
ABSTRACT
Membrane proteins are of great research interest, particularly because they are rich in targets for therapeutic application. The suitability of various membrane proteins as targets for therapeutic formulations, such as drugs or antibodies, has been studied in preclinical and clinical studies. For therapeutic application, however, a protein must be expressed and purified in as close to its native conformation as possible. This has proven difficult for membrane proteins, as their native conformation requires the association with an appropriate cellular membrane. One solution to this problem is to use extracellular vesicles as a display platform. Exosomes and microvesicles are membranous extracellular vesicles that are released from most cells. Their membranes may provide a favourable microenvironment for membrane proteins to take on their proper conformation, activity, and membrane distribution; moreover, membrane proteins can cluster into microdomains on the surface of extracellular vesicles following their biogenesis. In this review, we survey the state-of-the-art of extracellular vesicle (exosome and small-sized microvesicle)-based therapeutics, evaluate the current biological understanding of these formulations, and forecast the technical advances that will be needed to continue driving the development of membrane protein therapeutics.
Introduction
Exosomes and microvesicles are lipid bilayer-enclosed nano-sized extracellular vesicles (EVs) that exist in all body fluids; exosomes are formed by almost all cells that contain intracellular multivesicular bodies (MVBs), while microvesicles bud directly from the plasma membrane [Citation1–Citation5]. Exosomes are released into the extracellular space when MVBs fuse with the plasma membrane. Because of their inherently small size and status as a natural cellular product, EVs can be passively delivered everywhere in the body and should cause relatively few undesirable immune reactions (i.e., they are naturally biocompatible). EVs can have intrinsic targeting properties depending on their composition and origin [Citation6], and they may cross biological barriers and deliver their cargoes to recipient cells with virus-like efficiency [Citation7]. Given the problems associated with many of the current nanoparticle delivery systems, they hold great appeal as “nature’s delivery system” for the distribution of native biological molecules and as drug-delivery vehicles [Citation8–Citation11]. EVs have also been applied for the development of cancer vaccines [Citation12–Citation17] and cell-free therapeutics [Citation16–Citation19].
Crucially, EVs have the advantage of being able to display native membrane proteins on their surfaces (). Membrane proteins, which are encoded by approximately 30% of all open-reading frames [Citation20–Citation22], are very important in biology, drug discovery, and vaccination. Because they have important functions in various cellular processes, such as signal transduction, cell-to-cell interaction, membrane trafficking, and cellular metabolism, membrane proteins are targeted by more than 60% of drugs in clinical use today [Citation23,Citation24]. Remarkably, most membrane protein-related human diseases arise from unregulated signal transduction pathways or defective transport systems. A number of clinically relevant antagonists and agonists commonly target membrane proteins that are integral to such signalling pathways. However, the treatment of diseases related to membrane proteins (e.g., transporters) often requires that protein function be restored, which is a more difficult proposition.
Figure 1. Advantages of displaying membrane proteins on the surfaces of EVs. When membrane proteins are embedded naturally or artificially in the phospholipid bilayer of the EV surface, they can have various physiological properties, as indicated.
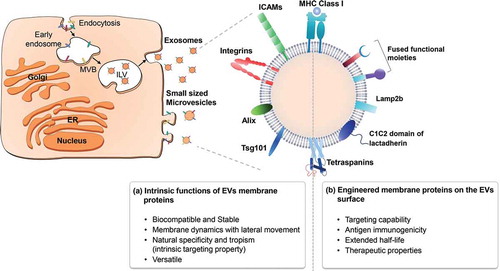
Even though membrane(-associated) proteins are critical for cellular processes and are considered to be important for the development of therapeutics, relatively little is known about their structure–function relationships compared to their water-soluble protein counterparts. Given that membrane proteins are embedded in membranes comprised of several kinds of lipids, a major challenge in restoring membrane protein function is the need to stably maintain the proteins in a lipid-like environment. Detergents, which are often used to purify membrane proteins, are associated with protein instability, show poor compatibility with biophysical/structural studies, and cannot provide the lipid bilayer-based environment required for the stability of membrane proteins [Citation25–Citation27]. Moreover, the methods for reconstituting purified membrane proteins in a lipid vesicle system (e.g., small unilamellar vesicles) are technically difficult [Citation28,Citation29]. These fundamental issues with expressing and purifying membrane proteins have hindered both the acquisition of proper structural information and the application of such proteins for therapeutic purposes [Citation30,Citation31].
Recently, emerging technologies have begun to enable researchers to delve into previously inaccessible areas of membrane protein research. EVs are relatively easy to control and highly versatile in terms of their surface engineering and cargo encapsulation [Citation32,Citation33]. They are expected to have therapeutic effects against various membrane defect-related diseases, and molecules attached to the EV surface have been shown to confer targeting ability, exhibit increased expression levels, offer enhanced solubility, and trigger antigen immunogenicity [Citation34,Citation35]. In this review, we focus primarily on recent progress in the study of membrane protein-harbouring EVs and discuss the hurdles that must be overcome if we hope to further develop EV-based therapeutics. Note that in this review, the term “EV” is used in reference to both small microvesicles and exosomes.
Advantages of EVs over other nanoparticles from the view of harbouring a native membrane protein
Structural features of EVs as a biocompatible membrane scaffold
Membranes are an integral part of life. They define the cellular structures that have evolved to compartmentalize metabolic activity, protect cells and organelles, and provide the means for communication over long and short distances. Membrane dynamics, which regulate cellular processes, depend on the function of membrane proteins. Thus, the dysregulation of the biological activity of such proteins is associated with various negative consequences, including inappropriate cellular responses to exogenous infection, cancers, and genetic diseases [Citation36]. Most membrane protein-related human diseases arise from dysregulation of signal transduction pathways or defective function of membrane proteins. Several antagonists and agonists commonly target membrane proteins to control signalling pathways. However, diseases related to defective or missing membrane proteins often require that protein function be restored, which is a more difficult proposition. In many cases, replacement of faulty proteins by gene therapy may be the solution for membrane defects; however, such strategies do not allow for the time- and dose-controlled expression of the exogenous protein [Citation37–Citation39]. Also, the DNA may integrate near an oncogene, leading to undesired immune responses and potential toxicity [Citation40–Citation42]. These risks and limitations have motivated researchers to explore alternative approaches for modifying cell functions. However, we still lack an appropriate platform to replace or recover the function of membrane proteins.
While many membrane proteins are considered to be high-priority targets for drug design, their therapeutic development has proven mechanistically difficult because many of the biochemical and biophysical approaches used to produce soluble protein drugs are not applicable for insoluble aggregates. One of the major challenges in the development of membrane protein therapeutics is the need to recover sufficient amounts of recombinant proteins from classical expression systems, such as those based in mammalian cells, yeast, and bacteria [Citation43,Citation44]. Despite continuing efforts to develop membrane protein therapeutics, the high content of hydrophobic amino acids in such proteins makes it difficult to generate a sufficient quantity of functional membrane proteins to enable structural studies and functional applications.
For membrane proteins to be used as therapeutics, they must first undergo solubilization. Historically, membrane proteins were solubilized by detergents to form mixed protein–lipid micelles [Citation45]. However, detergents may destabilize membrane proteins, and excess micelles can disrupt certain assay techniques (e.g., by resulting in non-ideal light scattering) [Citation46]. Detergents are also regarded as technical obstacles during the purification of membrane proteins, because they often co-concentrate with the proteins and cause their inactivation or denaturation [Citation47]. Furthermore, most membrane proteins require specific phospholipids to maintain their intrinsic activities, and such an environment cannot be mimicked by detergent-based micelles.
