
ABSTRACT
Bernard Soulier Syndrome is a genetically inherited platelet disorder that commonly presents with symptoms of impaired blood coagulation, such as epistaxis, menorrhagia, and petechiae formation. Here we present a case of Bernard Soulier Syndrome in which the individual has presented with melena, which is the appearance of black tarry stools due to bleeding from the upper gastrointestinal tract. This presentation is rare and should be discussed so that appearance of the less common symptoms can be caught early, leading to an early diagnosis and consequently earlier and more effective management options.
1. Introduction
Bernard Soulier Syndrome or BSS is a rare hematological disorder related to abnormal structure and function of platelets. It was initially described in 1948 by J Bernard and JP Soulier [Citation1]. Other names for the disease include giant platelet syndrome, hemorrhagiparous thrombocytic dystrophy, macrothrombocytopenia, platelet glycoprotein Ib deficiency or Von Willebrand factor receptor deficiency. BSS has an autosomal recessive inheritance pattern affecting 1 in 1,000,000 people [Citation2]. Characteristic lab findings include megakaryocytes, thrombocytopenia, increased bleeding time and impaired platelet agglutination in response to Ristocetin. It has also been classified as a giant platelet disorder, due to the appearance of abnormally large platelets. In addition to the typical clinical manifestations of epistaxis, cutaneous and mucosal bleeding, the disorder can also rarely present with menorrhagia and gastrointestinal tract bleeding. The dysfunction results from mutations which include nonsense, deletion, or missense mutations of the surface glycoprotein complex encoding genes [Citation3]. Once diagnosed, the medical opinion of a hematologist should be sought as the management plan of each patient must be tailored according to the severity and symptoms of each individual case. There is no cure or prophylactic measures that can be implemented for bleeding disorders, it is only possible to manage the risks and complications.
2. Case presentation
An 18-year-old male with a past medical history of Bernard-Soulier Syndrome presented with complaints of repeated episodes of black stools (melena). The current episode of melena had been occurring for the past 15 days. The patient also reported minor bruising following the occurrence of any trivial trauma. The patient had been taking oral iron supplements and had received occasional blood transfusions with no history of any other disease. His vital signs were normal with no fever. He was diagnosed with Bernard-Soulier Syndrome after developing excessive, persistent bleeding following circumcision about 15 years ago. His family history was positive for similar instances of bleeding in a first cousin on the paternal side of the family, who was also later diagnosed with Bernard-Soulier Syndrome.
On physical examination, the patient was in no active distress and was sitting comfortably on the bed. No fresh bleed was noted in the rectal vault. A few petechial rashes were noted in the lower limbs. Systemic examination was significant only for pallor and tachycardia. On abdominal examination, his liver span was normal, spleen was not palpable and there was no lymphadenopathy.
Baseline investigations were ordered which included a complete blood picture and coagulation profile. The significant lab test and their results are mentioned in the table ():
Table 1. Laboratory tests
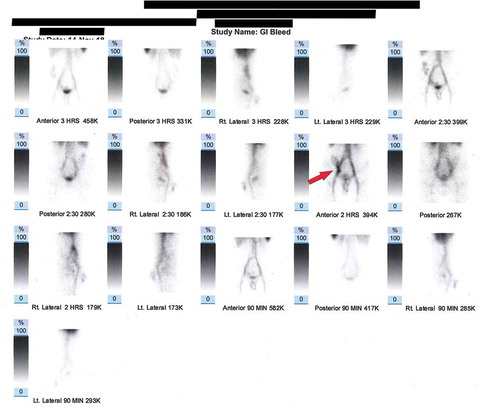
Peripheral blood film was also ordered which showed megakaryocytosis. All these findings were consistent with the diagnostic picture of Bernard Soulier Syndrome. An esophagogastroduodenoscopy as well as a colonoscopy was performed to identify the bleeding site but results were inconclusive. CT angiography had to be deferred because of a reported contrast allergy. As a result, RBC nuclear scan was performed which showed scintigraphic evidence of active bleeding at the region of the distal ileum and ileocecal junction (). Coil embolization was performed to stop the bleed.
