
ABSTRACT
Glioblastoma multiforme (GBM) are mortifying brain tumours that contain a subpopulation of tumour cells with stem-like properties, termed glioblastoma stem-like cells (GSCs). GSCs largely contribute to tumour initiation, propagation and resistance to current anti-cancer therapies. GSCs are situated in perivascular niches, closely associated with brain microvascular endothelial cells, thereby involved in bidirectional molecular and cellular interactions. Moreover, extracellular vesicles are suspected to carry essential information that can adapt the microenvironment to the tumour’s needs, including tumour-induced angiogenesis. In GBM, extracellular vesicles produced by differentiated tumour cells and GSCs were demonstrated to disseminate locally and at distance. Here, we report that the pro-angiogenic pro-permeability factor VEGF-A is carried in extracellular vesicles secreted from ex vivo cultured patient-derived GSCs. Of note, extracellular vesicle-derived VEGF-A contributes to the in vitro elevation of permeability and angiogenic potential in human brain endothelial cells. Indeed, VEGF-A silencing in GSCs compromised in vitro extracellular vesicle-mediated increase in permeability and angiogenesis. From a clinical standpoint, extracellular vesicles isolated from circulating blood of GBM patients present higher levels of VEGF-A, as compared to healthy donors. Overall, our results suggest that extracellular vesicle-harboured VEGF-A targets brain endothelial cells and might impact their ability to form new vessels. Thus, tumour-released EV cargo might emerge as an instrumental part of the tumour-induced angiogenesis and vascular permeability modus operandi in GBM.
Introduction
Glioblastoma multiforme (GBM) is the most common primary brain tumour in adults and one of the deadliest in humans [Citation1]. The current clinical management of GBM includes surgical resection followed by ionizing radiations and chemotherapies, among which is the alkylating agent temozolomide [Citation2]. However, this therapeutic regimen remains mostly inefficient and relapse is almost inevitable in a short window of time of about 7–10 months. In this context, it is now well documented that glioblastoma carries a subpopulation of rare but significant self-renewing cancer cells termed glioblastoma stem-like cells (GSCs) [Citation3,Citation4]. This reservoir of tumour-initiating cells has the capacity to sustain malignant properties including initiation, growth, resistance to treatments and recurrence of the disease [Citation5]. GSCs are localized in perivascular niches in close interaction with blood vessels [Citation6]. In this privileged microenvironment, vascular endothelial cells notably convey extracellular factors that help supervise the fate and survival of GSCs [Citation7–Citation9]. Although targeting the GBM perivascular niche with the anti-VEGF-A anti-angiogenic antibody bevacizumab leads to reduce vascularization along with the GSC population [Citation6], anti-VEGF strategies remain a matter of debate in the context of GBM treatment [Citation10–Citation12].
Reciprocally, GSCs might modulate endothelial behaviour and plasticity to foster tumour vascular development [Citation13,Citation14]. We recently found that the pro-permeability guidance molecule semaphorin 3A is transported in extracellular vesicles (EVs) isolated from ex vivo models, as well as from peripheral blood of tumour-bearing mice and from GBM patients [Citation15]. This GSC-liberated semaphorin 3A further operates on brain endothelial permeability [Citation15,Citation16]. In keeping with this idea, EVs are now recognized as potent means of intercellular communication, where nucleic acids, metabolites, proteins and lipids are transferred to target cells, in the vicinity of or at a distance from the source of secretion [Citation17]. EVs are highly heterogeneous vesicles, classified into three main sub-groups: exosomes, microvesicles and apoptotic bodies. These particles, under the unified term of EVs, are produced by a plethora of cell types, including non-malignant and malignant cells [Citation17,Citation18]. Thus, EVs have aroused growing interest in cancer research and could represent attractive new targets for anti-tumour therapies in the GBM microenvironment [Citation19–Citation21]. Indeed, pioneer work from Skog and colleagues revealed that both GBM bulk and isolated tumour cells produce EVs that are enriched in angiogenic factors [Citation22]. Furthermore, circulating EVs were proposed to be specific to the disease state and to reflect the tumour status [Citation22–Citation26]. Although VEGF-A is known to be enriched in the vascular niche and be transported in EVs [Citation6,Citation13,Citation22], the specific role of the angiogenic cargo remains to be fully elucidated. In this study, we now reported that patient-derived GSC-released EVs increase the angiogenic behaviour of human brain endothelial cells in vitro through VEGF-A. Importantly, VEGF-A levels were elevated in circulating EVs collected from blood samples of primary GBM patients upon diagnosis. Altogether, our results highlight the importance of EV-delivered VEGF-A for the properties of human brain endothelial cells.
Results
Characterization of patient-derived GSC-released extracellular vesicles in vitro
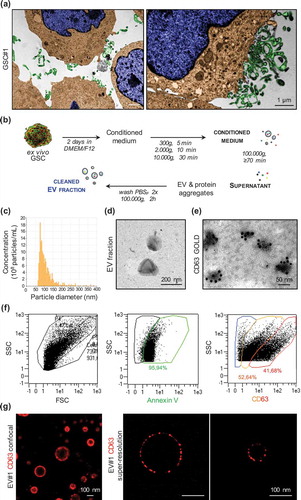
Electron microscopy analysis illustrates vesicular and membrane-derived structures decorating the surface of patient-derived glioblastoma stem-like cells (GSCs, Figure S1) [Citation15] that could be dispersed out in the milieu ()). These membranous particles were isolated from GSC-conditioned media through a differential ultracentrifugation protocol ()) [Citation15], and further characterized by qNano and electron microscopy (),()). Purified GSC-EVs exhibited a round-shaped and intact morphology with diameter ranging from 65 to 384 nm (mean 109 ± 44.4, measured concentration 8.2·109 particles/ml), confirming the nature of the purified EV fraction [Citation27,Citation28]. In addition, immunogold labelling and flow cytometry analysis indicate the expression of CD63 and Annexin V staining, frequently used as markers to identify EVs [Citation17]. Interestingly, the intensity of CD63 staining by cytometry analysis indicates the presence of two distinct populations that might correspond to exosome- and microvesicle-like particles (),()) [Citation17,Citation29]. This heterogeneity in size was further visualized by super-resolution confocal microscopy analysis for CD63 ()).