Methods for reconstituting membrane proteins in lipid-based nanoparticles (liposomes) or high-density apolipoprotein particles (nanodiscs) have been offered as a possible solution for these issues [Citation46,Citation48,Citation49]. Such approaches have been used routinely for biochemical, biophysical, and structural studies of membrane proteins. In particular, liposome systems have been used to reconstitute membrane proteins such as ion channels, which require compartmentalization of each side of the bilayer [Citation46]. It is difficult to precisely control the stability, size, and stoichiometry of reconstituted membrane proteins, however, and many techniques are currently being developed to overcome these challenges [Citation46,Citation50]. Moreover, it is relatively laborious to optimize the technologies for individual membrane proteins.
Compared to the above systems, EVs offer a number of potential benefits. They are more biocompatible than the relevant viruses, which have evolved mechanisms to fight or avoid the human immune system to infect the body. The greatest advantage offered by EVs is their ability to provide a perfect membrane environment for membrane proteins, in terms of both dynamics and stability. Membrane proteins can be expressed naturally on the surface of an EV during its biogenesis [Citation51], and thus there is no need for additional solubilizing processes. We now know that transmembrane domains (TMDs) and membrane-anchored domains are not simply passive membrane-spanning anchors for membrane proteins, but also play active roles in oligomerization/clustering and specifically drive protein–protein interactions within the plasma membrane [Citation52]. EVs enable such interactions, and thus are ideal vehicles for the purpose of supporting membrane proteins and enabling their study ().
EV biogenesis and membrane protein display
Especially, exosomes are nano-sized EVs that are secreted from MVBs. MVBs are generated by a two-step invagination process [Citation53–Citation55] that consists of the inward budding of the plasma membrane to create an endosome, and the subsequent invagination of parts of the endosomal membrane towards the endosome lumen to form an intraluminal vesicle (ILV) that selectively contains cytoplasmic molecules. When MVBs fuse with the plasma membrane, ILVs are released to the outside of cells, where they are termed exosomes [Citation56,Citation57]. As one might expect given this two-step invagination theory, an exosome will have the same membrane orientation as the plasma membrane of its producer cell. Exosomes comprise various membrane proteins, such as endosome-associated proteins (Alix, Tsg101, and Rab proteins), tetraspanins (CD63, CD81, CD82, CD53, and CD37), lipid raft-associated proteins (glycosylphosphatidylinositol and flotillin), and lipids that are highly enriched in cholesterol, sphingomyelin, and glycerophospholipids [Citation58–Citation60].
Recent papers have reported that exosomes also include the small-sized microvesicles (around 100 nm) that bud directly from plasma membranes [Citation61]. There is no molecular marker to strictly separate MVB-derived exosomes from small-sized microvesicles that have budded from the plasma membranes. In this review, we collectively refer to exosomes and small-sized microvesicles as “EVs”.
In addition to effectively presenting bioactive membrane proteins, naturally derived EVs may also allow these proteins to form functional domains. For example, both GPI-anchored proteins and membrane-associated ligand proteins were shown to cluster into microdomains on the exosome surface [Citation62]. Also, EGFRvIII proteins expressed on microvesicles were found to transfer oncogenic activity by activating signalling pathways (MAPK and Akt) [Citation63].
Compared to EVs, it is more difficult to add soluble domains to synthetic lipid nanoparticles because peptide ligands affect the stability and material properties of lipid-based nanoparticles and increase the complexity of their synthesis [Citation64,Citation65]. A related approach in which liposomes containing NTA-Ni lipids were used to display histidine-tagged proteins failed due to low dissociation constants associated with unstable immobilization [Citation66,Citation67].
Although challenges remain in terms of controlling the stability and expression level of proteins on EVs, protein ligands can be genetically fused to EV membrane proteins, making it relatively simple to engineer the display of functional domains on an EV. EV membrane can provide a suitable membrane scaffold to support the activity and membrane distribution of the membrane-bound proteins. Bioactive membrane proteins can be naturally located and expressed on EV surfaces, and their functions can be maximized in the EV membrane [Citation51].
Protein cargo loading through fusion with membrane proteins
Since the EV membrane is enriched with membrane proteins that associate with lipid rafts, any transmembrane protein on the surface of an EV can theoretically be used as a fusion partner. In order to deliver protein cargoes, researchers have fused cargo proteins with membrane proteins of EVs, such as tetraspanin and lactadherin [Citation68–Citation70]. Exosomal membrane proteins were also successfully fused with luciferase and fluorescent proteins to enable the tracking of exosomes in animal models and allow their fates to be monitored [Citation68,Citation71,Citation72]. The “XPack lenti-viral vector” developed by System Biosciences is a tool for delivering an encoded protein cargo for expression as a fusion protein that can be packaged into exosomes for secretion [Citation73].
Recently, Yim et al. reported a new system called EXPLORs (exosomes for protein loading via optically reversible protein–protein interactions) [Citation74]. The authors used the photoreceptor, cryptochrome 2 (CRY2), and the CRY-interacting protein (CIBN), which bind under blue light illumination. In the presence of blue light, the transient docking of CRY2-conjugated cargo proteins with CD9-conjugated CIBN was observed in the generated exosomes. When the blue light was removed, the proteins detached and the cargoes were released into the intraluminal space of the exosomes, enabling the cargo proteins to be delivered to target cells.
Therapeutic applications of surface-modified EVs
Functional membrane proteins can be expressed on the EV surface, and thus EV surface display has begun to emerge as a state-of-the-art therapeutic technique. Peptides directed to the surface of EVs can exhibit increases in their in vitro expression level, solubility, and activity [Citation75]. For example, exosomes carrying MHC-peptide complexes can trigger antigen-specific immunogenicity to improve vaccine efficacy [Citation76,Citation77]. Moreover, homing ligands addressed on the EV surface can enable EV targeting and have been used for the targeted delivery of drugs and RNA therapeutics [Citation78–Citation82]. In this chapter, we discuss the therapeutic uses of EVs, which depend on the type of membrane protein(s) found on the EV surface.
Membrane proteins on the EV surface for targeting
EVs are released from various cell lines, and these released EVs may be studied for the specificity of their uptake by different target cells. Such studies revealed that EVs can show natural specificity and tropism, as exemplified by the specific uptake of mantle cell lymphoma-derived exosomes by B lymphocytes [Citation83]. The cellular origin of an EV can guide its surface-expressed proteins, intrinsic targeting properties, and target cell tropism. For example, Hoshino et al. reported that tumour cell-derived exosomes display different integrins depending on the tumour origin, and these proteins mediate tumour metastasis to specific organ sites; exosomes expressing ITGαvβ5 specifically bind to Kupffer cells and thus mediate liver tropism, whereas exosomal ITGα6β4 and ITGα6β1 bind lung-resident fibroblasts and epithelial cells to govern lung tropism [Citation84]. Another study showed that tetraspanin-associated receptors on exosomal membranes can play an important role in target cell selection, as exosomes containing tetraspanin–integrin complexes (span8–integrin α4 complexes) could target CD54-expressing endothelial and pancreatic cells [Citation85].
Despite their tropic properties, exogenous EVs are limited therapeutic use because they are removed by macrophages of the liver and spleen [Citation86,Citation87]. Genetic engineering can be used to engineer the EV surface via the fusion of a moiety of interest (e.g., a ligand/homing peptide) with an EV transmembrane protein to enhance the targeting ability and therapeutic efficiency of the EVs. For example, targeting peptides have been fused with both the Lamp2b membrane glycoprotein and the pDisplay vector, which harbours the PDGFR transmembrane domain that allows exogenous proteins to be displayed on the extracellular side of a membrane [Citation88]. The lactadherin C1C2 domain, which can bind non-covalently to phospholipids that are present in membranes, has also been used as a fusion partner [Citation89] (). Since lactadherin is a membrane-associated protein, not a membrane-spanning protein, the tethering of fusion peptides on the exosomal membrane using the C1C2 domain of lactadherin would be less robust than that obtained using other exosomal transmembrane partners [Citation90].
Table 1. Functional peptides fused with transmembrane domains for EV surface display.
