
ABSTRACT
The lack of approved therapies for hepatic fibrosis seriously limits medical management of patients with chronic liver disease. Since extracellular vesicles (EVs) function as conduits for intercellular molecular transfer, we investigated if EVs from healthy individuals have anti-fibrotic properties. Hepatic fibrogenesis or fibrosis in carbon tetrachloride (CCl4)- or thioacetic acid-induced liver injury models in male or female mice were suppressed by serum EVs from normal mice (EVN) but not from fibrotic mice (EVF). CCl4-treated mice undergoing EVN therapy also exhibited reduced levels of hepatocyte death, inflammatory infiltration, circulating AST/ALT levels and hepatic or circulating pro-inflammatory cytokines. Hepatic histology, liver function tests or circulating proinflammatory cytokine levels were unaltered in control mice receiving EVN. As determined using PKH26-labelled EVN, principal target cells included hepatic stellate cells (HSC; a normally quiescent fibroblastic cell that undergoes injury-induced activation and produces fibrosis during chronic injury) or hepatocytes which showed increased EVN binding after, respectively, activation or exposure to CCl4. In vitro, EVN decreased proliferation and fibrosis-associated molecule expression in activated HSC, while reversing the inhibitory effects of CCl4 or ethanol on hepatocyte proliferation. In mice, microRNA-34c, -151-3p, -483-5p, -532-5p and -687 were more highly expressed in EVN than EVF and mimics of these microRNAs (miRs) individually suppressed fibrogenic gene expression in activated HSC. A role for these miRs in contributing to EVN actions was shown by the ability of their corresponding antagomirs to individually and/or collectively block the therapeutic effects of EVN on activated HSC or injured hepatocytes. Similarly, the activated phenotype of human LX-2 HSC was attenuated by serum EVs from healthy human subjects and contained higher miR-34c, -151-3p, -483-5p or -532-5p than EVs from hepatic fibrosis patients. In conclusion, serum EVs from normal healthy individuals are inherently anti-fibrogenic and anti-fibrotic, and contain microRNAs that have therapeutic actions in activated HSC or injured hepatocytes.
Abbreviations: ALT: alanine aminotransferase; AST: aspartate aminotransferase; CCl4: carbon tetrachloride; CCN2: connective tissue growth factor; E: eosin; EGFP: enhanced green fluorescent protein; EVs: extracellular vesicles; EVF: serum EVs from mice with experimental hepatic fibrosis; EVN: serum EVs from normal mice; H: hematoxylin; HSC: hepatic stellate cell; IHC: immunohistochemistry; IL: interleukin; MCP-1: monocyte chemotactic protein-1; miR: microRNA; mRNA: messenger RNA; NTA: nanoparticle tracking analysis; PCNA: proliferating cell nuclear antigen; qRT-PCR: quantitative real-time polymerase chain reaction; SDS-PAGE: sodium dodecyl sulphate – polyacrylamide gel electrophoresis; αSMA: alpha smooth muscle actin; TAA: thioacetic acid; TG: transgenic; TGF-β: transforming growth factor beta; TEM: transmission electron microscopy; TNFα: tumour necrosis factor alpha.
Responsible Editor Yong Song Gho, Pohang University of Science and Technology, Republic of Korea
Introduction
Chronic liver diseases such as hepatitis, alcoholic liver disease and non-alcoholic fatty liver disease are the cause of considerable morbidity and mortality and affect millions of individuals worldwide [Citation1]. The long-term nature of the inflammatory and injury processes in these diseases is frequently associated with the development of hepatic fibrosis, a pathology in which insoluble collagen and matrix components are deposited in excessive amounts in the interstitial spaces, which can severely compromise liver function [Citation2]. While fibrosis may reduce or resolve in some individuals after treatment of the underlying disease, many patients are non-responsive to this approach and are reliant on direct fibrosis therapy for which approved drugs are lacking [Citation3,Citation4]. A new lead in liver fibrosis research has come from studies of extracellular vesicles (EVs) which are derived from numerous cell types in the body and comprise exosomes, microvesicles and apoptotic bodies [Citation5]. Exosomes and microvesicles are able to mediate cell–cell transfer of their respective molecular payloads (microRNA [miR], messenger RNA [mRNA], proteins) resulting in transcriptional or translational modifications in the recipient cells [Citation5,Citation6]. EVs may either drive or dampen pathogenenic pathways in various liver cells depending on the phenotype of the donor and recipient cells and the nature of the molecular information transferred between them [Citation7].
After their cellular release, EVs enter the intercellular space and may be taken up by neighbouring cells or they may enter body fluids such as blood, urine or saliva and can potentially be delivered to target cells that reside at appreciable distances from their sites of release [Citation5]. EVs in plasma or serum are highly heterogeneous and originate from many circulating cell types (e.g. erythrocytes, leucocytes, platelets, megakaryocytes, monocytes, granulocytes, lymphocytes) as well as from cells in tissues and organs that have direct (e.g. endothelial cells) or indirect contact (via interstitial fluids) with the systemic circulation [Citation8]. Serum or plasma EVs are a readily accessible and rich source of biomarkers that have potential for assessing organ disease, including that of the liver [Citation9–Citation14], because the pathogenic changes involved may be reflected in an altered EV molecular payload that can be detected by comparison to EVs from healthy individuals or from individuals at a different disease stage. However, little attention has been paid to the functional properties of EVs from the body fluids of individuals in good health. In this study, we show that serum EVs from healthy individuals are therapeutic for liver fibrosis due to their ability to attenuate activation of hepatic stellate cells (HSC, the principal fibrosis-producing cell in the liver), hepatocyte injury and inflammation. The direct therapeutic properties of serum EVs on HSC or hepatocytes appear to be due, at least in part, to the action of EV miRs that are suppressed in serum EVs during fibrosis.
Materials and methods
Collection of human serum
Human blood was collected from healthy volunteers (9 males, 9 females; age range 23–28 years) using a protocol approved by the Institutional Review Board of Nationwide Children’s Hospital (Columbus, OH). Exclusion criteria for blood donation included the presence of known acute or chronic diseases, the taking of medications, the consumption of alcohol within the preceding 24 h, or age less than 21 years or greater than 30 years. Blood samples were de-identified prior to being received for processing. Blood was collected into SST™ tubes (BD Vacutainer™, ThermoFisher, Waltham, MA) from which serum was obtained by low speed centrifugation and stored for up to 1 month at -80oC prior to EV purification. Serum samples from hepatitis B virus patients with F3/4 fibrosis (n = 12) or age- and sex-matched healthy control subjects (n = 12) were obtained from The Liver Research Center, Beijing Friendship Hospital, Capital Medical University (Beijing, China). Samples were collected under an IRB protocol approved by the Ethical Committee of Beijing Friendship Hospital, Capital Medical University in support of clinical trial NCT01938781.
Collection of mouse serum
All animal protocols were approved by the Institutional Animal Care and Use Committee of Nationwide Children’s Hospital (Columbus, OH). Blood was collected by cardiac puncture or retro-orbital bleed in control male or female wild-type Swiss Webster or FVB mice (6–8 weeks) or in mice of the same strain in which liver fibrosis was induced, with or without EV therapy, as described below. Blood was allowed to clot and serum was collected in SST™ tubes (ThermoFisher), pooled within each strain or treatment group, and used for assay or EV isolation immediately or after storage at -80oC for up to 6 months [Citation15].
Serum EV purification
Human or mouse sera underwent low-speed centrifugation (300 × g, 15 min; 10,000 × g, 30 min, 4°C,) and the supernatants were passed through a 0.22 μm filter (Merck, Darmstadt, Germany) and centrifuged (10,000 × g, 30 min 4°C) to remove particulates. The supernatant was then subjected to ultracentrifugation (100,000 × g for 90 mins at 4°C) in a T-70i fixed-angle rotor (Beckman Coulter, Brea, CA) to pellet the small vesicles [Citation16–Citation18]. The pellet was then rinsed with PBS to remove contaminating proteins and ultracentrifuged using the same conditions after which the supernatant was discarded and the EV-containing pellet was recovered. For some experiments, mouse serum EVs were isolated using PureExo® kits (101Bio, Palo Alto, CA) [Citation19]. EVs from both purification procedures were resuspended in PBS. EVs purified from the serum of normal or fibrotic mice are hereafter termed “EVN” or “EVF”, respectively. For the therapeutic studies described, serum typically was pooled from 10–20 mice from which EVs were then isolated. Over the course of these experiments, 15 such pools were collected with consistent therapeutic outcomes irrespective of the actual pool used, with each pool being tested in at least triplicate among the various assays. For human EV characterization and biological assay, serum EVs from the 18 donors were individually purified and analysed after which some of the data were then combined for analysis.