Figure 1. Characterization of patient-derived glioblastoma stem-like cells (GSC)-released extracellular vesicles in vitro. (a) Representative transmission electronic microscopy (TEM) of GSCs cultivated under normal conditions. TEM images were artificially colourized with blue for nucleus, brown for cytoplasm and green for vesicle-like structures. (b) Schematic protocol of EV isolation by serial ultracentrifugation steps from the GSC-conditioned medium (serum-free and mitogen-free DMEM/F12). (c) Following purification, EVs were resuspended in 0.2-µm-filtered saline and characterized by qNano for particle concentration and diameter. (d,e) Representative TEM pictures of GSC#1-EVs either unlabelled or immunogold-labelled for CD63. (f) A MACSQuant Analyser Flow cytometer was used to evaluate Annexin V and CD63 on purified GSC-EVs. (g) Size diversity of GSC#1-derived EVs was analysed using confocal microscopy and super-resolution SIM microscopy. Representative pictures of CD63 staining are shown.
GSC-released extracellular vesicles exert pro-angiogenic activity on human brain endothelial cells
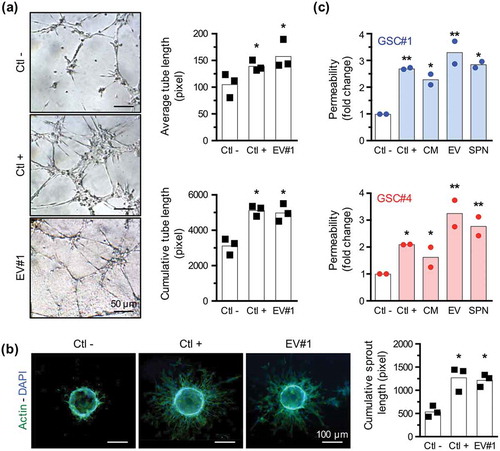
Because of their location in close proximity to endothelial cells within the tumour and their ability to release pro-angiogenic factors, GSCs could ignite tumour-based angiogenesis [Citation13]. Previous results from our lab established that GSC-derived EVs can be actively taken up by human brain microvascular endothelial cells [Citation15,Citation30]. To next evaluate the angiogenic potential of GSC-derived EVs, we employed two in vitro standard experimental models, namely tubulogenesis and sprouting assays [Citation31]. Purified GSC-EV fraction, re-suspended in serum-free and growth factor-free medium, induces an increase in average and cumulative tube length at a level similar to the effects of the VEGF-A positive control ()). Interestingly, the same holds true when measuring endothelial sprouting out of beads embedded in a fibrinogen matrix ()). Along with a stimulatory effect on angiogenesis, EVs derived from GSCs are also able to promote an increase in endothelial permeability, a typical hallmark of angiogenesis [Citation32] ()). Finally, permeability was found systematically heightened upon EV stimulation, as compared to either conditioned media or soluble 100,000×g supernatant, prepared from two patient-derived GSC lines ()). Altogether, our data provide compelling evidence that patient-derived GSC-EVs alone can foster both angiogenesis and permeability in human brain endothelial cells.
Figure 2. GSC-released extracellular vesicles exert pro-angiogenic activity on human brain endothelial cells. (a) In a tubulogenesis formation assay, brain microvascular endothelial cells (ECs) were embedded in a collagen-based matrix and stimulated with negative (serum-free mitogen-free DMEM/F12) and positive (VEGF, 50 ng/ml) controls, and 20 µg/ml of GSC#1-EVs resuspended in serum-free mitogen-free DMEM/F12. Pictures were acquired 8 h later and average and cumulative tube lengths were quantified (n = 3). *, p < 0.05. (b) ECs were coated on Cytodex microbeads and embedded in a fibrinogen matrix. Cells were incubated with serum-free mitogen-free DMEM/F12 (Ctl −), VEGF-A (Ctl +, 50 ng/ml) and GSC#1-EVs (20 µg/ml) for 2 days. EC-coated beads were then stained for phalloidin (actin, green) and DAPI (nucleus, blue) and pictures were quantified for sprout length (n = 3). *, p < 0.05. (c,d) Transwell permeability assay was performed treating ECs with VEGF-A (Ctl +, 50 ng/ml) or GSC#1, #4-EVs (20 µg/ml), to assess the passage of 40 kDa FITC-labelled dextran. Similarly, permeability assays were done with conditioned media (CM) or 100,000×g soluble supernatant fractions (SPN). Results are expressed as fold change relative to the negative condition (medium alone, Ctl −) (n = 2). *, p < 0.05; **, p < 0.01.