A number of targeting peptides have been used to date, including EBV glycoprotein 350, which targets CD19+ B cells [Citation91]; rabies viral glycoprotein (RVG) peptide fused with Lamp2b, which targets acetylcholine receptors on neurons [Citation78,Citation79,Citation92,Citation93]; Lamp2b-fused iRGD, which targets the ɑv-integrins and neuropilins of tumours [Citation81]; Lamp2b-fused interleukin 3 (IL3), which targets the IL3 receptor on chronic myeloid leukaemia cells [Citation94]; GE11 synthetic peptides inserted into the pDisplay vector, which target epidermal growth factor receptor (EGFR) [Citation95]; and antigens or soluble proteins fused with the C1C2 domain of lactadherin, which target immune cells or blood cells [Citation76,Citation89,Citation96–Citation98] (). In addition, GPI-anchored membrane proteins were used to express ligands such as anti-EGFR nanobodies on the surface of EVs [Citation99].
The published studies have shown that EV targeting may not change the biodistribution profile of unmodified EVs, but it can shorten the time required to reach the therapeutic concentration in targeted tissues and significantly decrease the off-target effects, leading to increased therapeutic efficacy. Recently, modified “iExosomes” were reported for the delivery of small RNAs to KRAS-mutant pancreatic cancer cells. These exosomes harbour CD47 (a fundamental “don’t eat me” signal) on their surface, and were found to evade phagocytosis by circulating monocytes. This strategy could contribute to limiting the clearance of exosomes from circulation and enhance their accumulation in target tumour tissues [Citation100].
EVs as cancer vaccines: delivery of vaccine peptides
Among the EVs, exosomes of intracellular origin often present signalling-related membrane proteins, such as MHC, HSP70, and tetraspanins. In addition, exosome-expressed tumour-specific antigens show strong antigen immunogenicity, and thus could be used to develop cancer vaccines and produce monoclonal antibodies [Citation76,Citation89].
In the late 1990s, it was reported that dendritic cell (DC)-derived exosomes could inhibit the growth of established tumours in mice. Similar to DCs, DC-derived exosomes bearing MHC-I complexed with tumour-derived peptides were found to activate T and/or B cells to induce anti-tumour immune responses [Citation17,Citation77,Citation101,Citation102]. To test the potential for exosomes to be used as cell-free vaccines against cancers, DC-derived exosomes have undergone phase I trial and are currently in phase II trial [Citation12,Citation103–Citation105]. In addition to MHC-bound antigens expressed on the exosomal surface, mature DC-derived exosomes also show surface expression of ICAM-1, which is important for the induction of immune responses, such as T-cell activation [Citation106], and the NKG2D ligand, which is a TNF superfamily ligand that binds directly to NK cells to induce their activation/proliferation and cause an anti-tumour immune response [Citation107,Citation108].
To improve vaccine potency, tumour-specific antigens expressed on the surface of a tumour cell-derived exosome can be used to induce DC priming and T cell activation, leading to anti-tumour immunity [Citation109,Citation110]. For example, the addition of exosome membrane-bound HSP70 has been suggested as a method to maximize the vaccination effect of tumour cell-derived exosomes and induce a stronger immune response [Citation111]. The engineering of exosomes for the surface expression of non-mutated tumour-associated antigen (TAA) has also been used to improve vaccine potency. Exosomes with surface expression of non-mutated TAA fused with C1C2 domain of lactadherin were found to deliver antigens to APC cells and thus trigger increased immunogenicity [Citation76] ().
Table 2. Vaccine peptides expressed on dendritic cell/tumour cell-derived exosomes.
EV-mediated therapeutic membrane protein delivery
Protein therapeutics
For therapeutic purposes, a protein needs to be expressed and purified in as close to its native conformation as possible. However, this can be difficult to achieve for membrane proteins, which must be associated with cellular membranes. Historically, recombinant membrane-associated enzymes could be produced only in truncated forms that were cleaved of the membrane-bound domain [Citation112–Citation114]. To circumvent this problem, researchers turned to using EVs as a display platform that can maintain the native conformation of a protein. EVs can provide a suitable membrane scaffold for membrane-bound proteins to exhibit their proper activity and membrane distribution. Bioactive membrane proteins can be naturally located to and expressed on EV surfaces [Citation51], and membrane-associated proteins known to cluster into microdomains, such as GPI-anchored proteins, are selectively sorted in EV membranes [Citation62,Citation115–Citation117]. Thus, it is expected that the biological activity of membrane (-associated) proteins will be maximized in the membrane environment offered by EVs.
Katsuda et al. reported that human adipose tissue-derived mesenchymal stem cells (ADSCs) secrete exosomes carrying enzymatically active neprilysin (NEP), which is a type 2 integral membrane protein that acts as an Aβ-degrading oligopeptidase enzyme in the brain. The authors showed that NEP-containing ADSC-derived exosomes had enzymatic activity and decreased the intracellular and secreted levels of Aβ in N2a cells [Citation118]. These results suggested that ADSC-derived exosomes could have therapeutic relevance for NEP protein delivery, potentially overcoming the unexpected risks of virus-mediated NEP gene delivery.
Recently, we developed an engineered exosome that harbours GPI-anchored PH20 hyaluronidase on its membrane surface. Our experiments revealed that the generated exosomes could effectively degrade HA both in vitro and in vivo. Remarkably, PH20 proteins were found to be highly enriched in the lipid raft fraction of these exosomes, and their enzymatic activity was 3-fold higher than that of truncated recombinant proteins [Citation119]. Using the GPI-anchor signal peptide of decay-accelerating factor (DAF), which can be selectively sorted to EVs during reticulocyte maturation, Kooijmans et al. further showed that GPI-anchored EGFR nanobodies could effectively bind EGFR-expressing tumour cells under static and flow conditions [Citation99].
Membrane proteins on the EV surface can also deliver signals to target cells. For example, exosomes harbouring membrane-bound TRAIL were demonstrated to deliver proapoptotic signals to cancer cells and inhibit growth in different tumour models [Citation120]. Given that the induction of tumour apoptosis by soluble TRAIL has been limited, it seems that TRAIL-armed exosomes could prove useful as an innovative tool for anti-cancer therapeutics.
EVs can be used as antagonists for delivering immune checkpoint blockade proteins that harbour a transmembrane domain (e.g., PD1, CTLA4, OX40, etc.). For example, Koh et al. developed exosomes harbouring signal-regulatory protein alpha (SIRPα), and found that they exhibited augmented antagonizing activity against CD47–SIRPα interactions [Citation121]. Emerging data suggest that the binding of tumour cell-surface CD47 to SIRPα on phagocytic cells can inhibit their phagocytic function [Citation122]. Because CD47 is expressed on most cancer cell types, it represents a potentially tractable and widely applicable target for therapeutic blockade in cancer patients. Recently, recombinant SIRPα proteins were developed as a competitive antagonist to human CD47; they showed blocking activity in vitro, but the higher-affinity variants did not eliminate tumours in vivo when applied as a mono-treatment [Citation123]. To address these issues, an exosome-based platform was developed to provide high avidity to CD47. SIRPα-exosomes were found to highly antagonize the “don’t eat me” signal of CD47 on cancer cells, thereby enhancing the phagocytosis of tumour cells by bone-marrow derived macrophages in vitro and suppressing tumour growth in xenografts [Citation121]. Remarkably, the therapeutic index of this exosome-mediated CD47 blockade against tumour growth was higher than that of the same dose of monomeric SIRPα. Given that native SIRPα proteins form and work as homodimers when they bind with CD47, the membrane scaffold offered by naturally derived exosomes could facilitate the formation of membrane-spanning SIRPα clusters, augmenting their effect against CD47 [Citation121].
These findings highlight the potential of EV-based platforms, as follows: (i) Naturally derived EVs can efficiently present bioactive membrane proteins, increasing the activity and avidity of particle-bound membrane proteins. (ii) EVs are considered to be well suited to improving the bioavailability and stability of therapeutic proteins.
