
ABSTRACT
Hyperammonemia can lead to serious outcomes including brain herniation, coma and death. It is often attributed to liver disease, specifically in association with alcohol use. However, in the absence of liver pathology, it can be difficult to diagnose the etiology. We present a case of a patient with a history of remote alcohol use disorder in remission and Roux-en-Y gastric bypass (RYGB) 20 years prior who was admitted for altered mental status, found to have hyperammonemia with normal liver function tests and a normal liver biopsy. An extensive workup was unremarkable until several weeks into her admission, where she was found to have osmotic demyelination syndrome on head MRI, which was obtained after she developed persistent myoclonus and opsoclonus. Her osmotic demyelination was speculated to be secondary to hyperammonemia, which itself was correlated to her history of RYGB. There have been multiple case reports on the association of late onset hyperammonemic encephalopathy after RYGB; however, no significant correlation has yet to be made between osmotic demyelination syndrome and hyperammonemia.
1. Introduction
Hyperammonemia is most commonly associated with liver disease. It can cause a multitude of neurotoxic effects such as cerebral edema, brain herniation and ultimately death. Multiple case reports have now shown that Roux-en-Y gastric bypass (RYGB) can be a causative factor of hyperammonemia in the absence of liver disease [Citation1–4]. The exact underlying mechanism is not entirely clear, but due to alteration of the gut microbiome in association with nutritional deficiencies, these are thought to be the two main reasons for increased ammonia level [Citation1]. Although more commonly hyperammonemia neurotoxicity would be expected to cause cerebral edema, we present a case of a patient with a history of RYGB who unfortunately developed osmotic demyelination syndrome. Osmotic demyelination syndrome was speculated to be secondary to hyperammonemia. Few case reports have made this association previously; however, there is a possible underlying relationship between the two [Citation5,Citation6].
2. Case description
A 58-year-old female with a history of polysubstance abuse including alcohol, cocaine and opiates in remission (sober for 4 years), and RYGB 20 years prior presented to the emergency department from her nursing facility due to altered mental status, which was noticed earlier that morning. The nursing facility had noted a progressive decrease in her memory over the last few days, and the morning of admission, she had notable body jerking. She was vitally stable in the emergency department. She was given both Narcan and Ativan for possible opioid overdose and seizure, respectfully; however, neither improved symptoms. She continued to have body jerking for approximately 10 minutes, which was deemed as status epilepticus. She was intubated, admitted to the ICU and started on propofol and phenytoin.
It was noted that the patient’s ammonia level was elevated to 206 mcmol/L on admission. She was started on lactulose and rifaximin; however, due to refractory hyperammonemia, she underwent continuous renal replacement therapy (CRRT) from day 7 to day 9 of admission (). She developed further seizures while on hemodialysis and was started on levetiracetam. It was initially thought that her hyperammonemia was due to cirrhosis secondary to her history of alcohol use disorder. Her liver function tests revealed that an AST was only mildly elevated to 49 IU/L (reference range 5–40 IU/L), ALT within the normal limit and alkaline phosphatase elevated to 128 IU/L (reference range 35–104 IU/L). Her INR was noted to be elevated at 2.9 (reference range 0.8–1.1). She underwent a liver biopsy, which was unremarkable and did not show advanced liver disease, thus ruling out this theory. The patient received both a head CT and MRI, which demonstrated stable multifocal cortical encephalomalacia as well as mild atrophic, chronic small vessel ischemic disease ()). No acute intracranial pathology was identified. EEG was also negative for epileptiform activity.