VEGF-A is among the pro-angiogenic cargo of GSC-released EVs
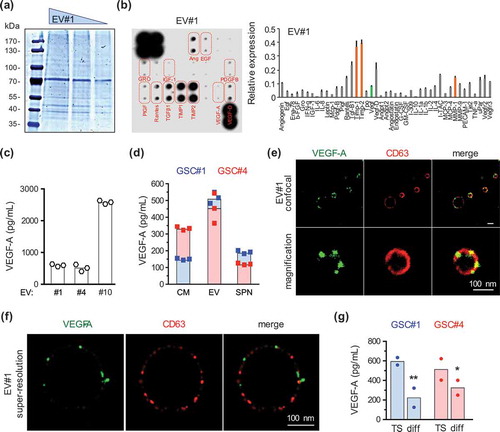
EVs have been described to convey cellular signalling by carrying metabolites, lipids, proteins and nucleic acids [Citation17,Citation21,Citation27]. First, the quality of the overall protein content of GSC-derived EVs was verified with SDS-PAGE Coomassie blue staining ()). An angiogenic protein array further suggested the presence of extracellular proteases and protease inhibitors, such as MMP-1 and TIMPs, known both for their modulatory role in angiogenesis and to accumulate in EVs (), orange bars) [Citation22,Citation33–Citation35]. Among the canonical angiogenic cytokines, VEGF-A was reproducibly detected in a similar relative amount, as compared to the conditioned media (), green bar and our unpublished observations). VEGF-D, EGF and IGF1 expression were, however, not confirmed (data not shown). VEGF-A is a crucial factor for angiogenesis operating through EC sprouting and vascular permeability [Citation32,Citation36,Citation37]. VEGF-A was consistently, although variably, detected in EVs prepared from three different patient-derived GSCs, exhibiting VEGF-A concentrations ranging from 500 to more than 2500 pg/ml ()). When compared to the soluble 100,000×g supernatant, ELISA detection of VEGF-A in two GSC lines, namely GSC#1 and GSC#4, was multiplied by nearly 2-fold, suggesting that VEGF-A is somehow enriched in the EV fraction ()). Furthermore, super-resolution confocal microscopy analysis showed that anti-VEGF-A antibodies indeed label discrete membranous subdomains, at either the inner or outer surface of vesicles (),(f)). However, the analysis of GSCs and their EV cargo failed to detect classic VEGF receptors (VEGF-R1, R2, R3 and neuropilin-1, Fig. S2). Interestingly, VEGF-A production in vesicles was reduced upon GSC differentiation (Fig. S1, )). Overall, our data suggest that VEGF-A is both free in the milieu and transported through GSC-EVs, and could therefore contribute to the EV-based pro-angiogenic activity observed in ECs.
Figure 3. VEGF-A is among the pro-angiogenic cargo of GSC-released EVs. (a) Decreasing amounts (50, 30, 10 µg) of purified EVs were separated by SDS-PAGE and further stained with Coomassie blue. (b) Protein content of purified EV#1 (50 µg) was analysed with a human angiogenesis array. Relative expression of the identified proteins is plotted on the graph. Orange bars indicate expression of matrix metalloproteases (MMP) and tissue inhibitor of metalloproteases (TIMPs), while the green bar is for VEGF-A (n = 2). (c) VEGF-A concentration was analysed by ELISA in purified EVs isolated from GSC#1, #4 and #10 (n = 3). (d) VEGF-A protein level was assessed by ELISA in conditioned media (CM), purified EV fractions or 100,000×g soluble supernatant fractions (SPN) from GSC#1 and #4 (n = 3). (e,f) Representative pictures of GSC#1-EVs stained for VEGF-A (green) and CD63 (red), and acquired with confocal and super-resolution SIM microscopy. Scale bar, 100 nm. (g) VEGF-A concentration was analysed by ELISA in purified-EVs isolated from GSC#1 and #4 cultured as either 3D spheres in defined medium or adherent differentiated cells (n = 2). *, p < 0.05; **, p < 0.01.
Extracellular vesicle-produced VEGF-A is pro-angiogenic
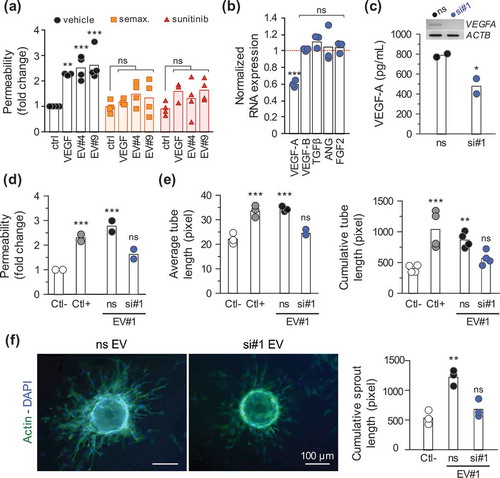
To next challenge the biological impact of EV-delivered VEGF-A on brain endothelial cells, two inhibitors of VEGFR signalling, namely semaxitinib and sunitinib, were employed [Citation38,Citation39]. GSC-isolated EVs failed to promote endothelial cell permeability when VEGF receptors were pharmacologically targeted, therefore arguing for a prominent role of EV-transported VEGF-A ()). To further provide evidence of VEGF-A involvement, RNA interference was employed to silence VEGF-A in GSCs. VEGF-A knockdown efficiency was first assessed in GSCs, while other pro-angiogenic factors expressed by GSCs, such as VEGF-B, TGFβ, angiogenin and bFGF, remain unaffected ()). VEGF-A-depleted EVs could then be prepared from transfected cells with decreased amounts of targeted protein ()). Interestingly, while VEGF-A silencing does not affect EV production (data not shown), VEGF-A-depleted EVs were no longer able to increase endothelial permeability and sprouting ()–()). Thus, our results indicate that GSC-emanating EV-transported VEGF-A remains functional and could directly trigger typical angiogenic responses in targeted human endothelial cells. However, the exact signalling pathway involved downstream would require more in-depth investigation.