Membrane protein delivery through fusogenic exosomes
As proteins are widely used to treat many diseases, extensive research has focused on developing techniques for transporting these biomolecules [Citation124–Citation126]. The purpose of a protein drug delivery system is to decrease nonspecific targeting, improve the effect of the drug, and reduce its side effects [Citation127]. Over the years, researchers have developed several techniques to deliver bio-macromolecules into the cytosol and nucleus of living cells [Citation128–Citation130].
A cell uses its membrane to communicate with the outside world; thus, most membrane protein-targeting therapeutics work to alter cellular signalling. In the case of membrane protein defects, replacement of the faulty proteins by gene therapy or protein replacement may be the only solution [Citation131]. However, there are potential risks and limitations associated with gene therapy, and the clinical application of protein therapy has been hampered by the lack of effective vehicles and issues with protein production [Citation132–Citation134]. For example, the viral vectors commonly used in gene therapy can trigger immune responses and may have off-target effects on other cells [Citation135,Citation136]. Moreover, despite a great deal of research effort, we largely lack an effective method to deliver membrane proteins to specific membrane environments. Thus, we need to explore alternative approaches for controlling the functions of membrane proteins and developing therapeutic vehicles for membrane protein delivery.
Engineered vesicles have been used to successfully deliver exogenous proteins into human cells. For example, an EV-based protein-transduction system was used to transport the membrane proteins, TetR transactivator and murine cationic amino acid transporter-1 (mCAT-1, the receptor for the murine leukaemia virus [MLV] envelope protein), as well as other cytoplasmic and nuclear proteins [Citation137]. Recently, Yang et al. developed a fusogenic exosome harbouring the viral fusogen, vascular stomatitis virus (VSV)-G protein, which can fuse with and modify plasma membranes [Citation138]. The presence of VSV-G was expected to facilitate the transfer of therapeutic proteins into the target cell membranes. Indeed, the generated fusogenic exosomes were found to effectively transfer GFP-tagged CD63 or glucose transporter 4 (GLUT4) to plasma membranes both in vitro and in vivo. Moreover, the transferred GLUT4 enhanced the glucose uptake of recipient cells both in vitro and in vivo. These findings highlight the potential of fusogenic exosomes to deliver membrane proteins [Citation138].
The highly efficient delivery of membrane proteins using fusogenic exosomes has appeal for both research and clinical applications. In genetic diseases, fusogenic exosomes could open new therapeutic avenues; in cystic fibrosis, for example, they could be used to transfer normal cystic fibrosis transmembrane conductance regulators into the affected epithelial cell membranes. Beyond such a membrane-editing function, fusogenic exosomes might also be used to modulate intracellular signalling by delivering signalling molecules directly into the cytosol, bypassing the need for endocytic trafficking.
Improving the methods for delivering proteins into living cells is of major interest for both research and medical purposes. Despite the continued evolution of transfer systems, such as the introduction of cell-penetrating peptides, virus-like particles, and proteoliposomes, the efficiencies of these approaches can still be limited by endosomal entrapment [Citation139–Citation141] and the techniques require the laborious purification of recombinant proteins. By contrast, when fusogenic exosomes are used, the loaded therapeutic cargoes can directly enter the cytosol of targeted cells via fusion, which bypasses the potential for becoming entrapped in an endosome. Thus, fusogenic exosomes loaded with soluble cargoes, such as transcription factors or cytosolic proteins, can be used to alter or supplement the biological pathways of recipient cells.
Limitations and factors that should be overcome for the therapeutic use of EVs
Although EVs are able to carry cargoes, we currently lack efficient loading techniques. For example, the electroporation-mediated loading of siRNAs or plasmid DNAs to EVs shows a very low efficiency [Citation142–Citation144]. We also do not yet have a reliable means to quantify the amount of cargo loaded to an EV [Citation79].
The therapeutic application of EVs is still in its infancy, and we need to improve our understanding of EV biogenesis and address issues with their large-scale production and in vivo biodistribution if we hope to develop a successful EV-based therapeutic platform. Moreover, our incomplete understanding of the pathophysiological role of EVs makes it difficult to predict their long-term safety and therapeutic effects.
The heterogeneity of EVs
To apply EVs to therapeutic drug delivery systems, particularly those for the delivery of membrane-associated therapeutic proteins, we require a better understanding of the molecular composition, biogenesis, and heterogeneity of EVs. For example, exosomes derived from the same cells were long thought to be similar in their protein, nucleic acid, and lipid compositions, but recent reports have shown that exosomes originating from the same parental cells can have different molecular compositions [Citation145–Citation147]. This heterogeneity occurs because the biogenesis of MVB and sorting of components to intraluminal vesicles require both the transport (ESCRT)-dependent and -independent pathways [Citation59,Citation60,Citation148].
Although EVs with targeting abilities are expected to apply their payloads to target cells for the induction or inhibition of a desired reaction, we found that not all EVs isolated from a given engineered cell line had the same targeting moiety. Therefore, gaining a better understanding of the heterogeneity and molecular composition of EVs could allow us to determine more suitable subpopulations for certain EV-based therapeutics (i.e., by identifying subpopulations that can exert particular effects without unwanted side-effects). The vesicle doses have varied across the existing studies, ranging from 1 to 500 μg per in vivo injection [Citation12,Citation104,Citation105,Citation149–Citation151], further emphasizing that the heterogeneity of EVs needs to be characterized to avoid the induction of adverse effects in patients. Although some methods have been developed to detect EV heterogeneity, their detection sensitivities and specificities must be improved to enable researchers to precisely characterize each subpopulation and the compositions of individual vesicles.
In vivo biodistribution of systemically administrated EVs
Exosomes and small-sized microvesicles are spherical 30–100 nm vesicles, and are thus small enough to passively diffuse into tumours via the enhanced permeability and retention (EPR) effect. Although the EPR effect has not yet been fully proven in humans, nanoparticles (including small EVs) can escape from the vasculature through leaky endothelial tissue via the EPR effect, and this constitutes an important mechanism for size-dependent “passive targeting” [Citation152,Citation153]. However, according to previous studies of their biodistribution, less than 5% of systemically administered nanoparticles reach tumour tissues [Citation154,Citation155]. Remarkably, most exosomes and small-sized microvesicles distribute to the liver, and less than 2% of the injected vesicles were found to accumulate in tumour tissues after systemic administration [Citation156]. A great deal remains unknown regarding the in vivo properties of EVs, including their tissue distribution, half-life, blood levels, and urine clearance. All of these parameters will be important to defining the therapeutic effectiveness and potential toxicity of EVs.
Recently, Wiklander et al. studied the biodistribution of systemically delivered EVs in mice, comparing them according to the delivery route (i.v., i.p., and s.c.), cell source (muscle cell-derived C2C12 cells, melanoma cell-derived B16F10 cells, and primary immature bone marrow-derived DCs), and injection dose [Citation157]. Indeed, the cellular origin, administration route, and injected dose of EVs all influenced their biodistribution pattern, highlighting the importance of considering these and other factors during the design of in vivo studies of exosome-based therapeutics.
Most of the clinical trials of EV-mediated therapies have focused on visualizing their treatment effects in the human body. Several EV-labelling methods involving the use of lipid fluorescence dyes or luciferase proteins have been applied to monitor the in vivo biodistribution of EVs in animal models; however, these methods act at fairly shallow depths, limiting their usefulness for clinical applications [Citation158,Citation159]. In addition, the application of indirect labelling methods that involve gene transduction into cells causes ethical issues for clinical applications [Citation160]. Nuclear imaging using direct labelling, which is safe and has no depth limitation, has recently emerged as a more useful option for clinical applications [Citation161,Citation162]. Radioisotopes such as radioiodine, 111In-oxine and 99mTc-hexamethylpropyleneamineoxime (HMPAO) can be used for clinical translation studies [Citation163–Citation165], although an appropriate and specific labelling strategy for EVs should be determined for each experimental setting. To measure the in vivo biodistribution of EVs, we need to further develop imaging techniques that can overcome the current technical issues.