EV characterization
Purified EVs were diluted to 106–107 particles/ml in PBS for nanoparticle tracking analysis (NTA) using a Nanosight 300 equipped with v3.2.16 analytical software (Malvern Instruments, Westborough, MA). Two videos (25 s each) were recorded for each sample and the software was used to estimate concentration and size of the particles. The recordings were performed at room temperature which was monitored manually. Camera gain was 15 and the shutter speed was 4.13 ms. For analysis, the detection threshold was set to 6. Calibration was carried out using 100 nm polystyrene latex microspheres (Magsphere Inc., Pasadena, CA) diluted to a known concentration in PBS and then two videos were recorded.
For analysis by transmission electron microscopy (TEM), 200-mesh, copper grids coated with a Formvar/Carbon support film (Ted Pella, Redding CA) were glow-discharged with a Pelco easiGlow discharge cleaning system (Ted Pella) prior to use. EV samples were fixed in 2.5% glutaraldehyde for 30 min and pelleted by ultracentrifugation and then placed on the grids which were subsequently negative-stained using 1% aqueous uranyl acetate. Grids were examined using a FEI Tecnai G2 Biotwin TEM (Atlanta, GA) operating at 80 kV and digital micrographs were captured on an AMT side-mount camera with FEI imaging software.
EV proteins (10–25 μg) were analysed by sodium dodecyl polyacrylamide gel electrophoresis (SDS-PAGE) or Western blot. Gels were stained with Coomassie Blue or subjected to Western blot using primary antibodies to CD81 (1:400; ProSci, Poway, CA), CD9 (1:300; Lifespan Bioscience Inc., Seattle WA), CD63 (1:400; Abcam, Cambridge, UK), flotillin-1 (1:500; Abcam) or asialoglycoprotein receptor 1 (ASGPR1) (1:500,GeneTex, Irvine, CA) [Citation16,Citation17,Citation19,Citation20]. For organ localization and cell binding studies, EVN were labelled with PKH26 lipophilic membrane red fluorescent dye (Sigma-Aldrich) as described [Citation17].
CCL4-induced hepatic fibrogenesis in mice
Male or female Swiss Webster wild-type or transgenic (TG) Swiss Webster mice (Stock TG (connective tissue growth factor-enhanced green fluorescent protein [CCN2-EGFP]) FX156GSat/Mmucd expressing EGFP under the control of the CCN2 promoter [Citation21]) (TG CCN2-EGFP) mice (4–5 weeks; n = 5 per group) received i.p. carbon tetrachloride (CCl4; 175 μl in 1325 μl corn oil/kg; Sigma-Aldrich, St Louis, MO) on Days 1, 3, 5, 7 and 9. Control mice received i.p. corn oil (1500 μl/kg) alone on the same days. Some mice received i.p. EVN (0–40 μg EVN protein per g body weight) on Days 2, 4, 6 and 8. Mice were sacrificed on Day 10 and liver lobes were either perfused with PBS, fixed in 4% paraformaldehyde and processed for histological analysis or immediately harvested for EGFP imaging using a Xenogen IVIS 200 (PerkinElmer, Waltham, MA).
CCL4- or TAA-induced hepatic fibrosis in mice
Wild-type male Swiss Webster mice or FVB (4–5 weeks; n = 5 per group) received i.p. CCl4 (175 μl in 1325 μl corn oil/kg) or corn oil (1500 μl/kg) three times per week for 5–6 weeks. During the last 2–3 weeks, some mice received i.p. EVN (0–40 μg/g) every other day 3 times per week, on alternative days to those used for CCl4 or oil administration. Mice were sacrificed 36 h after the last CCl4 or oil injection, or in non-treated littermates. Just prior to sacrifice in some animals, blood was collected by cardiac puncture and serum obtained for performing liver function tests, assessing cytokine levels or EV purification. Individual liver lobes were harvested and snap-frozen in liquid nitrogen for subsequent RNA extraction or perfused using PBS followed by 4% paraformaldehyde (Sigma-Aldrich, St Louis, MO) for histological analysis of fixed tissue. In some experiments, CCl4- or oil-treated FVB mice received a single i.v. injection of PKH26-labelled EVN (40 μg/g) 4 h prior to sacrifice after which PKH26 fluorescence was detected in (i) freshly isolated liver pieces by direct Xenogen imaging; (ii) 6 μm sections of frozen liver tissue by fluorescence microscopy (Axiovert 25, Zeiss, Obercochen, Germany) or (iii) in 24-h cultures of primary HSC or hepatocytes by confocal microscopy (Model 710, Zeiss) using our published procedures [Citation16]. As an alternative method of inducing liver fibrosis, wild-type male FVB mice (4–5 weeks; n = 10 per group) received i.p. thioacetic acid (TAA; 100 mg/kg; Sigma-Aldrich) in saline 3 times per week for 6 weeks. Control mice received i.p. saline alone. Over the last week, some mice received i.p. EVN (40 μg/g) every day for 6 days and were sacrificed 60 h later. Livers were processed as described above.
HSC or hepatocyte cultures
Livers from control or CCl4-treated male wild-type Swiss Webster mice (6–8 weeks) were used for isolation of primary HSC or hepatocytes. Livers were perfused in situ and then subjected to either collagenase digestion for isolation of hepatocytes [Citation16] or pronase/collagenase digestion and buoyant-density centrifugation for isolation of HSC [Citation16,Citation22,Citation23]. As we have previously described [Citation19], quiescent HSC isolated from control animals contained lipid droplets and were positive for desmin or Twist1, but not for CCN2, αSMA or collagen α(I) whereas activated HSC isolated from animals treated with CCl4 were positive for desmin, CCN2, αSMA or collagen α(I), but not for Twist1 (data not shown). Hepatocytes were maintained in complete William E medium (Gibco, Billings, MT) and used within the first 3 days of primary culture while HSC were split 1:3 in DMEM/F12/10% FBS medium (Gibco) every 5 days and used at up to passage 4 (P4). LX-2 human HSC (a kind gift from Dr Scott Friedman, Icahn School of Medicine at Mount Sinai, New York, NY) were cultured in DMEM/10% FBS as described [Citation19,Citation22]. AML12 mouse hepatocytes (American Type Culture Collection, Manassas, VA) were cultured in DMEM/F12/10% FBS supplemented with insulin, transferrin, selenium and dexamethasone [Citation19].
Histology
Perfused mouse livers were fixed and embedded in paraffin. Sections of 5 μm thickness were cut and stained with H and E. Collagen was detected by staining sections with 0. 1% Sirius Red (Sigma-Aldrich).
Immunohistochemistry (IHC)
Mouse liver sections were incubated with primary antibodies to albumin (1:400, Bethyl Laboratories, Inc., Montgomery, TX), Ki67 (1:500, Thermo Fisher, Grand Island, NY), F4/80(1:100, Abcam, Cambridge, MA). Ly6c (1:400, Abcam), CD45 (1:150, Abcam), monocyte chemotactic protein-1 (1:100, Abcam), TNFα (1:150, Abcam), IL1-β (1:100, Abcam), collagen α1 (1:250, Abcam), αSMA (1:1000; Dako Cytomation, Glostrup, Denmark) or CCN2 (in-house; 5 μg/ml [Citation21,Citation24]) followed by Alexa Fluor 488 goat-anti rabbit IgG and Alexa Fluor 568 goat-anti mouse IgG, or Alexa Fluor 647 goat-anti mouse IgG, or Alexa Fluor 568 goat-anti-chicken IgG (all at 1:1000; Thermo Fisher) for 1 h at room temperature. The slides were mounted with Vectashield Mounting Medium containing 4′,6-diamidino-2-phenylindole nuclear stain (Vector Laboratories, Burlingame, CA) and examined by confocal microscopy. Activated HSC were identified by positive immunostaining for CCN2, αSMA and/or collagen α(I).
Alanine aminotransferase (ALT) or aspartate aminotransferase (AST) assay
Mouse serum or hepatocyte conditioned medium were analysed using ALT or AST assay kits (Sigma) according to the manufacturer’s instructions.
Multiplex cytokine assay
Cytokine levels in mouse serum were determined using a Mouse Proinflammatory Panel 1 Kit (Meso Scale Discovery, Rockville, MD) according to the manufacturer’s instructions. All components of the kit were brought to room temperature and the antibody-coated plate was shaken with diluent for 30 min at 800 rpm. Two-fold dilutions of the serum samples were loaded in duplicate on to the plate alongside serial dilutions of the calibrator used to construct standard curves for each analyte. Plates were incubated with shaking for 2 h after which the wells were washed and incubated with labelled detection antibodies for 2 h. After washing, read buffer was added to develop the plate which was then read in a Sector Imager 2400 using Discovery Workbench Software (Meso Scale Discovery).
Mouse cytokine microarray
A mouse cytokine proteome profiler™ array (Panel A kit; R & D Systems, Minneapolis, MN) was used to detect 40 different cytokines or chemokines in liver extracts according to the manufacturers’ instructions. Each array was incubated with 267 μg/ml pooled liver tissue lysates from control or fibrotic Swiss Webster mice (5-week CCl4 model) that had or had not received EVN therapy as described above (n = 5 mice per group). Arrays were developed using chemiluminescence and the mean signal (pixel density) for each pair of duplicate component spots was quantified by scanning.