Figure 4. Extracellular vesicle-produced VEGF-A is pro-angiogenic. (a) Endothelial cells were seeded for permeability assays (105 cells, 4 days), treated for 6 h with semaxitinib (semax.; 5 µM) and sunitinib (100 nM) in serum-free media. The passage of FITC-dextran permeability was assessed in response to recombinant VEGF-A (50 ng/ml) and GSC#4 and GSC#9-prepared EVs (15 µg/ml), as previously described. **, p < 0.01. ***, p < 0.001. (b) GSC#4 were transfected with either non-silencing RNA (ns) or VEGF-A-targeting duplexes and knockdown efficiency was assessed by RT-PCR as compared to ACTB. Results were normalized to the expression levels of the ns condition, represented as the dashed red line. Relative expression levels of other angiogenic factors (VEGF-B, TGFβ, angiogenin (ANG) and FGF2) were also quantified (n = 3). ***, p < 0.001. (c) EVs were isolated from the conditioned media of GSC#4 transfected cells, and VEGF-A levels were measured by ELISA (n = 2). *, p < 0.05. (d) A confluent monolayer of ECs was exposed to control EV#1 (ns) or VEGF-A-depleted EVs (si#1) and permeability was assessed as described previously (n = 2). ***, p < 0.001. (e) Tubulogenesis and (f) sprouting were performed in ECs treated either with control (ns) or VEGF-A-depleted GSC#1-EVs. Representative pictures of EC-coated beads stained with phalloidin (actin, green) and DAPI (nucleus, blue) (n = 3). **, p < 0.01; ***, p < 0.001. Serum-free mitogen-free medium and VEGF-A (50 ng/ml) were used as negative and positive controls, respectively.
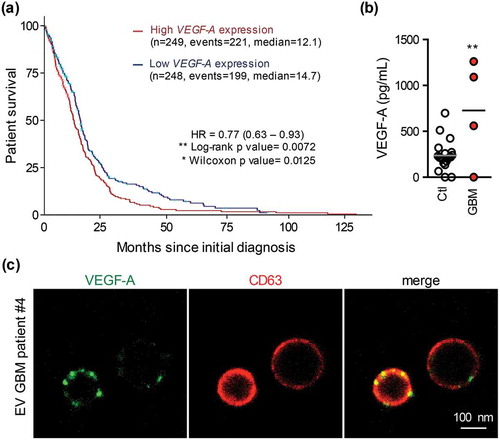
EVs isolated from GBM patient circulating blood contain abundant VEGF-A
VEGF-A chiefly orchestrates tumour angiogenesis, while its up-regulation correlates with poor prognosis in several solid cancers, including GBM [Citation40–Citation48]. Bioinformatics analysis of The Cancer Genome Atlas (TCGA) dataset on 498 primary GBM indeed confirmed that high VEGF-A expression is associated with lower probability of survival in GBM patients ()). Because previous works, including data from our lab, documented tumour-originated EVs in bloodstream [Citation15,Citation22,Citation49–Citation51], circulating EVs were isolated from blood samples of newly diagnosed primary GBM patients, as described and characterized previously [Citation15]. All patients were males, with an average age centred on 70.5 ± 8.7 and bearing primary brain tumour of 33.6 ± 4.9 mm average size. Notably, VEGF-A concentration was significantly higher in circulating EVs from GBM patients (n = 4) as compared to healthy donors (n = 20), with a mean of 237.1 ± 36.2 versus 727.5 ± 84.6 pg/ml ()). Interestingly, microscopy analysis shows that anti-VEGF-A antibodies illuminate CD63-positive vesicles isolated from GBM patients ()). Our data thus indicate that peripheral blood-isolated EVs from GBM patients display a higher level of VEGF-A.
Figure 5. EVs isolated from GBM patient circulating blood contain abundant VEGF-A. (a) Probability of survival after initial diagnosis in primary GBM patients expressing low (blue line) or high (red line) VEGF-A levels (n = 498). (b) Patient circulating EVs were isolated by successive ultracentrifugations from the serum of healthy donors (Ctl) or primary diagnosed GBM patients (GBM). VEGF-A levels were quantified by ELISA in circulating EVs from healthy (n = 20) or GBM patients (n = 4). **, p < 0.01. (c) Confocal microscopy of purified EVs from GBM patients stained for VEGF-A (green) and CD63 (red).
Discussion
In this study, we reported that in addition to differentiated and proliferating GBM cells [Citation22,Citation52], the self-renewing, slow cycling, tumour-initiating and therapy-resistant GSC population produces the pro-angiogenic pro-permeability VEGF-A cytokine within EVs. Interestingly, VEGF-A derived from GSC-secreted vesicles remains functionally active and can trigger angiogenic behaviour and permeability in human brain endothelial cells. Moreover, high levels of VEGF-A protein were measured in circulating EVs collected from GBM patient liquid biopsies.
First, we showed that VEGF-A carried by GSC-derived EVs retains the ability to promote angiogenesis of human brain endothelial cells. While the mechanism involved in cellular targeting was not investigated here, we can postulate that it might require endocytosis or pinocytosis mechanisms [Citation18,Citation27]. Our recent study suggests that it indeed involves an active process [Citation15]. One can question why VEGF-A needs to be embedded in EVs, in addition to being freely available and soluble in the milieu, to reach cells harbouring VEGF receptors. First, VEGF-A is accumulated in EVs, as compared to the soluble supernatant, and appeared tethered to membranous domains, at either the inner or the outer face of EVs. Second, EV transport might help protect the cytokine within the hostile tumour microenvironment, including hypoxia-derived acidification [Citation53,Citation54]. Third, this mode of secretion can also assist VEGF-A to evade decoy receptors and proteases, increasing further its pro-angiogenic potential on cell targets. In keeping with this idea, EV-presented VEGF-A might also facilitate fast signalling, as demonstrated in the context of IFNγ in neural precursor cells [Citation55]. However, while we failed to detect VEGF-R1, 2, 3 and neuropilin-1 within the GSC-EV cargo (Fig. S2), VEGF-A could be instead associated with yet to be identified proteoglycans. A recent study identified a specific high molecular weight of VEGFA (VEGF90K) as being transported in breast cancer cell-derived EVs. In this study, the authors nicely document how EV-harboured VEGF-A could escape anti-VEGF therapeutic compounds, such as bevacizumab [Citation56]. Altogether, EV-delivered VEGF-A from GSCs could potently prime endothelial cells in their vicinity for angiogenesis.