A pharmacokinetic analysis of blood concentration–time profiles revealed that B16-BL6 exosomes disappeared very quickly from the blood circulation, with a half-life of approximately 2 min [Citation72]. In contrast, other studies have found that EVs have many of the features desired for an ideal drug delivery system, including a long circulating half-life, the intrinsic ability to target tissues, good biocompatibility, and minimal or no inherent toxicity issues [Citation6,Citation8,Citation166]. Given these conflicting results, it remains unclear whether EVs will prove useful as universal biologics-transferring agents. For the future therapeutic applications of EVs in general, we urgently need the technological advances that will enable us to track them in vivo.
Large-scale production of EV-based therapeutics
Unlike shed large-sized microvesicles and apoptotic bodies, exosomes and small-sized microvesicles can be purified from biological fluids and cell culture-conditioned media using various techniques, including ultracentrifugation (UC), density-based separation, size-dependent methods such as ultrafiltration and size-exclusion chromatography, precipitation using polymers like ExoQuickTM, and immune-affinity capture methods [Citation167,Citation168]. To enable the therapeutic use of EVs, however, we urgently need more efficient and reliable methods for isolating large amounts of highly pure EVs.
The traditional and widely accepted method for isolating exosomes is differential UC. A low-speed centrifugation step (300 g for 10 min) is first used to eliminate dead cells and large apoptotic bodies, and a subsequent higher-speed centrifugation step (2000–10,000 g) is applied to remove larger microvesicles and debris. A final ultracentrifugation at over 100,000 g is then used to collect the precipitated exosomes and small-sized microvesicles [Citation169]. This protocol has been considered a gold standard because it yields UC-separated small EVs of high purity. However, the run time of the technique exceeds 6 h, the physicochemical functions of the vesicles can be damaged by the high relative centrifugal field (RCF), and the production yield is low [Citation170]. Thus, it is currently impractical to use UC-isolated vesicles in therapeutic studies.
Sucrose gradient separation, which resolves vesicles based on their flotation densities (exosomes, 1.08–1.22 g/ml), may yield high-purity exosomes [Citation171]. Recently, an iodixanol (OptiprepTM) gradient was used to separate the AChE-containing exosome fraction (8.4–12%) from viruses (at 15.6%) [Citation172].
EVs have also been isolated by size-based methods (filtration and size-exclusion chromatography) that take advantage of the size differences between small vesicles and other EVs. Filtration can rapidly separate small EVs of moderate purity using simple devices, but such techniques can run into problems with contamination of other proteins or vesicles, EV aggregation, and the trapping of vesicles in the filter pores [Citation167]. Size-exclusion chromatography has shown remarkable reproducibility in yielding moderate quantities of high-purity EVs [Citation173]. In contrast to the filtration method, however, the process is lengthy and not amenable to large-scale expansion [Citation174].
Recently, a readily scalable tangential flow filtration (TFF) system was developed for the high-enrichment isolation of EVs; however, the potential for contamination with large-sized microvesicles of similar size means that additional steps are needed, including (density gradient) UC [Citation175].
Several water-excluding polymers (e.g., ExoQuickTM) have been developed to providing a quick and easy way to isolate exosomes [Citation176]. However, little has been reported regarding the purity, quality, and clinical applicability of exosomes obtained using ExoQuickTM [Citation53]. Although System Bioscience has developed a product called ExoQuick-CGTM for the quick and easy production of clinical-grade (cGMP) exosomes, further confirmation is needed before this procedure applied for clinical use [Citation177].
Clearly, we need additional strategies for producing EVs that meet the GMP standards. Lamparski et al. produced exosomes for clinical trial I/II by properly combining filtration, UC, and sucrose-gradient methods. They also developed quality-control assays for the quantification and phenotypic characterization of cGMP exosomes [Citation178]. The ExoTESTTM kit, which uses double-sandwich ELISA, was also developed for quantitative and qualitative analysis of exosomes [Citation179]. However, the isolation of EVs is still a major technical challenge. In the future, we must develop an optimal technique for the large-scale production of clinical-grade EVs.
Conclusions and outlook
EVs function as natural carriers of biomacromolecules, which makes them attractive candidates for the therapeutic delivery of various synthetic and biological molecules. They have been shown to have advantages as delivery systems, owing to their nano-sized particles, low immunogenicity, lack of cytotoxicity, and long-term safety [Citation7,Citation82,Citation180–Citation183]. Moreover, many peptides capable of targeting specific tissues have been identified, offering an opportunity to improve EV-based strategies by reducing their unwanted homing/accumulation in the liver before reaching target sites [Citation10,Citation82]. In this review, we describe the state-of-the-art strategies for the therapeutic applications of engineered EVs. As one would expect from their biogenesis process, EVs provide an excellent biological scaffold for the display of membrane proteins [Citation51] that can be used for tissue targeting, as tumour vaccines, and as signalling molecules [Citation184]. EVs have intrinsic functional properties that can be harnessed by engineering them to express exogenous therapeutic proteins. EVs can thus be proposed as a bio-inspired alternative for membrane protein therapeutics.
We also highlight key translational challenges and opportunities in this rapidly growing field. Before engineered EVs can become a therapeutic reality, their components need to be characterized in the contexts of EV heterogeneity, the dose of EVs required for a given human patient, and immune reactions [Citation185–Citation187]. In addition, we need to develop and standardize protocols for the large-scale production of very pure high-quality EVs, as well for efficiently loading them with therapeutic payloads [Citation53,Citation188].
Despite these challenges, EVs clearly represent excellent candidates as therapeutic agents in vivo. Uncovering the pathophysiological roles of EVs may provide us with the tools we need to further improve EV-based therapeutics, and particularly their use for membrane protein delivery. Based on our current knowledge of EV technology, we expect to be able to explore these future directions moving towards clinical applications.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Keller S, Sanderson MP, Stoeck A, et al. Exosomes: from biogenesis and secretion to biological function. Immunol Lett. 2006;107(2):1–15.
- Madison MN, Roller RJ, Okeoma CM. Human semen contains exosomes with potent anti-HIV-1 activity. Retrovirology. 2014;11(1):102.
- Vella LJ, Greenwood DL, Cappai R, et al. Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet Immunol Immunopathol. 2008;124(3–4):385–393.
- Ogawa Y, Miura Y, Harazono A, et al. Proteomic analysis of two types of exosomes in human whole saliva. Biol Pharm Bull. 2011;34(1):13–23.
- Choi DS. Urinary extracellular vesicles for biomarker source to monitor polycystic kidney disease. Proteomics Clin Appl. 2015;9(5–6):447–448.
- Turturici G, Tinnirello R, Sconzo G, et al. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am J Physiol Cell Physiol. 2014;306(7):C621–33.
- Ha D, Yang N, Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B. 2016;6(4):287–296.
- Pashoutan Sarvar D, Shamsasenjan K, Akbarzadehlaleh P. Mesenchymal stem cell-derived exosomes: new opportunity in cell-free therapy. Adv Pharm Bull. 2016;6(3):293–299.
- Haney MJ, Klyachko NL, Zhao Y, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Control Release. 2015;207:18–30.
- Johnsen KB, Gudbergsson JM, Skov MN, et al. A comprehensive overview of exosomes as drug delivery vehicles - endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta. 2014;1846(1):75–87.
- Yang T, Martin P, Fogarty B, et al. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res. 2015;32(6):2003–2014.
- Escudier B, Dorval T, Chaput N, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. 2005;3(1):10.
- Tan A, De La Peña H, Seifalian AM. The application of exosomes as a nanoscale cancer vaccine. Int J Nanomedicine. 2010;5:889–900.
