia and thrombocytopenia [Citation3]. Indeed, our patients’ megaloblastic, macrocytic anemia, leukopenia, and thrombocytopenia reflect the cumulative effects of ongoing vitamin B12 deficiency. While thrombocytopenia and anemia are both classic findings in TTP, leukopenia is less commonly described. One case report highlighted leukopenia as a result of bone marrow necrosis in TTP, though blood smear revealed normal leukocyte morphology [Citation4], again differentiating true TTP from vitamin B12 deficiency.
4.2. Hemolysis
Both cases showed evidence of hemolytic anemia, with characteristically elevated LDH and low haptoglobin. The hemolytic anemia seen in vitamin B12 deficiency may be due to a combination of extramedullary and intramedullary hemolysis. The hypercoagulability and endothelial dysfunction associated with hyperhomocysteinemia may lead to erythrocyte fragmentation comparable to that observed in microangopathic hemolytic anemia/TTP [Citation5,Citation6]. In the cases described, however, bilirubin levels were normal to minimally elevated, suggesting an intramedullary process deviating from that of true TTP.
Intramedullary hemolysis secondary to pseudo-folate deficiency and subsequently impaired DNA synthesis and cellular division manifests in one of two mechanisms. One mechanism may be that immature reticulocytes arrest in development and are destroyed within the marrow [Citation7,Citation8]. An alternative mechanism is impaired marrow egression. Normally, RBCs leave the marrow by squeezing through sinusoids; however, a larger cell, as occurs with vitamin B12-associated nucleo-cytoplasmic asynchrony, would impede this process with resultant microcirculation entrapment and intramedullary cell lysis.
Both intra- and extra-medullary hemolysis are compounded by the increased erythrocyte membrane rigidity observed in vitamin B12 deficiency [Citation9]. While still under investigation, altered TCA and methionine cycles are thought to be involved [Citation9,Citation10]. Free radicals accumulate following inhibition of the mitochondrial respiratory chain due to reduced methylmalonyl CoA mutase activity. These free radicals damage the cellular phospholipid bilayer, particularly polyunsaturated fatty acids, a process that is further exacerbated by the expenditure of glutathione in free radical reduction: glutathionylcobalamin functions as a precursor to both physiologic coenzyme vitamin B12 forms, perpetuating the deficiency [Citation10]. Therefore, diversion of glutathione to reducing free radicals further reduces vitamin B12 availability. Impairment of the TCA cycle may also divert acetyl CoA, the final product of fatty acid catabolism, to cholesterol synthesis thereby increasing membrane cholesterol concentrations. Methylcobalamin, through its interaction with methionine synthase, is a regulator of methylation reactions including phospholipid methylation; a deficiency of this coenzyme would result in S-adenosylmethionine accumulation, an intermediate with a feedforward inhibitory role in methylation reactions. The combined impaired phospholipid production and increased cholesterol synthesis may additively decrease membrane fluidity, contributing to hemolysis [Citation9]. This multifactorial pathophysiology starkly contrasts the hemolysis of TTP, which is a direct consequence of erythrocytes shearing against platelet clots secondary to thrombotic microangiopathy.
4.3. Thrombotic microangiopathy
Like that of hemolysis, the mechanism of thrombotic microangiopathy in vitamin B12 deficiency is multifactorial, but most closely tied to hyperhomocysteinemia. This further distinguishes B12 deficiency from TTP, in which thrombotic microangiopathy occurs due to vWF accumulation and subsequent platelet aggregation. Homocysteine elevation is an independent risk factor for atherosclerotic disease, causing hypercoagulability through endothelial damage, coagulation activation, and impaired nitric oxide response [Citation11]. Hyperhomocysteinemia is associated with increased oxidative stress via the formation of superoxide and hydrogen peroxide radicals; those with nutritional deficiencies, as likely seen in the first patient case, will lack the antioxidant vitamins C and E required to reduce these radicals. Radicals cause damage directly to endothelium, facilitating the formation of atherosclerosis and a platelet plug. They further contribute to coagulation by inactivating nitric oxide. Homocysteine also impedes the anti-aggregating properties of nitric oxide indirectly, by reducing tissues’ responsiveness to l-arginine, a precursor to nitric oxide [Citation5]. These mechanisms were best appreciable in our first patient’s history of thrombosis in the setting of chronically depleted vitamin B12 levels.
4.4. Neurologic manifestations
Classically, vitamin B12 deficiency is associated with subacute combined degeneration, a condition characterized by damage to the dorsal columns of the spinal cord with resultant loss of vibration sense/proprioception, spastic paresis, and gait abnormalities. Mechanistically, this occurs due to lack of conversion of the three-carbon methylmalonyl-CoA to four-carbon succinyl-CoA, causing a subsequent accumulation of three-carbon proprionate. Some suggest this results in the formation and incorporation of 15- and 17-carbon fatty acids into neuronal lipids, predisposing to myelin and neuronal breakdown [Citation12]. Notably, individuals with inherited deficits in methylmalonyl coenzyme-A mutase have highly variable neurological phenotypes without evidence of predisposition to SCD [Citation13], purporting an alternative role of cobalamin in this symptomatology.
