
ABSTRACT
Introduction
Cardiac amyloidosis is a rare entity with a grave prognosis. Due to the low index of suspicion secondary to non-specific symptoms, it is often diagnosed at an advanced stage with multi-organ involvement.
Methods
We report a case of systemic AL amyloidosis with predominant cardiac and renal involvement associated with multiple myeloma.
Case Summary
A 60-year-old male presented with progressive anasarca, orthopnea and weight gain over 8 months. On clinical examination, 3+ pitting edema was found in bilateral extremities and scrotum. Serum N-type proBNP and troponin T were elevated, and EKG showed diffuse low voltage QRS, right axis deviation, and 1st degree AV block. Echocardiography revealed granular myocardium, biventricular hypertrophy, bi-atrial dilation and apical sparing pattern on global longitudinal strain which was suggestive of cardiac amyloidosis. Light chain assessment showed elevated kappa and lambda chains with kappa to lambda ratio of 16.2. Endomyocardial biopsy revealed AL type cardiac amyloidosis, and bone marrow biopsy confirmed the diagnosis of multiple myeloma. He received six cycles of bortezomib, cyclophosphamide, and dexamethasone but continued to deteriorate. He experienced an episode of cardiac arrest following which he had a return of spontaneous circulation but due to poor prognosis, the family opted for pursuing comfort measures only.
Conclusions
Cardiac involvement in AL type amyloidosis imparts significant morbidity and mortality. The management of cardiac amyloidosis entails a multidisciplinary approach with an emphasis on cardiology and oncology. Despite the novel diagnostic modalities and treatment regimens, the outcome for AL-type cardiac amyloidosis remains poor.
1. Introduction
Amyloidosis is a systemic disease involving the extracellular deposition of misfolded insoluble protein in tissues resulting in organ damage. Cardiac involvement is seen in more than 60% of AL type amyloidosis, which significantly increases mortality and morbidity. With untreated congestive failure, and the median survival time is less than 6 months [Citation1]. Thus, timely diagnosis and institution of therapy are integral for better outcomes. We report a case of AL type cardiac amyloidosis, and a review of its presentation, diagnosis, management, and prognosis.
2. Case
A 60-year-old male with a history of prostate cancer status post-prostatectomy and pelvic lymphadenectomy presented to the Emergency Department with complaints of generalized body swelling, orthopnea, and weight gain of 80 pounds in 6 months.
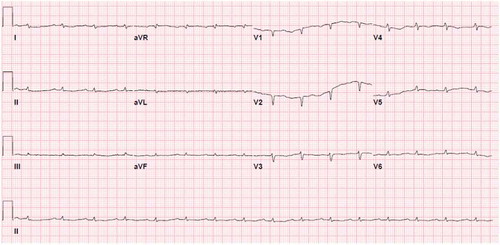
The patient reported that the generalized body swelling had started as bilateral lower extremity edema deteriorating into anasarca that was unresponsive to diuresis. Vitals at admission were temperature 36.7 degrees Celsius, blood pressure 124/81 mm Hg, heart rate 90 beats/minute, respiratory rate 16, and oxygen saturation 95% on room air. Examination revealed anasarca with significant scrotal edema and 3+ pitting edema involving bilateral upper and lower extremities. Significant laboratory findings included serum creatinine-1.68 mg/dl, elevated alkaline phosphatase-450 U/L, total bilirubin 5.4 mg/dl (Direct predominant), and troponin 0.12 ng/ml and pro-BNP 20,730 pg/ml, indicating multiorgan dysfunction. EKG showed sinus rhythm with 1st degree AV block, low voltage QRS in all leads, and right axis deviation, as shown in .
Figure 1. EKG showing diffuse low voltage QRS present in all leads
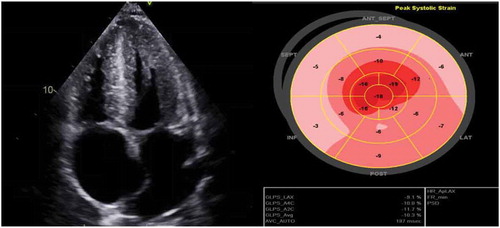
CT abdomen pelvis was obtained, which showed diffuse anasarca, and numerous pulmonary nodules in both lungs concerning for metastatic disease. Transthoracic echocardiography was completed which showed bi–atrial enlargement, biventricular hypertrophy, septal wall thickening, starry sky appearance of the tissue, grade one diastolic dysfunction (E/A ratio 3.1 and E/E’ ratio 11.71) and apical sparing of longitudinal systolic strain, all of which were suggestive of cardiac amyloidosis resulting in restrictive cardiomyopathy ().