EV binding assays
PKH26-labelled EVN were added for 16 h to Day 1 or P3 mouse HSC or to Day 2 primary mouse hepatocytes with or without 40-h exposure to CCl4 (20 mM in 0.1% DMSO) or DMSO carrier (0.1%). PKH26-labelled human serum EVs were added for up to 24 h to LX-2 cells. Cells were then washed in PBS and lysed in lysis buffer (ThermoFisher). Cell-associated PKH26 fluorescence was measured at 590/540 nm using a Spectra Max® M2 microplate reader (VWR, Atlanta, GA, USA) and normalized to total cellular protein determined by bicinchoninic acid assay (ThermoFisher).
Effect of serum EVs on HSC
P2-3 mouse HSC were seeded in 6-well plates for 24 h in DMEM/10% FBS, cultured in serum-free medium for 12–24 h and treated with EVN or EVF (0–8 μg/ml) for up to 24 h. LX-2 cells were grown for 24 h in DMEM/10% FBS and then cultured in serum-free medium for the next 24 h. Some LX-2 cells were treated with 0–10 ng/ml TGF-β for 36 h, the last 24 h of which included co-incubation with human serum EV. Expression of CCN2, αSMA or collagen α1(I) mRNA was determined by qRT-PCR; the presence of αSMA was determined by IHC and cell proliferation was determined using a CyQUANT® assay (Thermo Fisher).
Effect of serum EVs on hepatocytes
AML12 hepatocytes were incubated for 24 h in DMEM/F12/10% FBS medium (plus supplements) for 24 h, serum-starved for 12 h, treated with 0–20 mM CCl4 for 48 h in the presence or absence of EVN (8 μg/ml) for the last 24 h and assessed for cell proliferation using a CyQUANT® assay. Culture medium was assessed for ALT and AST as described above.
RNA extraction and RT-qPCR
Total RNA from liver tissues or cultured HSC was extracted using a microRNeasy Plus kit (Qiagen, Valencia, CA) and reverse transcribed using a miScript II RT kit (Qiagen) according to the manufacturers’ protocols. Resulting transcripts were analysed by qRT-PCR as described [Citation17] with primers for EGFP, CCN2, αSMA, collagen α1(I) or SMAD3 (). Each reaction was run in triplicate, and all samples were normalized to glyceraldehyde-3-phosphate dehydrogenase. Negative controls were a non-reverse transcriptase reaction and a non-sample reaction.
Table 1. Primers used for RT-PCR.
Differential EV miR analysis
Using a ABI-7900 qPCR machine (Applied Biosystems, Forster City, CA) miR profiling was performed on PureExo®-purified EVs obtained from pooled serum from Swiss Webster mice (200 μl serum per mouse; 5 mice per group (control, 5-week oil, 5-week CCl4) using a mouse miRnome Array kit (Qiagen). Differential comparison of the miRs that were up- or down-regulated in each group was performed using miR array software (Qiagen) to identify those miRs that were expressed at higher levels in EVN versus EVF, that underwent the greatest suppression of expression in EVF as compared to EVN, but that were unchanged between EVN and oil-treated mice. Data were confirmed by RT-PCR using primer sequences shown in . The same strategy, using a human miRnome Array (Qiagen), was used to establish differential serum EV miR expression between liver fibrosis patients and healthy control subjects. Serum (0.5 ml) from each patient (control, fibrosis) was individually processed to purify EVs from which small RNA was then isolated and reverse-transcribed. Samples in each group were then pooled for miR profiling.
Transfection of HSC or hepatocytes with miR mimics or antagomirs
P2-P3 mouse HSC or AML12 hepatocytes were transfected individually or collectively with 100 nM miR mimics (miRs-34c-3p, -151-3p, -483-5p, -532-5p, -687) or their corresponding antagomirs (Qiagen) by electroporation (Nucleofector Kit, Lonza, Koln, Germany) as described [Citation16]. Each antagomir preparation comprised chemically synthesized single-stranded modified RNAs that specifically inhibit the function of the mature miR target. Control transfections were performed using a commercial scrambled miR mimic or antagomir (Qiagen). MiR-transfected HSC were incubated for 24 h in DMEM/F12/10% FBS medium and then analysed by qRT-PCR. Antagomir-transfected HSC were incubated for 24 h in DMEM/F12/10% FBS medium, serum-starved for 12 h, treated with EVN (8 μg/ml) for 24 h and then analysed by RT-PCR or CyQUANT® cell proliferation. Antagomir-transfected AML12 hepatocytes were treated with 0–20 mM CCl4 for 48 h in the presence or absence of EVN (8 μg/ml) and assessed for cell proliferation as described above.
Statistical analysis
All experiments were performed at least three times with triplicate measurements, with data expressed as mean ± S.E.M. Fluorescence images were scanned and quantified using Image J software (NIH, Bethesda, MD). The data from qRT-PCR, NTA, imaging, multiplex cytokine analysis and ALT/AST assays were analysed by Student’s t-test using Sigma plot 11.0 software (SPSS Inc., Chicago, IL). P values < 0.05 were considered statistically significant.
Results
Characterization of serum EVs from normal or CCL4-treated mice
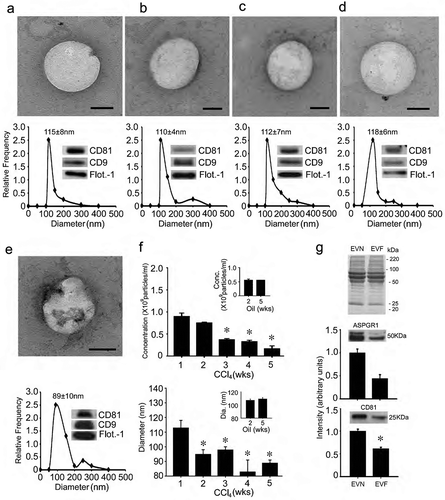
Vesicles isolated from the serum of normal wild-type Swiss Webster mice (“EVN”) using sequential ultracentrifugation were 115 ± 8 nm in diameter and positive for CD81, CD9 and flotillin-1 (()). As assessed by TEM, the isolated EVs were approximately 100 nm, had a spherical morphology and contained electron-dense material within a limiting membrane (()). Very similar characteristics were also evident when the serum of wild-type Swiss Webster mice was alternatively processed using ExoPure® isolation kits (()). Similar features were again observed for EVs isolated from the serum of TG CCN2-EGFP Swiss Webster mice or wild-type FVB mice ((, )). Serum EVs from wild type Swiss Webster mice that had been exposed to CCl4 for up to 5 weeks were also positive for CD81, CD9 or flotillin-1 but appeared to have membrane irregularities as assessed by TEM (()) and underwent a progressive decrease in their concentration and size during CCl4 treatment ((, )). Serum EVs collected from fibrotic mice after 5 weeks of CCl4 treatment (“EVF”) were approximately 10% fewer in number and 20% smaller than EVN from control animals ((, )). These changes were not evident for serum EVs isolated from control animals treated with oil (()). Although the predominant EV proteins did not appear to change qualitatively or quantitatively between EVN and EVF as assessed by SDS-PAGE analysis, Western blot analysis showed that the levels of ASPGR1 or CD81 were diminished in EVF to, respectively, 40% or 60% of their levels in EVN (()).
Figure 1. Characterization of mouse serum EVs. (a) Ultra-centrifugation or (b–e) PureExo® kits were used to isolate EVN from the serum of (a,b) normal male wild-type Swiss Webster mice; (c) normal male TG CCN2-EGFP Swiss Webster mice; (d) normal male wild type FVB mice or (e) male wild-type Swiss Webster mice that had been treated for 5 weeks with CCl4. EVs were characterized by TEM (upper panels; scale bar: 50 nm; representative images are shown), NTA (lower panels; mean ± S.E.M. for particle diameter (nm) is indicated) or Western blot (insets). (f) NTA was performed weekly on serum EVs purified by ultracentrifugation to determine their concentration (upper panel) or size (lower panel) in individual groups of male wild-type Swiss Webster mice that received CCl4 or oil (insets) 3 times per week for up to 5 weeks. n = 3 independent experiments (5 mice per group) performed in triplicate. *P < 0.01 versus Week 1. (g) SDS-PAGE analysis of EVN or EVF using Coomassie blue for protein detection (upper) and Western blot analysis of EVN versus EVF using anti-ASPGR1 (middle) or anti-CD81 (lower), with quantification of immunoreactive signals by scanning.