Besides its canonical pro-angiogenic pro-permeability action on endothelial cells, VEGF-A is now documented to operate on multiple cell types within the tumour microenvironment [Citation48]. For instance, abnormal elevated VEGF-A levels promote dedifferentiation of cancer cells, and induce their proliferation [Citation41,Citation46,Citation57], while its blockade attenuates tumour-initiating properties [Citation58,Citation59]. Because GSCs express the VEGF-A receptor neuropilin-1, albeit low levels of VEGF-R2 ([Citation16] and Fig. S2), VEGF-A derived from GSC-EVs could be employed in an autocrine fashion and regulate their stemness properties. Likewise, GBM-secreted vesicles could fuel and maintain the vascular niche, which in turn feeds back on GSC identity [Citation7–Citation9,Citation52,Citation60]. Furthermore, bFGF and TGFβ that were detected in GSC-released EVs are thought to be involved in GSC-maintenance processes [Citation9,Citation61–Citation64]. An extensive and comprehensive characterization of GSC-secreted vesicle cargo is therefore of high interest.
Using mice models and liquid biopsies, we and others previously reported that GSC-secreted EVs navigate through the cerebral blood microcirculation and the cerebrospinal fluid, and also disseminate in the body at distance from the primary tumour site [Citation15,Citation22,Citation49–Citation51]. One could propose that VEGF-A derived from GSC vesicles could co-opt circulating endothelial progenitors to regenerate/expand the tumour vasculature [Citation65,Citation66]. Therefore, by transporting VEGF-A, GSC-derived EVs could contribute to sustaining the vascular niche and, in turn, to fostering tumour angiogenesis and progression.
We report here that GBM patients present significantly elevated levels of VEGF-A in circulating EVs, as compared to healthy donors. However, this does not mean that the EV-trapped VEGF-A found in plasma arises solely from GSCs. Indeed, a predominant proportion could also be secreted by differentiated tumour cells, the tumour vasculature itself, as well as platelets and immune cells [Citation67]. It is nonetheless expected that non-GSC sources might vary with tumour size and treatments, while the GSC-derived pool of VEGF-A might be consistently produced and released, and therefore might remain indicative of the tumour status [Citation23,Citation68]. Collectively, these observations suggest that in patients, most likely GSC- and GBM cell-derived EVs have the ability to cross the blood-brain barrier and reach the bloodstream to disseminate, thus representing interesting biomarker perspectives [Citation22,Citation69].
Prognosis for GBM patients remains extremely poor, emphasizing the need for novel treatment strategies and improvement of quality of life in patients. Overall, it might be important to track both soluble and EV-embedded VEGF-A in the course of the disease progression and to estimate its theranostic value and to refine the therapeutic window.
Materials and methods
Ethics statement and clinical samples
This study abides by the rules of the Helsinki Protocol. All patients signed a written informed consent before sample collection for diagnostic purposes. Peripheral blood (3–5 ml) from glioblastoma (GBM) patients was collected in heparin-coated tubes (Hopital d’Instructions des Armées Percy, Clamart, France) and from healthy donors (Etablissement Français du Sang, Paris, France). Blood samples were stored at room temperature for less than 1 h before processing for EV isolation (please refer to the protocol described below and )). For each patient and healthy donor, serum-derived EVs were re-suspended in 500 µl of 0.2-µm-filtered saline vehicle or serum-free medium. Purified EVs were aliquoted and stored at −20°C if used in the following 2 weeks, or −80°C for longer storage.
Glioblastoma stem-like cell (GSC) culture and silencing RNA
Patient-derived GSCs were obtained from primary GBM resection and cultured ex vivo as previously described [Citation8,Citation15]. In brief, tumours were first gently dissociated using the MACsDissociator (Miltenyi) according to the manufacturer’s instructions. GSC spheres were maintained in suspension in DMEM/F12 supplemented with N2, G5 and B27, plus 1% penicillin/streptomycin (ThermoFisher Scientific). Conditioned media from GSCs were prepared from 1·106 cells cultivated in serum-free and mitogen-free DMEM/F12. Culture supernatants were collected 2 days later and processed for EV purification by ultracentrifugation steps (please refer to the protocol described below and )). Of note, GSCs were tested for mycoplasma contaminations (Invivogen). To induce differentiation, spheres were dissociated and cultured in DMEM + 10% FBS. Adherent cells were passaged using the same differentiation medium and analysed 2 weeks after.
Non-silencing (Low GC Duplex) negative control and the predesigned Stealth siRNA oligonucleotides targetting VEGFA (HSS111274) (ThermoFisher Scientific) were transfected in GSCs using Lipofectamine RNAiMax (ThermoFisher Scientific) at a final concentration of 25 nM. The preparation of conditioned medium from transfected cells was started 2 days post-transfection to ensure optimal protein depletion, and following the same protocol as with non-transfected GSCs.
Endothelial cell culture
Immortalized human brain microvascular endothelial cells [Citation30] were grown in complete EBM2 (Lonza) as described previously [Citation16].
Antibodies and reagents
The following antibodies were used: human CD63 (BD Biosciences), VEGF-A (Abcam) and fluorochrome-coupled species-specific secondary antibodies (ThermoFisher Scientific). Alexa488-conjugated phalloidin and AnnexinV were from ThermoFisher Scientific. VEGFR inhibitors semaxitinib and sunitinib were used at 5 µM and 100 nM, respectively (Selleckchem).