Figure 2. Echocardiogram demonstrating bi–atrial enlargement, biventricular hypertrophy, hypertrophied thickened interventricular septum and apical sparing on longitudinal strain
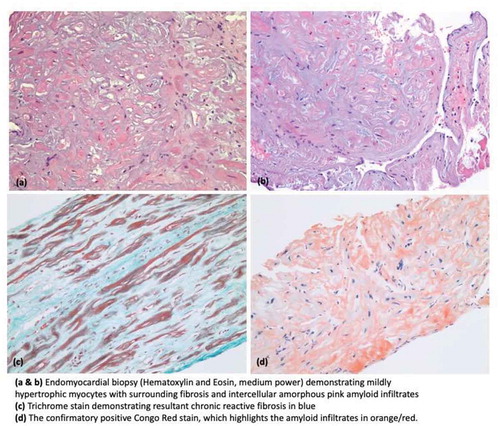
Further biochemical investigations for amyloidosis were sent including serum and urine protein electrophoresis and immunofixation. Light chain analysis showed Free Kappa Light Chains 24.12 mg/dl (reference range 0.33–1.94 mg/dl), Free Lambda Light Chains 1.46 mg/dl (reference range 0.57– 2.63 mg/dL), and Kappa/Lambda ratio 16.52 (reference range: 0.26- 1.65) with no evidence for monoclonal gammopathy. Interestingly, Technetium-99 m pyrophosphate (PYP) scan for transthyretin-related (ATTR) cardiac amyloidosis was unrevealing. For further work up, the patient underwent right heart catheterization and endomyocardial biopsy. A right heart catheterization revealed pulmonary capillary wedge pressure of 32 mm Hg, right atrial pressure of 27 mm Hg and mean pulmonary artery pressure of 42 mm Hg. Endomyocardial biopsy was notable for cardiac amyloidosis with positive Congo red staining ().
Figure 3. Histopathology images demonstrating cardiac amyloidosis
The patient subsequently underwent bone marrow biopsy confirming the diagnosis of AL type amyloidosis (Amyloid AL type) characterized by hypercellular bone marrow with trilineage maturation, findings confirmatory for multiple myeloma. The patient was transferred to the bone marrow transplant treatment floor (BMT) and initiated on chemotherapy with bortezomib, cyclophosphamide, and dexamethasone, along with aggressive intravenous (IV) diuresis. However, kidney function progressively declined requiring renal replacement therapy. On day 18 of admission, the patient became hemodynamically unstable with a blood pressure of 70/40 mm of Hg and heart rate of 52, which did not respond to IV fluids. The patient was transferred to the medical intensive care unit (ICU) for a higher level of care where he was treated with sepsis protocol due to sudden hemodynamic decompensation. Overnight the patient developed pulseless ventricular tachycardia and was defibrillated and emergently intubated. Despite being maintained on dual vasopressors, his condition continued to worsen leading to multi-organ failure. Due to guarded prognosis, a family meeting was held by the BMT team, and a shared decision was made by the family to pursue comfort measures only. The patient passed away shortly thereafter.
3. Discussion
Amyloidosis is a complex multi-organ disease with a variety of clinical presentations. It principally occurs due to unremitting extracellular deposition of misfolded beta-pleated protein known as amyloid. Amyloid deposition leads to cellular apoptosis, tissue architectural distortion, and fibrosis. Our primary focus of discussion is cardiac amyloidosis resulting from AL type amyloidosis. Cardiac amyloidosis (CA), albeit rare, is a grave presentation of systemic amyloidosis which manifests as restrictive cardiomyopathy. Amyloid can deposit anywhere in the heart, including the myocardium, valves, endocardium, endothelium, and pericardium, and this geographic diversity is the cause of the condition’s wide array of clinical presentations. The most important precursor proteins implicated in cardiac amyloidosis are summarized in [Citation2–4].
Table 1. Types of Cardiac Amyloidosis [Citation2–4]
Cardiac involvement with AL amyloidosis has been found in 60–80% of cases. It is more prevalent in African American males, often older than 50 years, similar to our patient [Citation5]. It is likely underdiagnosed, resulting in underestimation of disease burden, and delay in treatment increases the likelihood of disease-causing adverse outcomes [Citation6].