EVN reduce fibrogenesis and fibrosis in experimental models in vivo
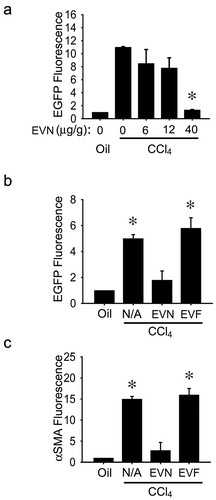
Swiss Webster TG CCN2-EGFP mice were treated on Days 1,3,5,7 and 9 with corn oil or CCl4 and received EVN or EVF i.p. on Days 4, 6 and 8, with EV dose normalized to EV protein concentration. As assessed on Day 10, expression of EGFP (a surrogate marker for CCN2 – a profibrogenic molecule that is synthesized downstream of transforming growth factor beta (TGF-β) and which is produced principally in activated HSC during liver injury [Citation25]) was dose-dependently inhibited in CCl4-injured liver by EVN as assessed by direct imaging of freshly harvested liver from female mice (()) or by IHC for EGFP in male mice (()), with EV doses of 40 μg/g reducing the fluorescence to levels that were not significantly different from control oil-treated animals. In contrast, CCl4-induced hepatic EGFP expression was not inhibited by the same dose (40 μg/g) of EVF (()). Moreover, immunostaining for CCl4-induced α-smooth muscle actin (αSMA) in activated HSC was reduced in male mice receiving EVN but not EVF (()). Since male and female mice responded to treatment with EVN and since EVF was not therapeutic, subsequent mouse studies were undertaken in male recipients treated with EVN from male donors.
Figure 2. Suppression of hepatic fibrogenesis in vivo by EVN. (a) Female TG CCN2-EGFP mice received corn oil (1500 μl/kg) or CCl4 (175 μl in 1325 μl corn oil/kg) i.p. on Days 1, 3, 5, 7 and 9 plus 0–40 μg/g EVN i.p. on Days 2, 4, 6 and 8 prior to analysis on Day 10 of direct EGFP fluorescence in pieces of whole liver. (b) EGFP or (c) αSMA were detected by IHC in fixed liver sections after i.p administration of 0 (“N/A”) or 40 μg/g EVN or EVF in CCl4-treated male TG CCN2-EGFP mice, essentially as described in (a). n = 3 independent experiments (5 mice per group) performed in triplicate. *P < 0.01 versus oil.
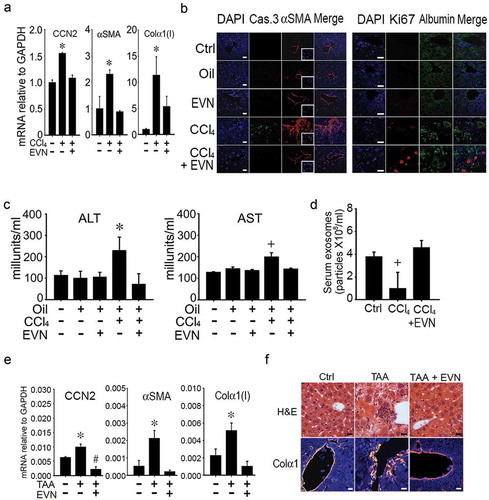
Experimental fibrosis was established in mice by chronic exposure (5 weeks) to CCl4 with EVN administered every other day for the last 2 weeks. In TG CCN2-EGFP mice, the CCl4-induced increase in hepatic EGFP was substantially reduced by EVN (()) as was the severity and extent of hepatic fibrosis as assessed by Sirius red staining for collagen (()). Hematoxylin and eosin (H and E) staining showed that CCl4-treated mice demonstrated significant levels of hepatocyte disorganization and cellular infiltration whereas these features were not evident in CCl4-treated mice that received EVN (()). EVN administration also resulted in suppressed hepatic mRNA expression of collagen α1(I), αSMA, or CCN2 in CCl4-treated TG CCN2-EGFP mice (data not shown) or their wild-type counterparts (()). Furthermore, the high level of caspase 3 and αSMA in activated HSC in fibrotic mice was diminished to control levels in EVN treated mice (()). In CCl4-treated mice, EVN treatment also resulted in increased hepatocyte proliferation and decreased liver injury as shown by, respectively, enhanced staining for Ki67 in albumin-positive cells (()) and decreased circulating ALT or AST levels (()). Moreover, EVN treatment caused restitution of serum EV number to pre-CCl4 treatment levels (()). In control animals, EVN had no effect on hepatic histology, the extent or pattern of Sirius red staining, expression of fibrosis-, apoptosis- or proliferation-associated molecule expression, or circulating levels of AST or ALT ((, ), (–)). Finally, in TAA-induced liver fibrosis in FVB mice, EVN attenuated hepatic collagen α1(I), αSMA or CCN2 mRNA levels as assessed by qRT-PCR (()) and reduced tissue damage and inflammation or collagen α1 production as assessed, respectively, by H and E staining or IHC (()).
Figure 3. Suppression by EVN of hepatic collagen deposition and inflammatory cell infiltration during CCl4-induced hepatic fibrosis in vivo. Male TG CCN2-EGFP mice were administered oil (1500 μl/kg) or CCl4 (175 μl in 1325 μl corn oil/kg) i.p. for 5 weeks, with or without EVN (40 μg/g; i.p.) every other day over the last 2 weeks. Animals were then sacrificed and examined (a) for EGFP fluorescence in whole liver pieces (typical staining from triplicate determinations (5 mice per group)); or (b) by Sirius red or (c) H and E staining of liver sections (typical staining from 5 independent experiments). Data are representative of five independent experiments. Scale bar: 20 μm.
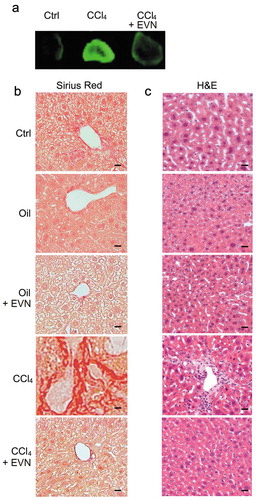
Figure 4. Normalization by EVN of hepatic gene expression or serum components during CCl4- or TAA-induced liver fibrosis in vivo. Male TG CCN2-EGFP mice were administered oil (1500 μl /kg) or CCl4 (175 μl in 1325 μl corn oil /kg) i.p. for 5 weeks, with or without EVN (40 μg/g; i.p.) every other day over the last 2 weeks. Animals were then sacrificed and examined by (a) qRT-PCR of hepatic mRNA for expression of CCN2, αSMA, or collagen α1(I) (n = 4 independent experiments performed in triplicate. * p < 0.01 versus no treatment)) or (b) IHC of liver sections for the presence of caspase 3 or αSMA (left panel) or Ki67 or albumin (right panel) (typical staining from 5 independent experiments). Serum from blood collected at the time of sacrifice was (c) assayed for ALT or AST or (d) subjected to NTA to determine EV frequency. Male FVB mice received TAA (i.p. 200μg/g; q.o.d.) for 6 wks with or without daily i.p. administration of EVN (40μg/g; isolated from serum of normal FVB mice) for 6 days during the last week after which (e) hepatic RNA underwent qRT-PCR for fibrosis-related gene expression in livers or (f) liver sections were stained with H and E or with anti-collagen α1 (orange) and DAPI (blue). n = 5 independent experiments performed in triplicate. *p < 0.01 versus no treatment. Scale bar: 20 μm.
Collectively, these data showed that reconstitution of three strains of male or female mice with EVN resulted in a reversal of fibrosis in two different experimental models. This response was associated with decreased expression of fibrogenic, activation or apoptotic genes in HSC, decreased liver injury, increased expression of proliferation markers in hepatocytes and restoration of serum EVs to pre-injury concentrations. Normal control animals exhibited no alterations in hepatic histology, inflammation or liver function following EVN administration.
EVN dampen CCL4-induced inflammation
Exposure of mice to CCl4 for 6 weeks caused a significant increase in the circulating levels of interferon-γ, TNFα, IL-10, IL-12p70, IL-2, IL-5, IL-4 or IL-6, all of which were reduced to baseline levels by treatment over the last 3 weeks with EVN (()). As assessed by IHC, CCl4-mediated infiltration into the liver of macrophages (F4/80+), monocytes (Ly6c+) and T cells (CD45+) was suppressed by EVN treatment (()), consistent with the EVN-mediated reduction of CCl4-induced infiltrating cells assessed by H and E staining (()). CCl4-induced hepatic interleukin (IL)-1β, monocyte chemotactic protein-1 (MCP-1) or tumour necrosis factor alpha (TNFα) which are produced by injured hepatocytes and infiltrating cells were also suppressed by EVN as assessed by IHC (()). Protein array analysis showed that EVN attenuated 5 of the same cytokines and chemokines in liver tissue (()) as in the circulation (()) (IFN-γ, IL-2, IL-4, IL-10, TNFα) and also that 22 additional components were attenuated by EVN in fibrotic mice (). EVN treatment of non-treated or oil-treated mice did not cause circulating cytokine levels to be altered (()), nor as assessed by staining did it cause any infiltration of inflammatory cells (() and ()) or expression of pro-inflammatory cytokines in the liver ((, )). As assessed by protein array, there appeared to be slight changes in hepatic cytokine or chemokine expression in control mice exposed to EVN but this has yet to be validated by independent analysis and in any case it was not associated with tissue damage or pathology. Thus, while EVN exerted profound anti-inflammatory actions in fibrotic mice, they did not elicit overt inflammatory reactions in control mice.