- Le Pecq JB. Dexosomes as a therapeutic cancer vaccine: from bench to bedside. Blood Cells Mol Dis. 2005;35(2):129–135.
- Altieri SL, Khan AN, Tomasi TB. Exosomes from plasmacytoma cells as a tumor vaccine. J Immunother. 2004;27(4):282–288.
- Taïeb J, Chaput N, Zitvogel L. Dendritic cell-derived exosomes as cell-free peptide-based vaccines. Crit Rev Immunol. 2005;25(3):215–223.
- Zitvogel L, Regnault A, Lozier A, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell derived exosomes. Nat Med. 1998;4(5):594–600.
- Phinney DG, Pittenger MF. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells. 2017;35(4):851–858.
- Kishore R, Khan M. More than tiny sacks: stem cell exosomes as cell-free modality for cardiac repair. Circ Res. 2016;118(2):330–343.
- Miles AJ, Wallace BA. Circular dichroism spectroscopy of membrane proteins. Chem Soc Rev. 2016;45(18):4859–4872.
- Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7(4):1029–1038.
- Krogh A, Larsson B, von Heijne G, et al. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580.
- Yildirim MA, Goh KI, Cusick ME, et al. Drug-target network. Nat Biotechnol. 2007;25(10):1119–1126.
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993–996.
- Frauenfeld J, Loving R, Armache JP, et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat Methods. 2016;13(4):345–351.
- Tate CG. Practical considerations of membrane protein instability during purification and crystallisation. Methods Mol Biol. 2010;601:187–203.
- Breyton C, Pucci B, Popot JL. Amphipols and fluorinated surfactants: two alternatives to detergents for studying membrane proteins in vitro. Methods Mol Biol. 2010;601:219–245.
- Serebryany E, Zhu GA, Yan EC. Artificial membrane-like environments for in vitro studies of purified G-protein coupled receptors. Biochim Biophys Acta. 2012;1818(2):225–233.
- Althoff T, Davies KM, Schulze S, et al. GRecon: a method for the lipid reconstitution of membrane proteins. Angew Chem Int Ed Engl. 2012;51(33):8343–8347.
- Lluis MW, Godfroy JI 3rd, Yin H. Protein engineering methods applied to membrane protein targets. Protein Eng Des Sel. 2013;26(2):91–100.
- Carpenter EP, Beis K, Cameron AD, et al. Overcoming the challenges of membrane protein crystallography. Curr Opin Struct Biol. 2008;18(5):581–586.
- Marcus ME, Leonard JN. FedExosomes: engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals (Basel). 2013;6(5):659–680.
- Ren J, He W, Zheng L, et al. From structures to functions: insights into exosomes as promising drug delivery vehicles. Biomater Sci. 2016;4(6):910–921.
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593.
- Shen B, Wu N, Yang JM, et al. Protein targeting to exosomes/microvesicles by plasma membrane anchors. J Biol Chem. 2011;286(16):14383–14395.
- Aperia A. Membrane transport proteins in health and disease. J Intern Med. 2007;261(1):2–4.
- Pfeifer A, Verma IM. Gene therapy: promises and problems. Annu Rev Genomics Hum Genet. 2001;2:177–211.
- Verma IM, Somia N. Gene therapy – promises, problems and prospects. Nature. 1997;389(6648):239–242.
- Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998;16(9):867–870.
- Orive G, Hernandez RM, Rodriguez Gascon A, et al. Drug delivery in biotechnology: present and future. Curr Opin Biotechnol. 2003;14(6):659–664.
- Sadelain M. Insertional oncogenesis in gene therapy: how much of a risk? Gene Ther. 2004;11(7):569–573.
- Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–3142.
- Briand L, Marcion G, Kriznik A, et al. A self-inducible heterologous protein expression system in Escherichia coli. Sci Rep. 2016;6:33037.
- Berlec A, Strukelj B. Current state and recent advances in biopharmaceutical production in Escherichia coli, yeasts and mammalian cells. J Ind Microbiol Biotechnol. 2013;40(3–4):257–274.
- Garavito RM, Ferguson-Miller S. Detergents as tools in membrane biochemistry. J Biol Chem. 2001;276(35):32403–32406.
- Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010;584(9):1721–1727.
- Bowie JU. Stabilizing membrane proteins. Curr Opin Struct Biol. 2001;11(4):397–402.
- Denisov IG, Sligar SG. Nanodiscs for structural and functional studies of membrane proteins. Nat Struct Mol Biol. 2016;23(6):481–486.
- Cappuccio JA, Blanchette CD, Sulchek TA, et al. Cell-free co-expression of functional membrane proteins and apolipoprotein, forming soluble nanolipoprotein particles. Mol Cell Proteomics. 2008;7(11):2246–2253.
- Akbarzadeh A, Rezaei-Sadabady R, Davaran S, et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):102.
- Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579.
- Yin H, Flynn AD. Drugging membrane protein interactions. Annu Rev Biomed Eng. 2016;18:51–76.
- Vlassov AV, Magdaleno S, Setterquist R, et al. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. 2012;1820(7):940–948.
- Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010;73(10):1907–1920.
- Schorey JS, Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic. 2008;9(6):871–881.
- Sahoo S, Losordo DW. Exosomes and cardiac repair after myocardial infarction. Circ Res. 2014;114(2):333–344.
- Denzer K, Kleijmeer MJ, Heijnen HF, et al. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000;113(Pt 19):3365–3374.
- Klein-Scory S, Tehrani MM, Eilert-Micus C, et al. New insights in the composition of extracellular vesicles from pancreatic cancer cells: implications for biomarkers and functions. Proteome Sci. 2014;12(1):50.
- Villarroya-Beltri C, Baixauli F, Gutierrez-Vazquez C, et al. Sorting it out: regulation of exosome loading. Semin Cancer Biol. 2014;28:3–13.
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383.
- Booth AM, Fang Y, Fallon JK, et al. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol. 2006;172(6):923–935.
- de Gassart A, Geminard C, Fevrier B, et al. Lipid raft-associated protein sorting in exosomes. Blood. 2003;102(13):4336–4344.
- Al-Nedawi K, Meehan B, Micallef J, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10(5):619–624.
- Kraft JC, Freeling JP, Wang Z, et al. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J Pharm Sci. 2014;103(1):29–52.
- Gallarate M, Battaglia L, Peira E, et al. Peptide-loaded solid lipid nanoparticles prepared through coacervation technique. Int J Chem Eng. 2011;2011:6.
- Knecht S, Ricklin D, Eberle AN, et al. Oligohis-tags: mechanisms of binding to Ni2+-NTA surfaces. J Mol Recognit. 2009;22(4):270–279.
- Colletier JP, Chaize B, Winterhalter M, et al. Protein encapsulation in liposomes: efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol. 2002;2:9.
- Stickney Z, Losacco J, McDevitt S, et al. Development of exosome surface display technology in living human cells. Biochem Biophys Res Commun. 2016;472(1):53–59.
- Kanuma T, Yamamoto T, Kobiyama K, et al. CD63-mediated antigen delivery into extracellular vesicles via DNA vaccination results in robust CD8+ T cell responses. J Immunol. 2017;198(12):4707–4715.
- Morishita M, Takahashi Y, Matsumoto A, et al. Exosome-based tumor antigens-adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA. Biomaterials. 2016;111:55–65.
- Meyer C, Losacco J, Stickney Z, et al. Pseudotyping exosomes for enhanced protein delivery in mammalian cells. Int J Nanomedicine. 2017;12:3153–3170.
- Takahashi Y, Nishikawa M, Shinotsuka H, et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol. 2013;165(2):77–84.
- SBI. XPack™ exosome protein engineering. Available from: https://www.systembio.com/xpack-exosomes/overview.
- Yim N, Ryu SW, Choi K, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun. 2016;7:12277.