Figure 1. (a) MRI on 21 August 2020, with stable bilateral frontal, parietal and right occipital encephalomalacia. Mild atrophy and chronic small vessel ischemic disease. (b) MRI on 16 September 2020 with trident-,shaped T2 hyperintense signal within the central pons consistent with osmotic demyelination. T2 hyperintense signal is seen within the splenium of the corpus callosum, bilateral corpus striatum and within bilateral insular cortices which are most likely secondary to hyperammonemia but can also be seen in extrapontine myelinolysis
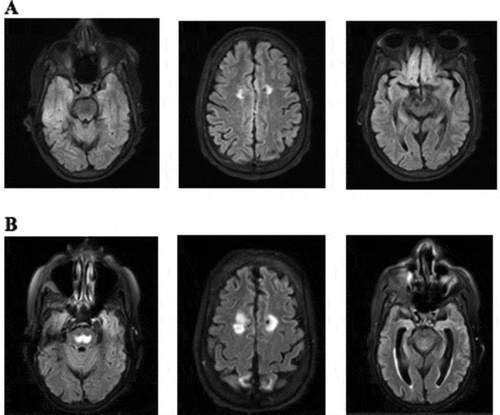
Table 1. Ammonia level concentrations over time
The patient was eventually extubated; however, her mental status continued to decline, prompting further investigation into her underlying diagnosis. Her infectious workup was unremarkable. Wilson’s disease was ruled out with normal urine copper levels. The patient underwent genetic testing to assess for urea cycle disorder; however, the results were all negative. It was noted that her zinc level was decreased at 21.1 mcg/dl (reference range 60–120 mcg/dl) and she had multiple amino acid deficiencies, which were thought to likely be related to her prior RYGB and altered gastrointestinal anatomy.
The patient’s seizures progressed to sustained myoclonus and opsoclonus. The head MRI was repeated and was remarkable for osmotic demyelination involving the central pons, splenium of the corpus callosum, bilateral corpus striatum and within bilateral insular cortices ()). These were all new findings compared to prior head imaging during the same admission. It was presumed that the osmotic demyelination was secondary to hyperammonemia. At this point, the patient’s prognosis was extremely poor. She eventually transitioned to hospice and passed away approximately 10 days after being discharged from the hospital.
3. Case discussion
3.1. Roux-en-Y gastric bypass and hyperammonemia
With the increasing rate of obesity in the USA, bariatric surgery continues to be offered as a weight loss treatment. However, over time, there have been multiple case reports on the correlation between RYGB and hyperammonemia in the absence of cirrhosis or liver disease [Citation1–4]. The onset of hyperammonemic encephalopathy after RYGB has been shown to present at various intervals, ranging from months to years [Citation1]. In this case, the patient had a RYGB approximately 20 years prior to presentation. RYGB hyperammonemia has been observed more so in women, and in some cases, women with X-linked heterozygous ornithine transcarbamylase deficiency who had previously been asymptomatic [Citation1]. Multiple nutritional deficiencies have been associated with this syndrome as well including hypoalbuminemia, multiple amino acid deficiencies, hypoglycemia and low zinc levels, many of which were seen in this patient [Citation2]. Nutritional deficiencies are thought to play a role in the urea cycle, interfering with the elimination of ammonia. In addition, RYGB alters the anatomy of the gastrointestinal system, which can cause intestinal overgrowth, leading to the production of ammonia from urease-producing bacteria () [Citation1,Citation2].
![Figure 2. Ammonia production as a result of urease [Citation1,Citation2]](/cms/asset/5eae196e-ab0f-49ce-900a-25fe75ffbc94/zjch_a_1952522_f0002_b.gif)
3.2. Hyperammonemia effects on the central nervous system
Ammonia is a nitrogen-containing compound, mainly produced in the small bowel by glutaminase, which converts glutamine into ammonia and glutamate. Ammonia is then metabolized by the liver, converted to urea – a nontoxic substance – and excreted through the kidneys [Citation4,Citation7]. Any disruption in this process can lead to excess ammonia, which is a potent neurotoxin as it can penetrate the blood–brain barrier by diffusion [Citation3]. This can manifest as changes in behavior, slurred speech, lethargy, coma and ultimately death [Citation4].