Table 2. Expression of hepatic cytokine and chemokines.
Figure 5. Suppression of inflammation by EVN. Wild-type male Swiss Webster mice received no treatment (“Ctrl”) or were administered oil or CCl4 i.p. for (a) 6 weeks or (b, c, d) 5 weeks, with or without i.p. administration of 40 μg/g EVN q.o.d for the last (a) 3 weeks or (b, c, d) 2 weeks. The figure shows (a) serum concentrations of IFN-γ, TNF-α, IL-10, IL-12p70, IL-2, IL-5, IL-4 and IL-6 which were assessed using a cytokine multiplex assay. n = 5 independent experiments performed in duplicate; (b) IHC for F4/80 (macrophages), Ly6c (monocytes) or CD45 (T cells) in liver sections; (c) IHC for IL-1β, MCP-1, or TNFα in liver sections (typical staining from five independent experiments) and (d) protein array showing altered expression of cytokines and chemokines (left) for which those that correspond to those in serum (a) are numbered and quantified (right). Signal intensities of all array components are shown in . *P < 0.01 versus no treatment. Scale bar: 20 μm.
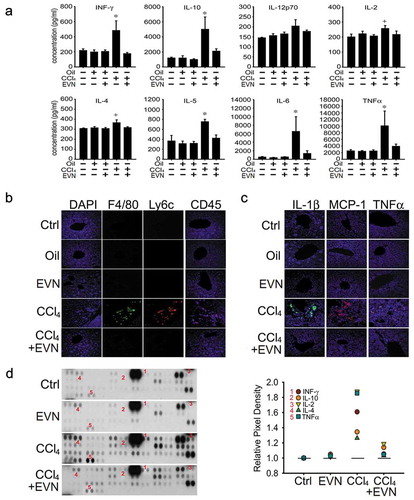
EVN are tropic for HSC or hepatocytes and show enhanced cellular binding after injury
To identify target cells in the liver, PKH26-stained EVN were injected once i.v. into mice at the end of a 5-week course of oil or CCl4 administration. Direct imaging of the livers after 4 h showed the presence of hepatic PKH26 fluorescence, for which the staining intensity was much greater in fibrotic livers than in control livers (()). This finding was also evident in liver tissue sections (()) and was attributable to the association of PKH26 with hepatocytes or HSC as shown by the presence of PKH26 staining in each cell type when isolated from the livers of control mice and by marked increase in PKH26 staining intensity in each cell type when they were isolated from CCl4-treated mice ((, )). To corroborate these findings, EV binding studies were performed on primary cultures of HSC or hepatocytes. In vitro binding of PKH26-stained EVN to P3 (activated) HSC was ~6-fold higher than to Day 1 (quiescent) HSC, while EVN binding to primary D2 hepatocytes was ~5-fold higher after in vitro exposure of the cells to CCl4 (()).
Figure 6. Binding of EVN to HSC or hepatocytes. PKH26-stained EVN were injected once i.v. (40 μg/g) into control or fibrotic male FVB mice (5-week CCl4 model) prior to sacrifice 4 h later (5 mice per group). Hepatic localization of PKH26 fluorescence was determined by (a) Xenogen imaging of freshly dissected liver slices or analysis of (b) frozen liver sections by fluorescent microscopy or (c) hepatocytes or (d) HSC after 24 h culture by confocal microscopy (blue is DAPI; red is EVN). Images shown are representative of five independent experiments. (e) In vitro binding of PKH26-stained EVN (8 μg/ml) was evaluated after 16hr-incubation with quiescent primary mouse HSC on Day 1 (“D1”), primary mouse hepatocytes on Day 2 (“Ctrl”), P3 mouse HSC (“P3), or Day 2 mouse hepatocytes after exposure to 20 mM CCl4 for the last 40 h of culture (“CCl4”). n = 3 independent experiments performed in triplicate. *P < 0.01 versus D1; +P < 0.05 versus control. Scale bar: 20 μm.
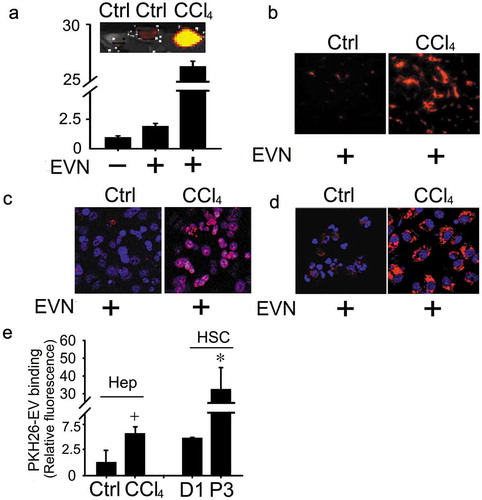
Attenuation of hepatocyte injury or HSC activation in vitro by EVN
The direct binding of EVN to hepatocytes or HSC in vivo or in vitro supported the possibility that EVN was beneficial for each cell type during fibrosing injury. To test this, in vitro experiments were first conducted on AML12 mouse hepatocytes that had been exposed to CCl4 or ethanol for 48 h (, )). Whereas hepatocyte proliferation was decreased by CCl4 or ethanol, this outcome was reversed in cells to which EVN were added for the final 24 h ((, )). Moreover, release of AST or ALT after exposure of hepatocytes to CCl4 in vitro reverted to baseline levels after exposure of the cells to EVN (, )). When tested on activated mouse HSC in vitro, EVN reduced the expression of collagen α1(I), αSMA or CCN2 and the rate of cell proliferation ((, )). Thus in vitro, EVN drive proliferation in injured hepatocytes and suppress fibrogenesis and proliferation in activated HSC, consistent with the outcomes seen in the livers of EVN-treated fibrotic mice. In contrast to the inhibitory effect of EVN, HSC proliferation trended upwards in response to EVF, but this was not statistically significant (()).
Figure 7. Effects of EVN on HSC or hepatocytes in vitro. AML12 cells were serum-starved for 12 h and then exposed for 48 h to (a,c,d) 20 mM CCl4 (in 0.1% DMSO) or DMSO carrier (0.1%) or (b) 50 mM ethanol for 48 h, with or without EVN (8 μg/ml) for the final 24 h after which (a,b) cell proliferation was assessed by CyQUANT® assay or (c,d) ALT and AST were assessed in the conditioned medium. (e) Effect on CCN2, αSMA or collagen α1(I) expression after 24 h treatment of serum-starved D9 HSC with EVN (8 μg/ml) (left) or EVF (right); (f) CyQUANT® assay of serum-starved P3 HSC after treatment of the cells for 24 h with EVN or EVF (8 μg/ml). n = 4 independent experiments performed in triplicate. *P < 0.01, +P < 0.05 versus control.
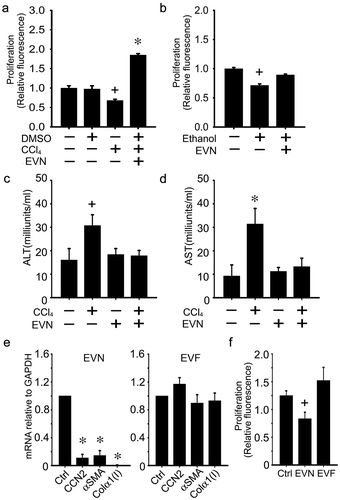
Identification of therapeutic miRs in EVN
Since miRs exert their effects over multiple targets and across many signalling pathways and exosomal miRs have been shown previously to regulate activated HSC [Citation16,Citation17,Citation19,Citation26], we focussed on the identity of candidate therapeutic miRs in EVN. Differential miRnome profiling was performed to identify EV miRs that were, as compared to EVN, the most differentially suppressed in EVF but not significantly changed in serum EV from oil-treated animals. The results of this analysis revealed that miR-34c-3p, -151-3p, -483-5p, -532-5p and -687 were among the candidate miRs identified (()). When independently assessed by RT-PCR in additional groups of control or fibrotic mice, each of these miRs was confirmed to be expressed at higher levels in serum EV from control versus fibrotic mice (()). Mimics of each miR attenuated expression of collagen α1(I), αSMA or CCN2 when tested on D9 mouse HSC (()), while Smad3 expression in HSC or hepatocytes was reduced by either EVN or a miR-532 mimic (()). To establish the role of these miRs as functional constituents of EVN, the responses of target HSC (activated) or hepatocytes (CCl4-injured) were evaluated using an antagomir approach. Whereas EVN exerted anti-fibrogenic or anti-proliferative effects in activated HSC ((, ), , )) or pro-proliferative effects on injured hepatocytes ((), ()), these effects were ablated when the antagomirs were added individually (()) or collectively ((, )). In the absence of EVN, the antagomirs did not affect proliferation of CCl4-injured hepatocytes (()). Thus the effects of EVN on hepatocytes or HSC were blocked by antagonism of miRs that were preferentially expressed in EVN (as compared to EVF) and that mimicked the biological actions of EVN on HSC or hepatocytes.