Figure 1. The scan shows an abnormal focus of increased radiotracer accumulation in right lower abdomen at 45 minutes post radiotracer injection. The focus increases in intensity and moves upward in cecum and ascending colon. Positive for scintigraphic evidence of active GI bleed – distal ileum/ileocecal junction
Once stable, the patient was discharged and was prescribed iron supplements, desmopressin, ascorbic acid and an anti-fibrinolytic agent. Patient was also advised to follow up after one month or in case of any emergency.
3. Discussion
Bernard Soulier syndrome is a congenital bleeding disorder caused by platelet dysfunction. Its inheritance is typically of an autosomal recessive pattern although rare cases following an autosomal dominant pattern due to mutations in the GP1BA or GP1BB gene have also been seen [Citation1]. It is characterized by an impairment of platelet adhesive function via a defect in the glycoprotein Ib/IX complex that binds endothelial VWF. It has a reported prevalence of one in one million individuals, and about 200 cases have been reported [Citation2]. The condition was first described in 1948 by two French hematologists, Jean Bernard and Jean Pierre Soulier who had described a male patient with a bleeding defect characterized by increased bleeding time, thrombocytopenia and appearance of megakaryocytes. Hemorrhagiparous thrombocytic dystrophy (Dystrophie thrombocytaire-hémorragipare congénitale) was the name given to the disorder by them [Citation3]. In this syndrome, thrombocytopenia is observed with abnormally large and functionally impaired platelets. The clinical manifestations are diverse and include purpura, epistaxis, menorrhagia, gingival and gastrointestinal bleeding, hematuria or hematoma [Citation4].
Bernard Soulier Syndrome has its molecular basis in defects in the GP IB-IX-V receptor complex on the platelet surface. The function of the GPIB-IX-V complex is to ensure primary hemostasis by initiating adhesion of platelets at sites of vascular injury which it accomplishes by adhesion to von Willebrand factor, itself captured from plasma by subendothelial collagen [Citation5]. Defects arise due to mutations in one of the following three genes encoding for the GPIB-IX-V receptor complex: 1) GPIBA 2) GPIBB 3) GP9. The defects maybe due to missense, nonsense or frameshift mutations [Citation6]. This receptor complex is responsible for the adhesion of platelets through interaction with Von Willebrand factor on the exposed sub endothelium, and its dysfunction leads to impaired platelet adhesion to the sub endothelium, leading to abnormal platelet function and bleeding [Citation7].
Bernard Soulier Syndrome is a frequently misdiagnosed condition. The clinical picture is similar to other platelet disorders and hence many cases have been misdiagnosed as idiopathic thrombocytopenic purpura (ITP), leading to unsuccessful treatment [Citation8]. It is therefore imperative that misdiagnosis be avoided through a thorough history taking with special emphasis on age, the initial presenting complaints and any positive family history. The subsequent lab evaluation must include a general assessment of coagulation parameters followed by platelet function specific testing [Citation9]. The additional specific tests include flowcytometry and platelet aggregation studies. Flowcytometry is the confirmatory test which demonstrates abnormalities of platelet membrane glycoprotein. Care should be taken to perform this test on a plasma sample with abundant platelets [Citation10]. Aggregation studies characteristically show platelets’ inability to aggregate with ristocetin and a slow response with low doses of thrombin. The addition of normal plasma doesn’t help aggregation, thus differentiating BSS from VWD. However, normal aggregation is seen with epinephrine, adenosine diphosphate, collagen, and arachidonic acid [Citation11]. Testing for genetic mutations can differentiate between functional or synthetic problems of the GPIB-IX-V receptor complex, and this approach greatly improves the diagnosis of rare inherited platelet disorders [Citation12].