The metabolism of ammonia is essentially linked to that of glutamate. Glutaminase initially cleaves glutamine in the gut into ammonia and glutamate. In the central nervous system, however, glutamine synthetase is responsible for catalyzing ammonia and glutamate into glutamine () [Citation7]. Although the mechanism is not entirely understood, multiple studies have demonstrated the occurrence of astrocyte swelling and subsequent cerebral edema when exposed to ammonia [Citation7,Citation8]. The ‘Trojan Horse’ hypothesis proposed by Albrecht et al. [Citation9] suggests that cerebral edema is in fact a consequence of both ammonia and glutamine. Ammonia is thought to induce glutamine production within the astrocyte by glutamine synthetase. However, excess glutamine as a result of excess ammonia is thought to be transported to the mitochondria where it is metabolized back to ammonia and glutamate by phosphate-activated glutaminase [Citation7–9]. The accumulation of ammonia can lead to oxidative stress and thus swelling of astrocytes [Citation7,Citation9].
Figure 3. Glutamate and ammonia metabolism [Citation7]
![Figure 3. Glutamate and ammonia metabolism [Citation7]](/cms/asset/eeec8f9e-9219-429b-888a-58571212dab0/zjch_a_1952522_f0003_b.gif)
The correlation between cerebral edema and hyperammonemia has been fairly well established; however, no significant correlation has yet been made between osmotic demyelination syndrome and hyperammonemia. There have been but a minor number of case reports associating such, mostly in the pediatric population. Most often, osmotic demyelination is associated with rapid correction of hyponatremia. Hyponatremia in itself can cause cerebral edema, but with rapid correction, this causes extracellular tonicity, which leads to osmotic fluid shifts that deplete cells essentially causing dehydration [Citation10]. Oligodendrocytes, the primary cell in the myelination of the nervous system, are susceptible to this type of damage, and this consequently leads to destruction of myelin [Citation10]. In this patient, her sodium level was normal throughout admission; however, ultimately ended up with osmotic demyelination.
The question we ask ourselves now is can hyperammonemia cause osmotic demyelination syndrome? A case report by Langer et al. [Citation5] described a pediatric patient with carbamoyl phosphate synthetase deficiency who developed osmotic demyelination and transient cortical blindness after rapid correction of hyperammonemia. The hypothesized underlying mechanism was due to disruption of the blood–brain barrier and re-equilibration of osmolytes, in particular glutamine. Similarly, another case had been reported earlier by Mattson et al. [Citation6] of a child with ornithine carbamoyl transferase deficiency who presented with hyperammonemic encephalopathy with a maximum ammonia level of 376 mmcol/L. Her ammonia was corrected with hydration and protein restriction; however, 5 days after correction of her hyperammonemia, she developed seizures and fell into a coma. MRI brain imaging ultimately revealed characteristic findings of central pontine myelinolysis.
While several studies suggest a possible correlation between ammonia and osmotic demyelination, we also have to question whether the rate of ammonia clearance plays a role. Dialysis is by far the quickest method for reducing serum ammonia levels. The clearance of ammonia in dialysis is dependent on the blood flow, dialysate flow rate and dialyzer membrane surface area. Ammonia is a small molecule (molecular mass 17 g/mol) and is not protein-bound, making it easy to remove by dialysis [Citation11]. The patient described in our case underwent hemodialysis on days 7 through 9, with a decrease in her ammonia level from 290 mmol/L to 125 mmol/L in 48 hours. Currently, there are no guidelines as to what serum ammonia level is appropriate to initiate dialysis [Citation11]. It has been proposed that when ammonia is three times the upper limit of normal, or when there are clinical symptoms of hyperammonemia, such as encephalopathy, hemodialysis can be considered [Citation11]. Unlike the clear correlation between rapid correction of hyponatremia and demyelination, it remains unclear if the rate of change in ammonia level can be correlated to demyelination.