Although SCD is highly associated with B12 deficiency, non-specific neurologic findings may also occur, including altered mental status and delirium. This presentation is comparable to that seen in TTP, wherein patients present with altered mental status, stroke, and seizures as a result of thrombosis of cerebral vasculature and focal vasoconstriction due to reversible cerebrovascular constriction syndrome [Citation14]. Neurologic issues were not observed in either case presented, with both patients demonstrating a benign neurologic exam. Few case studies discussing pseudo-TTP in the setting of vitamin B12 deficiency mention concomitant neurologic issues, including generalized weakness (potentially attributable to anemia), altered mental status [Citation15–18] and paresthesia [Citation11,Citation17]. These non-specific findings may be attributable to a combination of hypercoagulability and impaired myelin synthesis. We were unable to find any cases with frank descriptions of subacute combined degeneration with simultaneous pseudo-TTP, drawing into question individual factors influencing clinical presentation in the setting of vitamin B12 deficiency.
5. Conclusion
Vitamin B12 deficiency-induced pseudothrombotic microangiopathy is a rare condition that resembles the clinical features of TTP. This polysymptomatic presentation highlights the vitamin’s role in essential physiological cellular functions. Rapid recognition of the underlying etiology of microangiopathic hemolytic anemia is necessary as treatment approaches diverge greatly.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Stabler SP. Vitamin B12 deficiency. N Engl J Med. 2013;368(2):149–160.
- Farrelly SJ, O’Connor KA. Hypersegmented neutrophils and oval macrocytes in the setting of B12 deficiency and pancytopaenia. Case Rep. 2017; 2017.
- Bhattacharjee A, Samuel AE. Vitamin B12 deficiency in a patient presenting with dyspnea: a case report. Adv J Emergency Med. 2019;3(2):2.
- Parekh HD, Reese JA, Cobb PW, et al. Bone marrow necrosis discovered in a patient with suspected thrombotic thrombocytopenic purpura. Am J Hematol. 2015;90(3):264.
- Nappo F, De Rosa N, Marfella R, et al. Impairment of endothelial functions by acute hyperhomocysteinemia and reversal by antioxidant vitamins. Jama. 1999;281(22):2113–2118.
- Kollipara VK, Brine PL, Gemmel D, et al. A case of asymptomatic pancytopenia with clinical features of hemolysis as a presentation of pernicious anemia. J Community Hosp Intern Med Perspect. 2016;6(4):32493.
- Bailey M, Maestas T, Betancourt R, et al. Cause of thrombotic thrombocytopenia purpura-(TTP-) like syndrome, vitamin B12 deficiency: interpretation of significant pathological findings. Case Rep Hematol. 2019;2019. DOI:10.1155/2019/1529306
- Tran PN, Tran M-H. Cobalamin deficiency presenting with thrombotic microangiopathy (TMA) features: a systematic review. Transfus Apheresis Sci. 2018;57(1):102–106.
- Özcan Ö, İpçioğlu OM, Gültepe M, et al. Altered red cell membrane compositions related to functional vitamin B12 deficiency manifested by elevated urine methylmalonic acid concentrations in patients with schizophrenia. Ann Clin Biochem. 2008;45(1):44–49.
- Pezacka E, Green R, Jacobsen DW. Glutathionylcobalamin as an intermediate in the formation of cobalamin coenzymes. Biochem Biophys Res Commun. 1990 Jun 15;169(2):443–450.
- Vanoli J, Carrer A, Martorana R, et al. Vitamin B 12 deficiency-induced pseudothrombotic microangiopathy without macrocytosis presenting with acute renal failure: a case report. J Med Case Rep. 2018;12(1):1–5.
- Metz J. Cobalamin deficiency and the pathogenesis of nervous system disease. Annu Rev Nutr. 1992 Jul;12(1):59–79.
- Shevell MI, Matiaszuk N, Ledley FD, et al. Varying neurological phenotypes among mut° and mut− patients with methylmalonylCoA mutase deficiency. Am J Med Genet A. 1993;45(5):619–624.
- Xiao Z, Deng L, Steinberg L. Focal neurologic manifestation as initial presentation of thrombotic thrombocytopenic purpura. Ann Clin Case Rep. 2019;4:1725.
- Franks AM, Bannister T, Jarrell A, et al. Cobalamin deficient thrombotic microangiopathy: a case of TTP or pseudo-TTP. West Virginia Med J OA. 2018 Apr;26:3578.
- Kandel S, Budhathoki N, Pandey S, et al. Pseudo-thrombotic thrombocytopenic purpura presenting as multi-organ dysfunction syndrome: A rare complication of pernicious anemia. SAGE Open Med Case Rep. 2017 Jun;3(5):2050313X17713149.
- Walter K, Vaughn J, Martin D. Therapeutic dilemma in the management of a patient with the clinical picture of TTP and severe B 12 deficiency. BMC Hematol. 2015 Dec 1;15(1):16.
- Tadakamalla AK, Talluri SK, Besur S. Pseudo-thrombotic thrombocytopenic purpura: a rare presentation of pernicious anemia. N Am J Med Sci. 2011 Oct;3(10):472.