Clinical presentation of AL type cardiac amyloidosis is varied based on the involved site. Fatigue and weakness are the most common presenting symptoms. Restrictive cardiomyopathy presents with signs and symptoms of diastolic heart failure and decreased exercise tolerance. With progression of the disease, atrial dilation occurs, which predisposes patients to atrial fibrillation and further sequelae of clot formation and systemic embolization. Cardiac conduction may be disrupted by amyloid deposition, often causing a variety of heart blocks. Soft tissue involvement has been seen as periorbital ecchymosis and macroglossia in 12.5% and 27.2%, respectively [Citation7]. Renal AL amyloid can cause myeloma kidney and nephrotic syndrome. With underlying kidney disease, the patient may develop progressive renal failure requiring renal replacement therapy, as seen in the present case. Similar deposits in the liver and peripheral nerves can present as hepatomegaly, transaminitis, and peripheral neuropathy, respectively.
Suspicion of cardiac AL type amyloidosis should instigate a thorough workup for underlying pathology driving amyloidosis such as plasma cell dyscrasia. This includes urine and serum protein electrophoresis, immunofixation, and light-chain assessment, followed by confirmatory bone marrow biopsy.
A wide range of diagnostic modalities assist in identifying AL type cardiac amyloidosis. ECG is often the starting point of workup. A low voltage ECG (QRS < 5 mm in all peripheral leads) is the most common finding related to cardiac amyloidosis as seen in our patient [Citation8]. ECG changes become more evident with worsening cardiac disease burden and often include QRS axis deviation and left bundle branch block. An Echocardiography can provide ample findings that should raise the suspicion for cardiac amyloidosis, showing biventricular hypertrophy resulting in bi-atrial dilation, valvular thickening or regurgitation, and pericardial effusion. It can also illustrate increased myocardial echogenicity seen as granular sparkling, which has a sensitivity of 87% and a specificity of 81% for diagnosing cardiac amyloidosis; with concomitant increased atrial thickness, the specificity increases to 100% [Citation9]. ‘Cherry on Top’ description for the strain imaging is very characteristic in AL Amyloidosis as longitudinal strain is decreased in the basal wall and mid-wall regions with sparing of apical function [Citation9]. Similar findings were evident in our patient.
Cardiac magnetic resonance imaging (MRI) has been replaced by other diagnostic imaging modalities for cardiac amyloidosis. Excessive amyloid deposition in the myocardial extracellular matrix results in its expansion, which can be seen with late gadolinium enhancement. Prolonged relaxation time on Visual T1 mapping also supports the diagnosis of cardiac amyloidosis. The gold standard test for diagnosis remains the tissue biopsy, either endomyocardial or fat pad biopsy. Endomyocardial biopsy is an invasive procedure that is performed at expert centers; although safe, it bears a risk for free wall perforation in 0.4% of procedures, arrhythmias in 0.5–1.0% and conduction abnormalities in 0.2–0.4% [Citation9]. reviews diagnostic tests, their sensitivities, specificities, and notable findings [Citation6,Citation10].
Table 2. Diagnostic Modalities for AL-type Cardiac Amyloidosis [Citation6,Citation10]
The only way to halt the progression of AL type cardiac amyloidosis is to treat the underlying disease process. Chemotherapy is necessary to eliminate monoclonal proliferated plasma cells to limit the excessive production of light chains. Usually, patients require 6-12 cycles. Autologous stem cell transplantation can also be considered but it carries a high risk of mortality with cardiac disease. Different chemotherapeutic regimens are currently available, and their respective mechanisms of action are summarized in [Citation11]. A full detailed discussion about each medication is beyond the scope of this report.
Table 3. Summarizing the currently available chemotherapy agents and their mechanisms of action for plasma cell dyscrasias [Citation11]
The main goal of treatment of the cardiac component of AL type amyloidosis is managing the symptoms to improve quality of life while treating the underlying disease. Unlike conventional heart failure management, beta-blockers and calcium channel blockers are avoided due to the risk of hypotension as these patients are dependent on heart rate to maintain their adequate cardiac output [Citation12]. Angiotensin conversion inhibitors (ACE-I) and angiotensin receptor blockers can also cause significant hypotension; thus, both are avoided. Digoxin is also avoided as it has a strong affinity for amyloid fibrils, which increases the risk of toxicity. Loop diuretics and mineralocorticoid antagonists are mainstay treatments for symptom control. Advanced heart failure devices may be effective, but further data is needed to prove their efficacy. Ibutilide and amiodarone are commonly used for atrial fibrillation management in this group [Citation12]. Due to excessive amyloid deposition in cardiac conduction pathways, the patient can experience symptomatic bradycardia and heart block which can be managed with a pacemaker. summarizes symptomatic management of AL type cardiac amyloidosis [Citation5,Citation12,Citation13].