In regard to the treatment of hyperammonemia, lactulose, rifaximin and hemodialysis are common modalities used to lower ammonia levels, as were used in our patient. However, alternate forms of treatment are available. A recent study by Alimirah et al. [Citation12] demonstrated the use of novel therapies for hyperammonemia in the setting of hepatic encephalopathy. These included L-ornithine phenylacetate and glycerol phenylbutyrate, both of which are ammonia-scavenging agents used to improve cognition by decreasing ammonia levels. L-ornithine acts as a substrate for glutamine synthetase, while phenylacetate acts to excrete ornithine-related glutamine as phenylacetylglutamine in the kidneys [Citation13]. Glycerol phenylbutyrate is converted into phenylacetate that conjugates with glutamine to form phenylacetylglutamine, which provides as an alternative form of nitrogen waste excretion [Citation5]. For patients specifically with urea cycle disorders, nitrogen scavengers such as sodium benzoate and arginine are administered for excretion of ammonia [Citation11].
4. Conclusion
Hyperammonemia can be confidently labeled as a neurotoxic risk. It has been proven to be associated with cerebral edema and seizures, and without treatment, it can be fatal. It seems that hyperammonemia may also be correlated with osmotic demyelination syndrome; however, further studies and evidence must be available to ascertain the association.
Disclosure statement
All authors listed have contributed sufficiently to the project to be included as authors, and all those who are qualified to be authors are listed in the author byline. To the best of our knowledge, no conflict of interest, financial or other, exists.
The authors declare no potential conflicts of interest with respect to research, authorship, and/or publication of this article.
Additional information
Funding
References
- Nagarur A, Fenves AZ. Late presentation of fatal hyperammonemic encephalopathy after Roux-en-Y gastric bypass. Proc (Bayl Univ Med Cent). 2017 Jan;30(1):41–43.
- Kofman BM, Greenfeld LJ, Ali II, et al. Neurologic complications after surgery for obesity. Muscle Nerve. 2006;33(2):166–176.
- Kromas ML, Mousa OY, John S. Hyperammonemia-induced encephalopathy: a rare devastating complication of bariatric surgery. World J Hepatol. 2015 May 8;7(7):1007–1011.
- Salcedo JD, Goldstein JS, Quinonez JM, et al. Nonfatal hyperammonemic encephalopathy as a late complication of Roux-en-Y gastric bypass. Case Rep Gastrointest Med. 2019;2019:9031087.
- Langer JE, Wilson WG, Raghavan P, et al. Extrapontine myelinosis resulting in transient cortical blindness. Pediatr Neurol. 2010 Feb 1;42(2):154–156.
- Mattson LR, Lindor NM, Goldman DH, et al. Central pontine myelinolysis as a complication of partial ornithine carbamoyl transferase deficiency. Am J Med Genet. 1995 June 19;60(3):210–213.
- Scott TR, Kronsten VT, Hughes RD, et al. Pathophysiology of cerebral oedema in acute liver failure. World J Gastroenterol. 2013 Dec 28;19(48):9240–9255.
- Rao KVR, Jayakumar AR, Norenberg MD. Induction of the mitochondrial permeability transition in cultured astrocytes by glutamine. Neurochem Int. 2003 Sept–Oct;43(4–5):517–523.
- Albrecht J, Norenberg MD. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology. 2006;44:788–794.
- Yuridullah R, Kumar V, Nanavati S, et al. Clinical resolution of osmotic demyelination syndrome following overcorrection of severe hyponatremia. Case Rep Nephrol. 2019;2019:Article ID 1757656, 4 pages.
- Gupta S, Fenves AZ, Hootkins R. The role of RRT in hyperammonemic patients. Clin J Am Soc Nephrol. 2016 Oct 7;11(10):1872–1878.
- Alimirah M, Sadiq O, Gordon SC. Novel therapies in hepatic encephlopathy. Clin Liver Dis. 2020 May;24(2):303–315.
- Jalan R, Wright G, Davies NA, et al. L-ornithine phenylacetate (OP): a novel treatment for hyperammonemia and hepatic encephalopathy. Med Hypotheses. 2007;69(5):1064–1069.