Figure 8. Identification of therapeutic miRs in EVN. (a) Small RNA was purified from EVN or EVF (200 μl serum/mouse, n = 5) and subjected to miRnome miR profiling. The heat map shows those miRs that were differentially expressed in EVN versus Exo F (left panel). Ranking of differential miR expression from highest (“H”) to lowest (“L) identified miR-34c-3p, -151-3p, -483-5p, -532-5p and -687 as the most highly differentially expressed in EVN versus EVF (right panel). Differential array showed expression of these five miRs to be unchanged between serum EVs from oil-treated animals and EVN (data not shown). (b) RT-PCR validation of reduced expression of selected miRs in EVF versus EVN. (c) Suppression of mRNA expression of activation or fibrogenesis markers in D9 primary mouse HSC after 24 h transfection of the cells with 100 nM mimics of miR-34c-3p, -151-3p, -483-5p, -532-5p or -687. “Ctrl” is negative control mimic. (d) RT-PCR detection of Smad3 in HSC or AML12 hepatocytes treated for 24 h with EVN (8 μg/ml) or transfected with miR-532-5p (100 nM). P3 HSC were transfected with either (e) individual antagomirs to miRs-34c-3p, -151-3p, -483-5p, -532-5p or -687 (100 nM) or (f) a mixture containing all five antagomirs (100 nM each) for 24 h, serum-starved for 12 h, treated with EVN (8 μg/ml) for 24 h and then assayed for (e) cell proliferation by CyQUANT® assay or (f) gene expression by qRT-PCR. (g) CyQUANT® cell proliferation assay for AML12 hepatocytes after being transfected with a mixture of all five antagomirs (100nM each) for 24 h, serum-starved for 12 h, and treated with or without 20 mM CCl4 for 48 h in the presence or absence of EVN (8 μg/ml) for the last 24 h. n = 3 independent experiments performed in triplicate. P < 0.01 versus control.
Human serum EVs attenuate fibrogenic gene expression in human HSC
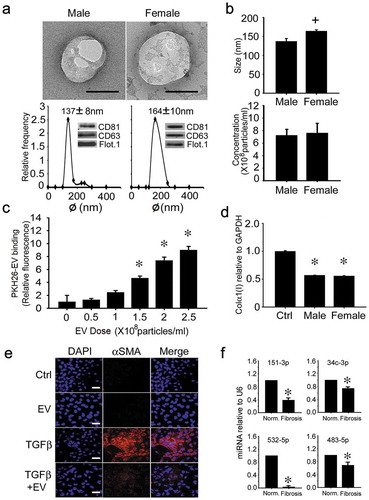
Male and female human blood donors did not significantly differ in age (males: 25.5 ± 1.5 years; females: 25.5 ± 2.3 years). Based on TEM, NTA and Western blot, human serum EVs were highly comparable within and between gender, except that those from females were ~20% larger than those from males (164 ± 10 nm vs. 137 ± 8 nm) ((, )). Even so, as determined by individual analysis of each human serum sample, there was clear heterogeneity in their respective mean size, size-range and relative expression of CD81 or flotillin (Supplemental (–)). Pooled human serum EVs demonstrated dose-dependent binding to LX-2 cells (a human activated HSC line) (()) and caused an attenuation of collagen α1(I) mRNA expression (()) and of TGF-β-induced αSMA protein production (()). The biological activity of serum EVs from individual donors was again heterogeneous, with EVs from 7 of 9 females or from 6 of 9 males causing an inhibition of collagen expression of between 25% and 60% (Supplemental (, )). Although miR-687 was not detected, expression of miR-34c-3p, -151-3p, -483-5p and -532-5p was higher in EVs from serum of normal human subjects as compared to those of patients with F3/4 fibrosis (()).
Figure 9. Characterization and actions of human serum EVs. (a) TEM (upper panel; scale bar: 100 nm; representative images are shown), NTA (lower panel) and Western blot analysis (lower panel, inset) of EVs purified by ultracentrifugation of serum of normal male or female human blood. Data are from individual donors and are representative of each gender (n = 9 males, 9 females). (b) size (upper panel) and concentration (lower panel) of serum EVs from individual NTA analysis of all human donors (n = 18), with data grouped by gender (n = 9 per group). (c) LX-2 cells were incubated for 16 h with purified human serum EVs that had been labelled with PKH26 dye. Cell-associated PKH26 was determined by fluorescence measurement in cell lysates. n = 3 independent experiments performed in triplicate. *P < 0.01 versus no treatment. (d) suppression of collagen α1(I) gene expression in LX-2 cells that were serum-starved for 24 h and then treated for 24 h with 5 × 108 particles/ml pooled human serum EVs from healthy male or female donors. n = 9 independent experiments performed in triplicate. *P < 0.01 versus control. (e) αSMA IHC in LX-2 cells that were serum-starved for 24 h and then incubated in the presence of TGFβ for 36 h, with or without human serum EVs for the last 24 h. Images are representative of five independent experiments. Scale bar: 20 μm (f) RT-PCR of serum EV miRs in control subjects versus F3/F4 fibrosis patients (*P < 0.05).
Discussion
There is growing interest in understanding the function of EVs in hepatic intercellular communication and their potential as carriers of biomarkers to aid disease diagnosis and prognosis [Citation7,Citation27]. While analysis of cargo molecules in circulating EVs has begun to identify potential candidate markers for disease assessment in chronic liver diseases [Citation9–Citation14], our findings for EVN provide an important distinction from these prior studies in that they show that EVs from healthy individuals have inherent therapeutic properties that drive anti-fibrotic outcomes in in vitro or in vivo models. EV treatment resulted in gender-independent therapeutic effects in three strains of mice using in vivo fibrogenesis (short-term CCl4) or fibrosis models (chronic CCl4 or TAA). HSC were found to be among the principal cells involved, with EVN demonstrating greater binding to activated HSC than to quiescent HSC and exerting suppressive effects on activated HSC function, including fibrogenesis and proliferation. As compared to mouse serum EVs, human serum EVs exhibited comparable biophysical and biochemical properties (TEM, NTA, Western blot) and, importantly, they similarly attenuated fibrogenic- and activation-associated gene expression in human HSC and also contained the same differentially expressed miRs between normal and fibrotic subjects. This suggests the existence of evolutionarily conserved actions of serum EVs in regulating HSC, and possibly of some of the miR mediators involved, although analysis of other hepatic fibrosis patient populations (e.g. non-alcoholic steatohepatitis, alcoholic liver disease, hepatitis C, etc.) in addition to the fibrotic HBV patients studied here will be necessary to fully answer this question. Additionally, we showed that hepatocytes are also a target of EVN and that, following injury in vitro or in vivo, EVN bind more strongly to hepatocytes, reduce cellular damage and drive proliferation. The therapeutic effects of EVN in vivo were further manifested by a decreased incidence in proinflammatory cells in the liver and by normalization of the levels of circulating proinflammatory cytokines. Importantly, as assessed in normal mice, EVN administration did not cause hepatic inflammation or histopathology, nor did it result in overt alterations in cytokines or inflammatory mediators in the circulation or liver. The absence of pathophysiological sequelae in normal animals treated with EVN attests to the intrinsic safety of EVN as a therapeutic tool, consistent with the origin of EVN from normal mice. We are not aware of prior studies demonstrating that serum EVs from healthy individuals are intrinsically anti-fibrotic, although plasma exosomes protected the myocardium against ischemia-reperfusion injury [Citation28], serum-derived EVs were therapeutic in a mouse model of acute hind limb ischemia that was functionally associated with angiogenic, matrigenic, mitogenic and immunomodulatory components of their payload [Citation29], and serum exosomes were proposed to play a role in maintenance of homeostasis under normal conditions [Citation30]. Collectively these findings suggest that serum EVs have rich therapeutic value for treating diverse pathologies. In the future, it will be of interest to establish if shared EV populations or cargo components exist between EVN and (i) mesenchymal stem cell-derived EVs which are therapeutic for fibrosis, ischemia-reperfusion injury or drug-induced injury in the liver [Citation31–Citation36]; (ii) exosomes from quiescent HSC which suppress CCN2 expression and fibrogenic signalling in activated HSC [Citation16,Citation17,Citation19]; or (iii) hepatocyte-derived exosomes which promote hepatocyte proliferation in models of hepatectomy or hepatic ischemia-reperfusion injury [Citation37] and which we recently showed to reverse fibrogenesis in HSC and ethanol-induced damage in hepatocytes downstream of integrin- or heparin-dependent binding interactions [Citation38]. This latter possibility is supported by the fact that expression of ASPGR1, which is produced by hepatocytes, was diminished in EVF as compared to EVN, suggesting a possible loss of hepatocyte-derived EVs from the circulation during fibrosis. The possible regulation of hepatocyte EVs release by serum EV as well as their potential functional interactions in the liver will be interesting areas for future study. Previous studies have shown that liver-associated miRs or proteins are present in serum or plasma EVs under normal conditions [Citation30] and/or during various hepatic pathologies (reviewed in [Citation39]), the latter of which may facilitate disease assessment.