The management of Bernard Soulier Syndrome is largely supportive. For potentially life-threatening hemorrhage, platelet transfusion therapy is indicated, but it should be kept in mind that the patient may develop anti platelet antibodies to the donor’s GPIB-IX-V receptor complex. Desmopressin acetate (DDAVP) shortens the bleeding time in a few patients, by increasing levels of von Willebrand factor which binds to some residual GPIB in individuals who don’t have absolute deficiency of GPIB-IX-V [Citation10]. Recombinant factor VII is also used by virtue of its action to increase thrombin production and the fibrin deposition at bleeding sites. In order to avert the danger of disastrous bleeding, patients should be adequately counseled to avoid trauma and anti-platelet medications like aspirin.
Our patient was a diagnosed case of Bernard Soulier syndrome who presented with complaints of recurrent melena, a manifestation of gastrointestinal bleeding which was later found to have its origin at distal ileum and ileocecal junction. Gastrointestinal bleeding in Bernard Soulier syndrome is a rare occurrence. In a study conducted in a tertiary care hospital in Pakistan, none of the Bernard Soulier cases evaluated for a duration of over eight years had presented with hematemesis or melena [Citation13]. There have been only a handful of Bernard Soulier cases in literature who have presented with gastrointestinal hemorrhage, with a majority of them due to angiodysplasia [Citation14]. It is therefore necessary to keep congenital bleeding disorders like Bernard Soulier syndrome in mind while evaluating a patient with gastrointestinal bleeding of unknown origin.
4. Conclusion
Focus on the most commonly presenting clinical symptoms in any ailment can lead to a misdiagnosis of easily manageable diseases. Bernard Soulier, while having a very typical clinical appearance, can present with some atypical symptoms. Melena, black tarry stools as a result of a GI bleed, is one of those rarely occurring symptoms which can often be misleading and result in a late or inaccurate diagnosis. The prevalence of such rare cases needs to be further studied, so that more effective treatment methods can be implemented in each unique clinical case.
Disclosure statement
The authors report no conflict of interest.
References
- Bernard soulier syndrome; 2020 [cited 2020 Jan 22]. Available from: https://ghr.nlm.nih.gov/condition/bernard-soulier-syndrome.
- Rare diseases-bernard soulier syndrome; 2018 [cited 2020 Jan 22]. Available from: https://rarediseases.org/rare-diseases/bernard-soulier-syndrome/.
- Lanza F. Bernard-soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphanet J Rare Dis. 2006;1:46.
- Pham A, Wang J. Bernard-Soulier syndrome: an inherited platelet disorder. Arch Pathol Lab Med. 2007;131:1834–1836.
- Clemetson KJ. Platelet receptors and their role in diseases. Clin Chem Lab Med. 2003;41:253–260.
- Macêdo MB, Brito JDEM, PDAS M, et al. Primigravida with bernard-soulier syndrome: a case report. BMC Res Notes. 2015;8:178.
- Ruggeri ZM. Von willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. 2003;1:1335–1342.
- Moiz B, Rashid A. BSS misdiagnosed as ITP. Blood. 2013;122:1693.
- Linden MD, Al Iii F, Barnard MR, et al. Application of flow cytometry to platelet disorders. Semin Thromb Hemost. 2004;30:501–511.
- Bernard soulier syndrome workup; 2018 [cited 2020 Jan 25]. Available from: https://emedicine.medscape.com/article/954877-workup.
- Balduini CL, Iolascon A, Savoia A. Inherited thrombocytopenias: from genes to therapy. Hematologica. 2002;87:860–880.
- Sánchez-Guiu I, Antón AI, Padilla J, et al. Functional and molecular characterization of inherited platelet disorders in the iberian peninsula: results from a collaborative study. Orphanet J Rare Dis. 2014;9:213.
- Farhan S, Iqbal I, Ahmed N. Bernard soulier syndrome: 10 years’ experience at a tertiary care hospital. Pak J Med Sci. 2019;35:705–708.
- xOkita R, Hihara J, Konishi K, et al. Intractable gastrointestinal bleeding from angiodysplasia in a patient of bernard-soulier syndrome--report of a case. Hiroshima J Med Sci. 2005;54:113–115.