EV purification
EVs were isolated as described previously [Citation15] through differential ultracentrifugation steps ()) using the L-70 ultracentrifugation with SW55 rotors and appropriate tubes (Beckman). Fresh samples were collected at room temperature and then successively centrifuged at 300×g (5 min), 2000×g (10 min), 10,000×g (30 min) and 100,000×g (≥70 min). To obtain a cleaned EV fraction, pellets were then washed twice in PBS and re-suspended in 0.22-µm-filtered vehicle (saline or serum-free media). All centrifugation steps were performed at 4°C. EV preparations were checked with the qNanoGold Instrument (Izon Science).
Flow cytometry analysis
Samples were analysed with a MACSQuant Analyzer (Miltenyi) as previously described in [Citation15]. In brief, 100 µl of purified EVs re-suspended in 0.22-µm-filtered PBS were incubated 1 h at room temperature in the dark with 15 µl PE-coupled CD63 antibody (BD Biosciences) or 12 µl AlexaFluor488-conjugated Annexin V (ThermoFisher Scientific) with the provided Annexin V buffer. PE-conjugated isotype and 1X Annexin V Buffer (ThermoFisher Scientific) were used as control for CD63 and Annexin V staining, respectively.
Elisa
EVs were then subjected to a 10 min incubation at 72°C, in 0.5% SDS final concentration. VEGF-A protein levels were then quantified as per the manufacturer’s instruction (R&D Systems).
Human angiogenesis protein array
Prior to incubation with each membrane antibody array, 50 µg of GSC-EVs were pre-treated with 0.5% SDS for 10 min, 72°C. Subsequently, analysis was performed as per the manufacturer’s instruction (RayBiotech, Clinisciences) and membranes were scanned using the Fusion imager (Vilber Lourmat). Membranes were quantified by densitometry analysis using the ImageJ software (NIH) and normalized to the intensity of their internal positive controls. Identified proteins were then depicted in graph bars for their relative expression.
Electron microscopy and immunogold labelling
For whole-cell analysis, GSCs were imaged as previously described [Citation8]. GSCs were added on poly-L-lysine-coated slides and fixed for 1 h with 3% glutaraldehyde/PBS. After incubation in 1% OsO4, cells were dehydrated in graded dilutions of ethanol, embedded in artificial resin (Epon, Momentive Specialty Chemicals) and processed for electron microscopy performed at 60–80 kV on unstained thin sections (EM10CR, Zeiss, Imagery facility, Institut Cochin, Paris, France). GSC-derived EVs were analysed as previously described [Citation15]. In brief, EV fraction was either re-suspended in 0.22-µm-filtered PBS for direct acquisition, or fixed for 6 h, 4°C, in filtered PBS PFA 4% for immunogold labelling. EVs were then added on the formvar/carbon-coated grid overnight, 4°C. EVs were incubated with 0.1% bovine serum albumin (BSA), 10% normal donkey serum for 20 min, followed by anti-CD63 antibody (dilution 1:100) diluted in PBS, 0.1% BSA and 4% normal donkey serum. After extensive washes, sections were incubated with gold-labelled secondary antibody with a gold particle size of 10 nm (GAM 10, British Biocell International), washed again, stained with 2% uranyl acetate (10 min) and air-dried. Vesicles were examined using a transmission electron microscope (EM10CR, Zeiss, Imagery facility, Institut Cochin, Paris, France).
Immunostaining and confocal analysis
Immunofluorescence on EVs was performed as described previously in [Citation15], using 0.2 µm filtered solutions. Purified EVs (100 µl) were added overnight on poly-L-lysine slides (Thermo Fisher Scientific), fixed in 4% paraformaldehyde and further permeabilized in 0.5% Triton. After a blocking step in PBS-BSA 3%, slides were stained overnight at 4°C with CD63 and/or VEGF-A antibodies (dilution 1:100). After washes and secondary antibody incubation (ThermoFisher Scientific), slides were mounted with Prolong gold anti-fade mounting medium (ThermoFisher Scientific). Pictures were acquired with a Nikon SIM super-resolution confocal microscope (Micropicell microscopy platform, SFR Sante Francois Bonamy, Nantes, France).
Tubulogenesis assay
The tubulogenesis assay was adapted from previously described protocols [Citation70,Citation71]. Matrigel together with 1·104 ECs were added to a 96-well plate and allowed to polymerize for 30 min at 37°C. Cells were then treated for 8 h with 20 µg/ml of GSC-EVs re-suspended in DMEM/F12 serum-free media. At least five fields of view per condition were randomly acquired (Motic, AE21 microscope). For tube length quantifications, images were processed in a blind manner using ImageJ software (NIH).
Sprouting assay
Sprouting of ECs was described previously in [Citation70]. Four thousand Cytodex3 microcarrier beads (Sigma) were first coated with collagen (BD Biosciences) and mixed with 1·106 ECs in EBM2 for 4 h at 37°C, with regular shaking. Coated beads were transferred to a new culture dish overnight to remove unattached cells, then washed with PBS and re-suspended in a 2.5 mg/ml fibrinogen-aprotinin (0.15 units/ml, Sigma) solution + bFGF2 (200 ng/ml, Sigma). The mixture was then distributed in an eight-well plate (Ibidi Biovalley) containing 0.625 U/ml of thrombin, and allowed to clot (15 min, 37°C). EBM2 medium alone or 20 µg/ml of EVs re-suspended in serum-free EBM2 medium were added on top of the fibrinogen matrix and sprouting was allowed for 2 days. For imaging, matrix-containing beads were fixed (PFA 4%, 30 min) and permeabilized (Triton 0.5%, 10 min). Phalloidin and DAPI (ThermoFisher Scientific) labelling was performed to visualize ECs. At least five fields of view were randomly acquired using a Leica fluorescence microscope (Imagery facility, Institut Cochin, Paris). For sprout length quantifications, images were processed in a blind manner using ImageJ software (NIH).