In this study, several miRs were found to be expressed at relatively higher levels in serum EVs from experimental or clinical fibrosis as compared to their normal controls. In the mouse system, these miRs mimicked the beneficial effects of EVN on HSC or hepatocytes in vitro. Importantly, the use of antagomirs provided powerful evidence that these miRs contribute functionally to the therapeutic actions of EVN. The identification of miR-483-5p is of particular interest because, through the targeting of platelet-derived growth factor and tissue inhibitor of metalloprotease-2, miR-483-5p and -3p (non-EV) were shown to cooperatively suppress HSC activation or CCl4-induced liver fibrosis [Citation40]. While the miR-34 family was reported to promote liver fibrosis [Citation41], our data suggests a suppressive effect on fibrogenesis and activation in HSC and this is supported by its ability to diminish epithelial to mesenchymal transition in the kidney [Citation42]. Tissue levels of miR-151-3p were reported to be suppressed in experimental pulmonary fibrosis [Citation43], suggestive of its possible role as a fibrosis suppressor. While miR-151 in liver fibrosis has not been reported, high levels of miR-151 are associated with patient survival in choriocarcinoma [Citation44] but, conversely, with increased tumorigenicity and metastasis in hepatocellular carcinoma [Citation45]. MiR-687 has been studied in acute kidney injury [Citation46], but its role in fibrosis has not been reported. Computer algorithms do not predict that CCN2, αSMA or Col 1α1 to be direct targets of the identified miRs, suggesting that they are likely secondary downstream targets. In the case of miR-532-5p, one such primary target is predicted to be SMAD3, an important mediator of TGF-β action, which we showed to be down-regulated by either EVN or miR-532-5p in either hepatocytes or HSC. During fibrosing injury, SMAD3 is phosphorylated and associates with SMAD4 after TGF-β binds and activates its cognate receptors. SMAD3/4 then migrates into the nucleus where it modulates gene transcription [Citation47–Citation50], including that of CCN2 [Citation50]. We showed that SMAD3 binding to the CCN2 promoter is required for TGF-β-stimulated CCN2 gene transcription in HSC and that αSMA or collagen 1α(I) are produced downstream of CCN2 [Citation22,Citation51]. Furthermore, the TGF-β-SMAD3 pathway in hepatocytes is activated during CCl4 injury and is associated with decreased cell proliferation and apoptosis [Citation49,Citation52,Citation53]. We thus speculate that, in the context of chronic fibrosing liver injury, the suppression of SMAD3 by miR-532 in EVN results in a dual therapeutic action by favouring hepatocyte proliferation and by decreasing HSC fibrogenesis.
While the study of cellular and EV miRs in liver fibrosis clearly requires further study, we speculate that the EV miRs identified may constitute part of an intrinsic anti-fibrotic regimen (comprising additional but as yet unidentified miRs, as well as other EV cargo molecules such as mRNAs, proteins, etc.) that limits aberrant fibrogenesis at sites of injury in otherwise healthy individuals. Since unbiased array analysis was an effective means of identifying EV miRs with novel anti-fibrotic actions in the liver, similar strategies could be used in the future to identify a more complete slate of anti-fibrotic components in EVN that might also include mRNAs or proteins. Given the complexities of the many EV subpopulations in serum and the multitude of producer and target cells, future studies will also need to address whether EVN represents a mixture of EV subpopulations from a variety of donor cells (one of which may be hepatocytes as discussed above), each possibly carrying distinct anti-fibrotic moieties.
Recent studies have begun to identify an EV hepatic cellular communication network which contributes to hepatic pathophysiology via EV-mediated functional reprogramming in target cells [Citation7]. Transmission of hepatitis C virus between hepatocytes is mediated by liberation of virus-containing EVs from infected cells [Citation54] while EV communication between hepatocarcinoma cells drives tumour progression and chemoresistance [Citation55,Citation56]. EVs obtained from hepatocytes during non-alcoholic fatty liver disease, non-alcoholic steatohepatitis or alcoholic hepatitis drive activation or chemotaxis in macrophages as well as features of angiogenesis or activation in, respectively, endothelial cells or HSC [Citation26,Citation57–Citation61]. Migratory responses and expression of activation markers in HSC are also stimulated by exosomes from, respectively, endothelial cells or other activated HSC [Citation62,Citation63]. In these prior studies, the observed responses were attributed to the uptake into the recipient cells and subsequent action of a variety of EV cargo molecules including CXCL10 [Citation58] S1P [Citation59,Citation64], TRAIL [Citation32], vanin-1 [Citation60], CD40L [Citation57], SK1/2[Citation59,Citation64], CCN2[Citation25] or miRs [Citation26]. An important feature of many of these findings is that the pathophysiology-related pathways were driven by EVs derived from cells that were injured (e.g. hepatocytes) or that were phenotypically or functionally altered in an injury-related manner (e.g. activated HSC) whereas the therapeutic EVs identified in our study were from healthy individuals. Indeed, we showed that, unlike EVN, EVF do not suppress proliferation or expression of fibrosis- or activation-related genes in HSC. Since inflammatory pathways in the liver have been linked to EV communication between hepatocytes and monocytes [Citation65], an interesting area for future study will be to determine whether the therapeutic actions of EVN in fibrosis are in part attributable to the action of EV miRs and/or other EV constituents on pro-inflammatory gene expression and regulation of inflammatory cell function.
In conclusion, serum EVs from healthy donors have anti-fibrogenic properties that are attributable, at least in part, to specific miR constituents that have therapeutic actions in activated HSC or injured hepatocytes. Further studies of these miRs, other therapeutic molecules in the EV payload and EV homing will help to establish mechanisms of EV-mediated innate fibrosis resistance and the utility of leveraging these components for developing new anti-fibrotic strategies.
Supplemental_Figures.zip
Download Zip (1.4 MB)Acknowledgments
We thank Cindy McAllister (Biomorphology Core) for help with histology and David Dunaway and Victoria Valquerez (Flow Cytometry Core) for help with cell sorting and NTA. We also thank Dr. Angela Blissett at the Ohio State University Campus Microscopy and Imaging Facility for help with TEM sample preparation and imaging.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Rowe IA. Lessons from epidemiology: the burden of liver disease. Dig Dis. 2017 May 3;35(4):1–19.
- Rockey DC, Bell PD, Hill JA. Fibrosis–a common pathway to organ injury and failure. N Engl J Med. 2015 Mar 19;372(12):1138–1149.
- Yoon YJ, Friedman SL, Lee YA. Antifibrotic therapies: where are we now? Semin Liver Dis. 2016 Feb;36(1):87–98.
- Rockey DC. Translating an understanding of the pathogenesis of hepatic fibrosis to novel therapies. Clin Gastroenterol Hepatol. 2013 Mar;11(3):224–31 e1-5.
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013 Feb 18;200(4):373–383.
- Thery C. Exosomes: secreted vesicles and intercellular communications. F1000 Biol Rep. 2011;3:15.
- Hirsova P, Ibrahim SH, Verma VK, et al. Extracellular vesicles in liver pathobiology: small particles with big impact. Hepatology. 2016 Dec;64(6):2219–2233.
- Teixeira JH, Silva AM, Almeida MI, et al. Circulating extracellular vesicles: their role in tissue repair and regeneration. Transfus Apher Sci. 2016 Aug;55(1):53–61.
- Bala S, Petrasek J, Mundkur S, et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology. 2012 Nov;56(5):1946–1957.
- Eguchi A, Lazaro RG, Wang J, et al. Extracellular vesicles released by hepatocytes from gastric infusion model of alcoholic liver disease contain a microRNA barcode that can be detected in blood. Hepatology. 2017 Feb;65(2):475–490.
- Povero D, Eguchi A, Li H, et al. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PloS One. 2014;9(12):e113651.
- Sohn W, Kim J, Kang SH, et al. Serum exosomal microRNAs as novel biomarkers for hepatocellular carcinoma. Exp Mol Med. 2015 Sep;18(47):e184.
- Fornari F, Ferracin M, Trere D, et al. Circulating microRNAs, miR-939, miR-595, miR-519d and miR-494, identify cirrhotic patients with HCC. PloS One. 2015;10(10):e0141448.
- Murakami Y, Toyoda H, Tanaka M, et al. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PloS One. 2011;6(1):e16081.
- Lorincz AM, Timar CI, Marosvari KA, et al. Effect of storage on physical and functional properties of extracellular vesicles derived from neutrophilic granulocytes. J Extracell Vesicles. 2014;3:25465.