- Estelles A, Sperinde J, Roulon T, et al. Exosome nanovesicles displaying G protein-coupled receptors for drug discovery. Int J Nanomedicine. 2007;2(4):751–760.
- Hartman ZC, Wei J, Glass OK, et al. Increasing vaccine potency through exosome antigen targeting. Vaccine. 2011;29(50):9361–9367.
- Andre F, Chaput N, Schartz NE, et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol. 2004;172(4):2126–2136.
- Alvarez-Erviti L, Seow Y, Yin H, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29(4):341–345.
- El-Andaloussi S, Lee Y, Lakhal-Littleton S, et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012;7(12):2112–2126.
- Ohno S, Takanashi M, Sudo K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21(1):185–191.
- Tian Y, Li S, Song J, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–2390.
- El Andaloussi S, Lakhal S, Mager I, et al. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev. 2013;65(3):391–397.
- Hazan-Halevy I, Rosenblum D, Weinstein S, et al. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015;364(1):59–69.
- Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–335.
- Rana S, Yue S, Stadel D, et al. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol. 2012;44(9):1574–1584.
- Imai T, Takahashi Y, Nishikawa M, et al. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J Extracell Vesicles. 2015;4:26238.
- Feng D, Zhao WL, Ye YY, et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11(5):675–687.
- Xitong D, Xiaorong Z. Targeted therapeutic delivery using engineered exosomes and its applications in cardiovascular diseases. Gene. 2016;575(2 Pt 2):377–384.
- Delcayre A, Estelles A, Sperinde J, et al. Exosome Display technology: applications to the development of new diagnostics and therapeutics. Blood Cells Mol Dis. 2005;35(2):158–168.
- Hung ME, Leonard JN. Stabilization of exosome-targeting peptides via engineered glycosylation. J Biol Chem. 2015;290(13):8166–8172.
- Ruiss R, Jochum S, Mocikat R, et al. EBV-gp350 confers B-cell tropism to tailored exosomes and is a neo-antigen in normal and malignant B cells–a new option for the treatment of B-CLL. PLoS One. 2011;6(10):e25294.
- Liu Y, Li D, Liu Z, et al. Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci Rep. 2015;5:17543.
- Yang J, Zhang X, Chen X, et al. Exosome mediated delivery of miR-124 promotes neurogenesis after ischemia. Mol Ther Nucleic Acids. 2017;7:278–287.
- Bellavia D, Raimondo S, Calabrese G, et al. Interleukin 3- receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth. Theranostics. 2017;7(5):1333–1345.
- Ohno SI, Takanashi M, Sudo K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21(1):185–191.
- Rountree RB, Mandl SJ, Nachtwey JM, et al. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer Res. 2011;71(15):5235–5244.
- Zeelenberg IS, Ostrowski M, Krumeich S, et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68(4):1228–1235.
- Sedlik C, Vigneron J, Torrieri-Dramard L, et al. Different immunogenicity but similar antitumor efficacy of two DNA vaccines coding for an antigen secreted in different membrane vesicle-associated forms. J Extracell Vesicles. 2014;3:24646.
- Kooijmans SA, Aleza CG, Roffler SR, et al. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J Extracell Vesicles. 2016;5:31053.
- Kamerkar S, LeBleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498–503.
- Romagnoli GG, Zelante BB, Toniolo PA, et al. Dendritic cell-derived exosomes may be a tool for cancer immunotherapy by converting tumor cells into immunogenic targets. Front Immunol. 2014;5:692.
- Näslund TI, Gehrmann U, Qazi KR, et al. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J Immunol. 2013;190(6):2712–2719.
- Besse B, Charrier M, Lapierre V, et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology. 2016;5(4):e1071008.
- Morse MA, Garst J, Osada T, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3(1):9.
- Dai S, Wei D, Wu Z, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther. 2008;16(4):782–790.
- Segura E, Nicco C, Lombard B, et al. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood. 2005;106(1):216–223.
- Munich S, Sobo-Vujanovic A, Buchser WJ, et al. Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. Oncoimmunology. 2012;1(7):1074–1083.
- Viaud S, Terme M, Flament C, et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One. 2009;4(3):e4942.
- Wolfers J, Lozier A, Raposo G, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7(3):297–303.
- André F, Schartz NE, Chaput N, et al. Tumor-derived exosomes: a new source of tumor rejection antigens. Vaccine. 2002;20(Suppl 4):A28–31.
- Xie Y, Bai O, Zhang H, et al. Membrane-bound HSP70-engineered myeloma cell-derived exosomes stimulate more efficient CD8(+) CTL- and NK-mediated antitumour immunity than exosomes released from heat-shocked tumour cells expressing cytoplasmic HSP70. J Cell Mol Med. 2010;14(11):2655–2666.
- Rigi G, Beyranvand P, Ghaedmohammadi S, et al. Comparison of the extracellular full-length and truncated recombinant protein A production in Escherichia coli BL21 (DE3). J Paramed Sci. 2015;6:3.
- Sørensen HP, Sperling-Petersen HU, Mortensen KK. Production of recombinant thermostable proteins expressed in Escherichia coli: completion of protein synthesis is the bottleneck. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;786(1–2):207–214.
- Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin Drug Deliv. 2007;4(4):427–440.
- Ashiru O, López-Cobo S, Fernández-Messina L, et al. A GPI anchor explains the unique biological features of the common NKG2D-ligand allele MICA*008. Biochem J. 2013;454(2):295–302.
- Valapala M, Vishwanatha JK. Lipid raft endocytosis and exosomal transport facilitate extracellular trafficking of annexin A2. J Biol Chem. 2011;286(35):30911–30925.
- Théry C, Regnault A, Garin J, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999;147(3):599–610.
- Katsuda T, Tsuchiya R, Kosaka N, et al. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci Rep. 2013;3:1197.
- Hong Y, Nam G-H, Koh E, et al. Exosome as a vehicle for delivery of membrane protein therapeutics, PH20, for enhanced tumor penetration and antitumor efficacy. Adv Funct Mater. 2017;28(5):170374.
- Rivoltini L, Chiodoni C, Squarcina P, et al. TNF-related apoptosis-inducing ligand (TRAIL)-armed exosomes deliver proapoptotic signals to tumor site. Clin Cancer Res. 2016;22(14):3499–3512.
- Koh E, Lee EJ, Nam GH, et al. Exosome-SIRPalpha, a CD47 blockade increases cancer cell phagocytosis. Biomaterials. 2017;121:121–129.
- Willingham SB, Volkmer JP, Gentles AJ, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–6667.
- Weiskopf K, Ring AM, Ho CCM, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341(6141):88–91.
- Chen Y, Zhang M, Jin H, et al. Intein-mediated site-specific synthesis of tumor-targeting protein delivery system: turning PEG dilemma into prodrug-like feature. Biomaterials. 2017;116:57–68.
- Lu Y, Sun W, Gu Z. Stimuli-responsive nanomaterials for therapeutic protein delivery. J Control Release. 2014;194:1–19.
- Yun Y, Cho YW, Park K. Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev. 2013;65(6):822–832.
- Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56(11):1649–1659.
- Capecchi MR. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell. 1980;22(2 Pt 2):479–488.
- Kaczmarczyk SJ, Sitaraman K, Young HA, et al. Protein delivery using engineered virus-like particles. Proc Natl Acad Sci U S A. 2011;108(41):16998–17003.
- Melikov K, Chernomordik LV. Arginine-rich cell penetrating peptides: from endosomal uptake to nuclear delivery. Cell Mol Life Sci. 2005;62(23):2739–2749.
- Guan X, Goddard MA, Mack DL, et al. Gene therapy in monogenic congenital myopathies. Methods. 2016;99:91–98.
- de Planque MR, de Planque MR, Mendes GP, et al. Controlled delivery of membrane proteins to artificial lipid bilayers by nystatin-ergosterol modulated vesicle fusion. IEE Proc Nanobiotechnol. 2006;153(2):21–30.
- Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. J Pharm Sci. 2010;99(6):2557–2575.
- Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014;5:172.
- Sack BK, Herzog RW. Evading the immune response upon in vivo gene therapy with viral vectors. Curr Opin Mol Ther. 2009;11(5):493–503.
- Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–358.
- Mangeot PE, Dollet S, Girard M, et al. Protein transfer into human cells by VSV-G-induced nanovesicles. Mol Ther. 2011;19(9):1656–1666.
- Yang Y, Hong Y, Nam GH, et al. Virus-mimetic fusogenic exosomes for direct delivery of integral membrane proteins to target cell membranes. Adv Mater. 2017;29:13.
- El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. Aaps J. 2009;11(1):13–22.
- Erazo-Oliveras A, Muthukrishnan N, Baker R, et al. Improving the endosomal escape of cell-penetrating peptides and their cargos: strategies and challenges. Pharmaceuticals (Basel). 2012;5(11):1177–1209.
- Fu A, Tang R, Hardie J, et al. Promises and pitfalls of intracellular delivery of proteins. Bioconjug Chem. 2014;25(9):1602–1608.
- Shahabipour F, Banach M, Sahebkar A. Exosomes as nanocarriers for siRNA delivery: paradigms and challenges. Arch Med Sci. 2016;12(6):1324–1326.
- Lamichhane TN, Raiker RS, Jay SM. Exogenous DNA loading into extracellular vesicles via electroporation is size-dependent and enables limited gene delivery. Mol Pharm. 2015;12(10):3650–3657.
- Kooijmans SA, Stremersch S, Braeckmans K, et al. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release. 2013;172(1):229–238.
- Ferguson SW, Nguyen J. Exosomes as therapeutics: the implications of molecular composition and exosomal heterogeneity. J Control Release. 2016;228:179–190.
- Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659.
- Zhang J, Li S, Li L, et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinf. 2015;13(1):17–24.
- Willms E, Johansson HJ, Mäger I, et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci Rep. 2016;6:22519.
- Xiu F, Cai Z, Yang Y, et al. Surface anchorage of superantigen SEA promotes induction of specific antitumor immune response by tumor-derived exosomes. J Mol Med (Berl). 2007;85(5):511–521.
- Yang Y, Xiu F, Cai Z, et al. Increased induction of antitumor response by exosomes derived from interleukin-2 gene-modified tumor cells. J Cancer Res Clin Oncol. 2007;133(6):389–399.
- Chaput N, Schartz NE, Andre F, et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol. 2004;172(4):2137–2146.
- Shi J, Kantoff PW, Wooster R, et al. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37.
- Tan S, Wu T, Zhang D, et al. Cell or cell membrane-based drug delivery systems. Theranostics. 2015;5(8):863–881.
- Park K. Facing the truth about nanotechnology in drug delivery. ACS Nano. 2013;7(9):7442–7447.
- Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1(5):16014.
- Smyth T, Kullberg M, Malik N, et al. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015;199:145–155.
- Wiklander OP, Nordin JZ, O’Loughlin A, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316.
- Leblond F, Davis SC, Valdés PA, et al. Pre-clinical whole-body fluorescence imaging: review of instruments, methods and applications. J Photochem Photobiol B. 2010;98(1):77–94.
- Ntziachristos V, Bremer C, Weissleder R. Fluorescence imaging with near-infrared light: new technological advances that enable in vivo molecular imaging. Eur Radiol. 2003;13(1):195–208.
- Luker KE, Luker GD. Applications of bioluminescence imaging to antiviral research and therapy: multiple luciferase enzymes and quantitation. Antiviral Res. 2008;78(3):179–187.
- Miller CG, Krasnow J, Schwartz LH. Medical imaging in clinical trials. London: Springer; 2014.
- Gangadaran P, Hong CM, Ahn BC. Current perspectives on in vivo noninvasive tracking of extracellular vesicles with molecular imaging. Biomed Res Int. 2017;2017:9158319.
- Bulte JW. In vivo MRI cell tracking: clinical studies. AJR Am J Roentgenol. 2009;193(2):314–325.
- Correa PL, Mesquita CT, Felix RM, et al. Assessment of intra-arterial injected autologous bone marrow mononuclear cell distribution by radioactive labeling in acute ischemic stroke. Clin Nucl Med. 2007;32(11):839–841.
- Giaffer MH. Labelled leucocyte scintigraphy in inflammatory bowel disease: clinical applications. Gut. 1996;38(1):1–5.
- Aryani A, Denecke B. Exosomes as a nanodelivery system: a key to the future of neuromedicine? Mol Neurobiol. 2016;53(2):818–834.
- Li P, Kaslan M, Lee SH, et al. Progress in exosome isolation techniques. Theranostics. 2017;7(3):789–804.
- Momen-Heravi F, Balaj L, Alian S, et al. Current methods for the isolation of extracellular vesicles. Biol Chem. 2013;394(10):1253–1262.
- Thery C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006:3.22 1-3.22. 29.
- Helwa I, Cai J, Drewry MD, et al. A comparative study of serum exosome isolation using differential ultracentrifugation and three commercial reagents. PLoS One. 2017;12(1):e0170628.
- Raposo G, Nijman HW, Stoorvogel W, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183(3):1161–1172.
- Cantin R, Diou J, Belanger D, et al. Discrimination between exosomes and HIV-1: purification of both vesicles from cell-free supernatants. J Immunol Methods. 2008;338(1–2):21–30.
- Nordin JZ, Lee Y, Vader P, et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine. 2015;11(4):879–883.
- Zeringer E, Barta T, Li M, et al. Strategies for isolation of exosomes. Cold Spring Harb Protoc. 2015;2015(4):319–323.
- Safdar A, Saleem A, Tarnopolsky MA. The potential of endurance exercise-derived exosomes to treat metabolic diseases. Nat Rev Endocrinol. 2016;12(9):504–517.
- Taylor DD, Zacharias W, Gercel-Taylor C. Exosome isolation for proteomic analyses and RNA profiling. Methods Mol Biol. 2011;728:235–246.
- Antes TJ, Kwei K, Wu F. Methods for microvesicle isolation and selective removal. Unites States patent US 9,005,888 2015 Apr 14.
- Lamparski HG, Metha-Damani A, Yao JY, et al. Production and characterization of clinical grade exosomes derived from dendritic cells. J Immunol Methods. 2002;270(2):211–226.
- Logozzi M, De Milito A, Lugini L, et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS One. 2009;4(4):e5219.
- Yeo RW, Lai RC, Zhang B, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 2013;65(3):336–341.
- Tan A, Rajadas J, Seifalian AM. Exosomes as nano-theranostic delivery platforms for gene therapy. Adv Drug Deliv Rev. 2013;65(3):357–367.
- Lakhal S, Wood MJ. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays. 2011;33(10):737–741.
- Momen-Heravi F, Bala S, Bukong T, et al. Exosome-mediated delivery of functionally active miRNA-155 inhibitor to macrophages. Nanomedicine. 2014;10(7):1517–1527.
- Sun D, Zhuang X, Zhang S, et al. Exosomes are endogenous nanoparticles that can deliver biological information between cells. Adv Drug Deliv Rev. 2013;65(3):342–347.
- van Dommelen SM, Vader P, Lakhal S, et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release. 2012;161(2):635–644.
- Hao S, Moyana T, Xiang J. Review: cancer immunotherapy by exosome-based vaccines. Cancer Biother Radiopharm. 2007;22(5):692–703.
- Johnstone RM. Exosomes biological significance: A concise review. Blood Cells Mol Dis. 2006;36(2):315–321.
- Lasser C. Exosomes in diagnostic and therapeutic applications: biomarker, vaccine and RNA interference delivery vehicle. Expert Opin Biol Ther. 2015;15(1):103–117.