Permeability assays and RT-PCR
In vitro brain endothelial cell permeability was performed as described previously [Citation15,Citation72,Citation73]. Standard protocol was used for RT-PCR [Citation73].
TCGA analysis
The Cancer Genome Atlas GBM dataset was interrogated for the survival analysis based on VEGFA expression (Affymetrix HT HG U133A). Median preset thresholds were applied to discriminate between High and Low expression levels of VEGFA (http://www.gliovis.com/) [Citation74]. The Kaplan-Meier curve results from the analysis on 498 samples from primary GBM only. The log-rank test and Wilcoxon tests were conducted, and the hazard ratio was depicted.
Supplementary_figures.pdf
Download PDF (2.2 MB)Acknowledgements
The authors are grateful to present and past members of the SOAP team in the Institut Cochin, Paris, France, and CRCINA, Nantes, France. We wish to thank Alain Schmitt (electron microscopy facility, Institut Cochin, Paris, France) and Steven Nedellec (Micropicell, SFR Sante Francois Bonamy, Nantes, France).
LT is supported by a doctoral fellowship from Université Paris Descartes and the Bettencourt Schueller Foundation. RP is supported by a fellowship from The Fondation ARC contre le Cancer.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol. 2014;9:1–12.
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996.
- Yuan X, Curtin J, Xiong Y, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23(58):9392–9400.
- Lathia JD, Mack SC, Mulkearns-Hubert EE, et al. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203–1217.
- Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291.
- Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69–82.
- Galan-Moya EM, Le Guelte A, Fernandes EL, et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011;12(5):470–476.
- Galan-Moya EM, Treps L, Oliver L, et al. Endothelial secreted factors suppress mitogen deprivation-induced autophagy and apoptosis in glioblastoma stem-like cells. PLoS One. 2014;9(3):e93505.
- Fessler E, Borovski T, Medema JP. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol Cancer. 2015;14:157.
- Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709–722.
- Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708.
- Batchelor TT, Reardon DA, De Groot JF, et al. Antiangiogenic therapy for glioblastoma: current status and future prospects. Clin Cancer Res. 2014;20(22):5612–5619.
- Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66(16):7843–7848.
- Folkins C, Shaked Y, Man S, et al. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009;69(18):7243–7251.
- Treps L, Edmond S, Harford-Wright E, et al. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene. 2016;35(20):2615–2623.
- Le Guelte A, Galan-Moya E-M, Dwyer J, et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J Cell Sci. 2012;125(Pt 17):4137–4146.
- Yanez-Mo M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066.
- Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289.
- Bronisz A, Godlewski J, Chiocca EA. Extracellular vesicles and MicroRNAs: their role in tumorigenicity and therapy for brain tumors. Cell Mol Neurobiol. 2016;36(3):361–376.
- Chistiakov DA, Chekhonin VP. Extracellular vesicles shed by glioma cells: pathogenic role and clinical value. Tumour Biol. 2014;35(9):8425–8438.
- Andre-Gregoire G, Gavard J. Spitting out the demons: extracellular vesicles in glioblastoma. Cell Adh Migr. 2016;13:1–9.
- Skog J, Würdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–1476.
- Kucharzewska P, Christianson HC, Welch JE, et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A. 2013;110(18):7312–7317.
- Halin Bergström S, Hägglöf C, Thysell E, et al. Extracellular vesicles from metastatic rat prostate tumors prime the normal prostate tissue to facilitate tumor growth. Sci Rep. 2016;6:31805.
- Chow A, Zhou W, Liu L, et al. Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κB. Sci Rep. 2015;4:5750.
- Yang N, Li S, Li G, et al. The role of extracellular vesicles in mediating progression, metastasis and potential treatment of hepatocellular carcinoma. Oncotarget. 2017;8:3683–3695.
- Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232.
- Witwer KW, Buzas EI, Bemis LT, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2: DOI:10.3402/jev.v2i0.20360.
- Yoshioka Y, Konishi Y, Kosaka N, et al. Comparative marker analysis of extracellular vesicles in different human cancer types. J Extracell Vesicles. 2013;2: DOI:10.3402/jev.v2i0.20424.
- Weksler BB, Subileau EA, Perrière N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19(13):1872–1874.
- Goodwin AM. In vitro assays of angiogenesis for assessment of angiogenic and anti-angiogenic agents. Microvasc Res. 2007;74(2–3):172–183.
- Le Guelte A, Dwyer J, Gavard J. Jumping the barrier: VE-cadherin, VEGF and other angiogenic modifiers in cancer. Biol Cell. 2011;103(12):593–605.
- Muller L, Muller-Haegele S, Mitsuhashi M, et al. Exosomes isolated from plasma of glioma patients enrolled in a vaccination trial reflect antitumor immune activity and might predict survival. Oncoimmunology. 2015;4(6):e1008347.
- Vallabhaneni KC, Penfornis P, Dhule S, et al. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget. 2015;6(7):4953–4967.
- Lo Cicero A, Majkowska I, Nagase H, et al. Microvesicles shed by oligodendroglioma cells and rheumatoid synovial fibroblasts contain aggrecanase activity. Matrix Biol. 2012;31(4):229–233.
- Dvorak HF, Brown LF, Detmar M, et al. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146(5):1029–1039.
- Nagy JA, Benjamin L, Zeng H, et al. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11(2):109–119.
- Fong TA, Shawver LK, Sun L, et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59(1):99–106.
- Faivre S, Delbaldo C, Vera K, et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24(1):25–35.