- Chen L, Chen R, Velazquez VM, et al. Fibrogenic signaling is suppressed in hepatic stellate cells through targeting of connective tissue growth factor (CCN2) by cellular or exosomal microRNA-199a-5p. Am J Pathol. 2016 Nov;186(11):2921–2933.
- Chen L, Charrier A, Zhou Y, et al. Epigenetic regulation of connective tissue growth factor by microRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology. 2014 Mar;59(3):1118–1129.
- Chen L, Brigstock DR. Integrins and heparan sulfate proteoglycans on hepatic stellate cells (HSC) are novel receptors for HSC-derived exosomes. FEBS Lett. 2016 Dec;590(23):4263–4274.
- Chen L, Chen R, Kemper S, et al. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: role of exosomes in horizontal transfer of Twist1. Am J Physiol Gastrointest Liver Physiol. 2015 Sep 15;309(6):G491–9.
- Chen L, Brigstock DR. Cellular or exosomal microRNAs associated with ccn gene expression in liver fibrosis. Methods Mol Biol. 2017;1489:465–480.
- Charrier A, Chen R, Kemper S, et al. Regulation of pancreatic inflammation by connective tissue growth factor (CTGF/CCN2). Immunol. 2014;141:564–576.
- Chen L, Charrier AL, Leask A, et al. Ethanol-stimulated differentiated functions of human or mouse hepatic stellate cells are mediated by connective tissue growth factor. J Hepatol. 2011 Aug;55(2):399–406.
- Ghosh A, Sil PC. A 43-kDa protein from the leaves of the herb Cajanus indicus L modulates chloroform induced hepatotoxicity in vitro. Drug Chem Toxicol. 2006;29(4):397–413.
- Charrier AL, Brigstock DR. Connective tissue growth factor production by activated pancreatic stellate cells in mouse alcoholic chronic pancreatitis. Lab Invest. 2010 Aug;90(8):1179–1188.
- Huang G, Brigstock DR. Regulation of hepatic stellate cells by connective tissue growth factor. Front Biosci. 2012;17:2495–2507.
- Povero D, Panera N, Eguchi A, et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-gamma. Cell Mol Gastroenterol Hepatol. 2015 Nov 1;01(6):646–663 e4.
- Maji S, Matsuda A, Yan IK, et al. Extracellular vesicles in liver diseases. Am J Physiol Gastrointest Liver Physiol. 2017 Mar 1;312(3):G194–G200.
- Vicencio JM, Yellon DM, Sivaraman V, et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J Am Coll Cardiol. 2015 Apr 21;65(15):1525–1536.
- Cavallari C, Ranghino A, Tapparo M, et al. Serum-derived extracellular vesicles (EVs) impact on vascular remodeling and prevent muscle damage in acute hind limb ischemia. Sci Rep. 2017 Aug 15;7(1):8180.
- Zhou X, Jiao Z, Ji J, et al. Characterization of mouse serum exosomal small RNA content: the origins and their roles in modulating inflammatory response. Oncotarget. 2017 Jun 27;8(26):42712–42727.
- Li T, Yan Y, Wang B, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013 Mar 15;22(6):845–854.
- Haga H, Yan IK, Borelli D, et al. Extracellular vesicles from bone marrow derived mesenchymal stem cells protect against murine hepatic ischemia-reperfusion injury. Liver Transpl. 2017;23(6):791–803.
- Qu Y, Zhang Q, Cai X, et al. Exosomes derived from miR-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J Cell Mol Med. 2017;21(10):2491–2502.
- Yan Y, Jiang W, Tan Y, et al. hucMSC exosome-derived GPX1 is required for the recovery of hepatic oxidant injury. Mol Therapy. 2017 Feb 1;25(2):465–479.
- Nong K, Wang W, Niu X, et al. Hepatoprotective effect of exosomes from human-induced pluripotent stem cell-derived mesenchymal stromal cells against hepatic ischemia-reperfusion injury in rats. Cytotherapy. 2016 Dec;18(12):1548–1559.
- Tan CY, Lai RC, Wong W, et al. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Res Ther. 2014 Jun 10;5(3):76.
- Nojima H, Freeman CM, Schuster RM, et al. Hepatocyte exosomes mediate liver repair and regeneration via sphingosine-1-phosphate. J Hepatol. 2016 Jan;64(1):60–68.
- Chen L, Chen R, Kemper S, et al. Pathways of production and delivery of hepatocyte exosomes. J Cell Commun Signal. 2018;12(1):343–357.
- Szabo G, Momen-Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat Reviews Gastroenterol Hepatol. 2017 Aug;14(8):455–466.
- Li F, Ma N, Zhao R, et al. Overexpression of miR-483-5p/3p cooperate to inhibit mouse liver fibrosis by suppressing the TGF-beta stimulated HSCs in transgenic mice. J Cell Mol Med. 2014 Jun;18(6):966–974.
- Li WQ, Chen C, Xu MD, et al. The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats. Febs J. 2011 May;278(9):1522–1532.
- Morizane R, Fujii S, Monkawa T, et al. miR-34c attenuates epithelial-mesenchymal transition and kidney fibrosis with ureteral obstruction. Sci Rep. 2014;4:4578.
- Ji X, Wu B, Fan J, et al. The anti-fibrotic effects and mechanisms of microRNA-486-5p in pulmonary fibrosis. Sci Rep. 2015;5:14131.
- McNally ME, Collins A, Wojcik SE, et al. Concomitant dysregulation of microRNAs miR-151-3p and miR-126 correlates with improved survival in resected cholangiocarcinoma. HPB. 2013 Apr;15(4):260–264.
- Chen CL, Wu JC, Chen GY, et al. Baculovirus-mediated miRNA regulation to suppress hepatocellular carcinoma tumorigenicity and metastasis. Mol Therapy. 2015 Jan;23(1):79–88.
- Zhou P, Chen Z, Zou Y, et al. Roles of non-coding RNAs in acute kidney injury. Kidney Blood Press Res. 2016;41(6):757–769.
- Fabregat I, Moreno-Caceres J, Sanchez A, et al. TGF-beta signalling and liver disease. Febs J. 2016 Jun;283(12):2219–2232.
- Gressner AM, Weiskirchen R, Breitkopf K, et al. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002 Apr;1(7):d793–807.
- Xu F, Liu C, Zhou D, et al. TGF-beta/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016 Mar;64(3):157–167.
- Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004 May;18(7):816–827.
- Leask A, Chen S, Pala D, et al. Regulation of CCN2 mRNA expression and promoter activity in activated hepatic stellate cells. J Cell Commun Signal. 2008 Jun;2(1–2):49–56.
- Niu L, Cui X, Qi Y, et al. Involvement of TGF-beta1/Smad3 signaling in carbon tetrachloride-induced acute liver injury in mice. PloS One. 2016;11(5):e0156090.
- Sato M, Flanders KC, Matsubara T, et al. Smad3 deficiency counteracts hepatocyte apoptosis and portal fibrogenesis induced by bile duct ligation. J Liver. 2014;3:145.
- Ramakrishnaiah V, Thumann C, Fofana I, et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci USA. 2013 Aug 6;110(32):13109–13113.
- Kogure T, Lin WL, Yan IK, et al. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011 Oct;54(4):1237–1248.
- Takahashi K, Yan IK, Kogure T, et al. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio. 2014;4:458–467.
- Hirsova P, Ibrahim SH, Krishnan A, et al. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. 2016;150(4):956–967.
- Ibrahim SH, Hirsova P, Tomita K, et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology. 2016 Mar;63(3):731–744.
- Kakazu E, Mauer AS, Yin M, et al. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J Lipid Res. 2016 Feb;57(2):233–245.
- Povero D, Eguchi A, Niesman IR, et al. Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci Signal. 2013 Oct 8;6(296):ra88.
- Verma VK, Li H, Wang R, et al. Alcohol stimulates macrophage activation through caspase-dependent hepatocyte derived release of CD40L containing extracellular vesicles. J Hepatol. 2016 Mar;64(3):651–660.
- Wang R, Ding Q, Yaqoob U, et al. Exosome adherence and internalization by hepatic stellate cells triggers sphingosine 1-phosphate-dependent migration. J Biol Chem. 2015 Dec 25;290(52):30684–30696.
- Charrier A, Chen R, Chen L, et al. Exosomes mediate intercellular transfer of pro-fibrogenic connective tissue growth factor (CCN2) between hepatic stellate cells, the principal fibrotic cells in the liver. Surgery. 2014 Sep;156(3):548–555.
- Xiao Y, Zhou Y, Chen Y, et al. The expression of epithelial-mesenchymal transition-related proteins in biliary epithelial cells is associated with liver fibrosis in biliary atresia. Pediatr Res. 2015 Feb;77(2):310–315.
- Momen-Heravi F, Bala S, Kodys K, et al. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific miRNA-122 and sensitize monocytes to LPS. Sci Rep. 2015 May 14;5:9991.