Figure 4. Summarizing the pharmacological agents for management of complications of AL type cardiac amyloidosis [Citation5,Citation12,Citation13]
![Figure 4. Summarizing the pharmacological agents for management of complications of AL type cardiac amyloidosis [Citation5,Citation12,Citation13]](/cms/asset/a88822d6-4765-471b-8d91-38b34cdc3656/zjch_a_1915547_f0004_oc.jpg)
Despite advancements supporting early diagnosis and a variety of treatment regimens, AL-type cardiac amyloidosis still carries a poor prognosis (median overall survival 12 months), particularly with severe impairment of left ventricle function [Citation8]. The median survival of 12 months without therapy can be prolonged to 17 months with immunomodulators and steroids [Citation14]. Only 5% of patients live for more than ten years [Citation15]. The median survival rate from the onset of congestive heart failure is only six months. Left atrial enlargement, right ventricular dysfunction, or syncope indicates a poor prognosis and is often a precursor of sudden cardiac death.
In terms of prognostic assessment, troponin, adrenomedullin, and pro-BNP have been found to be useful as it indicates toxic myocardial. In 2010, a study demonstrated that all three markers predicted median survival, but the best baseline prognostic marker was found to be hs-cTnT [Citation16]. The trend of proBNP can be used to gauge response to treatment. Echo findings of LA enlargement and RV dysfunction are thought to confer a poor prognosis. T1 mapping and cardiac MRI can predict survival at two years [Citation17]. These variables have been used to develop different models and tools to predict survival and prognosis. For instance, Mayo clinic staging, which relies on NT-proBNP, serum troponin T, and free light chains, is validated for risk stratification [Citation18].
4. Conclusion
Cardiac involvement in AL-type amyloidosis imparts significant morbidity and mortality to patients suffering from plasma cell dyscrasias. The management of cardiac amyloidosis entails a multidisciplinary approach, with significant input from cardiology and oncology. Despite novel diagnostic modalities and treatment regimens, the outcomes for AL-type cardiac amyloidosis remain poor. In no small part, the prognosis depends on a high degree of suspicion from the physicians who encounter these patients at the initial stages of the disease.
Prior presentation
Society of Hospital Medicine local chapter Pittsburgh, Pennsylvania 2020
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Xu ZW, Li YQ, Liu LX, et al. Light-chain cardiac amyloidosis with neuropathy: a case report. Clin Interv Aging. 2015;10:1219–1222.
- Mohty D, Damy T, Cosnay P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528–540.
- Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis. Clin Med (Lond). 2018;18(Suppl 2):s30–s5.
- Banypersad SM, Moon JC, Whelan C, et al. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364.
- Giorgetti A, Genovesi D, Milan E, et al. Cardiac amyloidosis. Clin Transl Imaging. 2019;7(1):21–32.
- Bhogal S, Ladia V, Sitwala P, et al. Cardiac amyloidosis: an updated review with emphasis on diagnosis and future directions. Curr Probl Cardiol. 2018;43(1):10–34.
- Meng L, Wh D, Lb S, et al. [The clinical features and outcomes of immunoglobulin light-chain amyloidosis with heart involvement]. Zhonghua Xin Xue Guan Bing Za Zhi. 2007;35(4):340–343.
- Cheng Z, Zhu K, Tian Z, et al. The findings of electrocardiography in patients with cardiac amyloidosis. Ann Noninvasive Electrocardiol. 2013;18(4):157–162.
- Falk RH, Plehn JF, Deering T, et al. Sensitivity and specificity of the echocardiographic features of cardiac amyloidosis. Am J Cardiol. 1987;59(5):418–422.
- Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142(1):e7–e22.
- Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29(14):1924–1933.
- Hassan W, Al-Sergani H, Mourad W, et al. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Tex Heart Inst J. 2005;32(2):178–184.
- Giancaterino S, Urey MA, Darden D, et al. Management of arrhythmias in cardiac amyloidosis. JACC Clin Electrophysiol. 2020;6(2):351–361.
- Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336(17):1202–1207.
- Kyle RA, Gertz MA, Greipp PR, et al. Long-term survival (10 years or more) in 30 patients with primary amyloidosis. Blood. 1999;93(3):1062–1066.
- Palladini G, Barassi A, Klersy C, et al. The combination of high-sensitivity cardiac troponin T (hs-cTnT) at presentation and changes in N-terminal natriuretic peptide type B (NT-proBNP) after chemotherapy best predicts survival in AL amyloidosis. Blood. 2010;116(18):3426–3430.
- Karamitsos TD, Neubauer S. T1 mapping and amyloid cardiomyopathy: how much better can it get?. Eur Heart J. 2014;36(4):203–205.
- Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–995.