- Zhang S-D, Leung KL, McCrudden CM, et al. The prognostic significance of combining VEGFA, FLT1 and KDR mRNA expressions in brain tumors. Journal of Cancer. 2015;6(9):812–818.
- Hamerlik P, Lathia JD, Rasmussen R, et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209(3):507–520.
- Bhattacharya R, Ye X-C, Wang R, et al. Intracrine VEGF signaling mediates the activity of prosurvival pathways in human colorectal cancer cells. Cancer Res. 2016;76(10):3014–3024.
- Lichtenberger BM, Tan PK, Niederleithner H, et al. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell. 2010;140(2):268–279.
- Schoeffner DJ, Matheny SL, Akahane T, et al. VEGF contributes to mammary tumor growth in transgenic mice through paracrine and autocrine mechanisms. Lab Invest. 2005;85(5):608–623.
- Bachelder RE, Crago A, Chung J, et al. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res. 2001;61(15):5736–5740.
- Cao Y, Guangqi E, Wang E, et al. VEGF exerts an angiogenesis-independent function in cancer cells to promote their malignant progression. Cancer Res. 2012;72(16):3912–3918.
- Zhou R, Curry JM, Roy LD, et al. A novel association of neuropilin-1 and MUC1 in pancreatic ductal adenocarcinoma: role in induction of VEGF signaling and angiogenesis. Oncogene. 2016;35(43):5608–5618.
- Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13(12):871–882.
- Garcia-Romero N, Carrion-Navarro J, Esteban-Rubio S, et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget. 2016.
- Shao H, Chung J, Balaj L, et al. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat Med. 2012;18(12):1835–1840.
- Setti M, Osti D, Richichi C, et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget. 2015;6(31):31413–31427.
- Giusti I, Delle Monache S, Di Francesco M, et al. From glioblastoma to endothelial cells through extracellular vesicles: messages for angiogenesis. Tumour Biol. 2016;37(9):12743–12753.
- Taraboletti G, D’Ascenzo S, Giusti I, et al. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia. 2006;8(2):96–103.
- Parolini I, Federici C, Raggi C, et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem. 2009;284(49):34211–34222.
- Cossetti C, Iraci N, Mercer TR, et al. Extracellular vesicles from neural stem cells transfer IFN-gamma via Ifngr1 to activate Stat1 signaling in target cells. Mol Cell. 2014;56(2):193–204.
- Feng Q, Zhang C, Lum D, et al. A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumour angiogenesis. Nat Commun. 2017;8:14450.
- Xu C, Wu X, Zhu J. VEGF promotes proliferation of human glioblastoma multiforme stem-like cells through VEGF receptor 2. Scientific World J. 2013;2013:417413.
- Li Q, Qiao G, Ma J, et al. Downregulation of VEGF expression attenuates malignant biological behavior of C6 glioma stem cells. Int J Oncol. 2014;44(5):1581–1588.
- Grun D, Adhikary G, Eckert RL. VEGF-A acts via neuropilin-1 to enhance epidermal cancer stem cell survival and formation of aggressive and highly vascularized tumors. Oncogene. 2016;35(33):4379–4387.
- Liu S, Sun J, Lan Q. Glioblastoma microvesicles promote endothelial cell proliferation through Akt/beta-catenin pathway. Int J Clin Exp Pathol. 2014;7(8):4857–4866.
- Rinkenbaugh AL, Cogswell PC, Calamini B, et al. IKK/NF-kappaB signaling contributes to glioblastoma stem cell maintenance. Oncotarget. 2016.
- Anido J, Saez-Borderias A, Gonzalez-Junca A, et al. TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18(6):655–668.
- Zhang M, Kleber S, Rohrich M, et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011;71(23):7155–7167.
- Podergajs N, Brekka N, Radlwimmer B, et al. Expansive growth of two glioblastoma stem-like cell lines is mediated by bFGF and not by EGF. Radiol Oncol. 2013;47(4):330–337.
- Rafat N, Beck G, Schulte J, et al. Circulating endothelial progenitor cells in malignant gliomas. J Neurosurg. 2010;112(1):43–49.
- Chen X, Fang J, Wang S, et al. A new mosaic pattern in glioma vascularization: exogenous endothelial progenitor cells integrating into the vessels containing tumor-derived endothelial cells. Oncotarget. 2014;5(7):1955–1968.
- Tseng J-C, Chang L-C, Jiang B-Y, et al. Elevated circulating levels of tissue factor-positive microvesicles are associated with distant metastasis in lung cancer. J Cancer Res Clin Oncol. 2014;140(1):61–67.
- Wendler F, Favicchio R, Simon T, et al. Extracellular vesicles swarm the cancer microenvironment: from tumor-stroma communication to drug intervention. Oncogene. 2017;36:877–884.
- Westphal M, Lamszus K. Circulating biomarkers for gliomas. Nat Rev Neurol. 2015;11(10):556–566.
- Azzi S, Treps L, Leclair HM, et al. Desert hedgehog/patch2 axis contributes to vascular permeability and angiogenesis in glioblastoma. Front Pharmacol. 2015;6:281.
- Dwyer J, Azzi S, Leclair HM, et al. The guanine exchange factor SWAP70 mediates vGPCR-induced endothelial plasticity. Cell Commun Signal. 2015;13(1):11.
- Treps L, Gavard J. Assaying the action of secreted semaphorins on vascular permeability. Methods Mol Biol. 2017;1493:417–427.
- Leclair HM, Andre-Gregoire G, Treps L, et al. E3 ubiquitin ligase MARCH3 controls the endothelial barrier. FEBS Lett. 2016;590(20):3660–3668.
- Bowman RL, Wang Q, Carro A, et al. Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017;19(1):139–141.