
ABSTRACT
Foetal calf serum (FCS) is a common supplement of cell culture medium and a known source of contaminating extracellular vesicles (EV) containing RNA. Because of a high degree of sequence similarity among homologous non-coding RNAs of mammalian species, residual FCS-RNA in culture medium may interfere in the analysis of EV-RNA released by cultured cells. Recently, doubts have been raised as to whether commonly used protocols for depletion of FCS-EV efficiently remove FCS-RNA. Moreover, technical details in FCS-EV depletion protocols are known to vary between labs, which may lead to inter-study differences in contaminating FCS-RNA levels. Here, we investigated how technical modifications of EV-depletion protocols affect the efficiency with which bovine RNAs are depleted from FCS, and determined the contribution of contaminating bovine RNA to EV-RNA purified from cell cultures. Our data show differences in depletion efficiency between and within various classes of small non-coding RNA. Importantly, we demonstrate that variations in FCS-EV depletion protocols affect both the quantity and type of residual FCS-RNAs in EV-depleted medium. By using optimised FCS-EV depletion protocols combined with methods for high-grade purification of EV the levels of contaminating bovine RNA in EV populations isolated from cell culture medium can be reduced. With illustrative datasets we also demonstrate that the abundance of a specific RNA in cell culture EV can only be determined if measured relative to background levels of this RNA in medium control samples. These data highlight the need for optimisation and validation of existing and novel FCS-EV depletion methods and urge for accurate descriptions of these methods in publications to increase experimental reproducibility.
Introduction
The characterisation of EV-enclosed RNA has gained enormous interest over the last decade, because of its involvement in cell-cell communication and potential use as biomarkers for disease [Citation1–Citation5]. For analysis of EV released by in vitro cultured cells, it is important to deplete contaminating EV present in foetal calf serum (FCS), which is a common supplement of cell culture medium [Citation6–Citation9]. Although depletion of FCS-EV may affect the phenotype and behaviour of cultured cells [Citation7,Citation8,Citation10], applying an FCS-EV depletion protocol is particularly important in studies addressing the RNA content of cell culture EV. In such studies, residual bovine small non-coding RNAs in FCS may be erroneously mapped to human or murine genomes due to high sequence homology, thereby confounding sequencing analyses of EV-RNA [Citation11,Citation12]. Additionally, the presence of homologous FCS-derived RNAs in culture medium may affect RT-qPCR-based quantification of RNA in EV released by cultured cells.
RNA in serum is not only enclosed in EV but can also be associated with other macromolecular structures, such as ribonucleoprotein particles (RNPs) and lipoprotein particles. These structures overlap in size and/or density with EV and may be co-isolated in commonly used EV isolation procedures, such as ultracentrifugation [Citation9,Citation13–Citation16]. A number of studies have addressed the removal of contaminating FCS-EV and FCS-derived RNA by ultracentrifugation and/or ultrafiltration [Citation7,Citation8,Citation10,Citation11], and doubts were raised on whether these methods allow effective depletion of extracellular RNA from bovine serum [Citation11]. Furthermore, it is known that technical details in FCS-EV depletion protocols vary between labs. It is unclear to what extent differences in FCS-EV depletion methods introduce unwanted variation in EV-RNA data.
Here, we investigated technical modifications of EV depletion protocols that affect the efficiency with which different classes of small non-coding RNAs are depleted from FCS. In illustrative experiments, we furthermore determined the extent to which residual FCS-RNA in EV-depleted medium affects analysis of the RNA content of cell culture-derived EV purified by density gradient ultracentrifugation. Our data show that variations in FCS EV-depletion protocols influence the quantity and type of residual FCS-derived RNA in culture medium. Additionally we show that accurate analysis of small non-coding RNAs in cell culture EV requires parallel assessment of residual levels of these RNAs in medium control samples.
Materials and methods
FCS-EV depletion
FCS (lot BDC-12270, Bodinco, Alkmaar, the Netherlands) was left undiluted (100%) or was diluted with Iscove’s Modified Dulbecco’s Medium (IMDM, Lonza, Verviers, Belgium) to 30% or 10% (vol/vol %) (), panel I). (Diluted) FCS was spun overnight for 15–18 h at 100,000× g in a SW28 rotor (k-factor 334.2) or SW40 rotor (k-factor 381.5) (Beckman Coulter, Brea, CA, USA). Supernatant was collected using a pipette, leaving at least 5 mL (SW28 tubes) or 1.5 mL (SW40 tubes) of fluid on top of the pellet. The pellet was resuspended in the residual fluid by pipetting up and down until a homogeneous suspension was obtained. This “pellet fraction” contained all material that was depleted from the FCS and which would normally be discarded. RNA was subsequently isolated from both the pellet and the EV-depleted supernatant.
Figure 1. (a) Schematic overview of the experimental setup. Critical methodological parameters that are compared are indicated in red. (I) Suspensions of undiluted, 3.3× and 10× diluted FCS (100%, 30%, 10% FCS) were depleted from EV by overnight ultracentrifugation (15–18 h) at 100,000× g. EV-depleted supernatant was recovered from ultracentrifugation tubes by pipetting. EV-depleted supernatant fractions were diluted 3× or 10× before RNA isolation to correct for differences in FCS concentration. RNA was additionally isolated from pellet fractions and non-depleted medium (containing 10% FCS). (II) A suspension of diluted FCS (30%) was depleted from EV by overnight ultracentrifugation (15–18 h) at 100,000× g. EV depleted medium was recovered from ultracentrifugation tubes by pipetting or decanting and diluted three times before RNA isolation. RNA was isolated from EV-depleted supernatant and from non-depleted medium (10% FCS). (III) EV-depleted supernatant containing 30% FCS was used to prepare EV-depleted medium in which HEK293T and A20 cells were cultured 20 h for EV production. Identical volumes of cell conditioned EV-depleted medium or non-conditioned EV-depleted medium were subjected to differential (ultra)centrifugation and density gradient centrifugation to purify cell-derived and/or residual bovine EV. RNA was isolated from EV-containing density fractions (1.12–1.18 g/mL). (b) Representative Bioanalyser Pico chip electropherograms of RNA isolated from non-depleted medium (I), supernatant (II), and pellet (III) fractions of EV-depleted medium. (c) Suspensions of 100%, 30%, or 10% FCS were depleted from EV by overnight ultracentrifugation (15–18 h) at 100,000× g. EV-depleted supernatant was recovered from ultracentrifugation tubes by pipetting. Supernatant fractions were diluted 3x and 10x before RNA isolation to correct for differences in FCS concentrations. The concentrations of RNA in supernatants and pellets were assessed on Bioanalyser. Total RNA quantity in the entire ultracentrifuge tube was calculated by adding up the total RNA quantities in pellet and supernatant fractions, corrected for the dilution factor before RNA isolation. Indicated is the distribution of RNA over supernatant and pellet fractions for each of the FCS dilutions. Mean ± SD of n = 3 experiments, * p < 0.05 ANOVA with Dunnett’s one-sided post-hoc test (control condition: 100% EV-depleted supernatant). (d) Equal volumes of RNA isolated from EV-depleted supernatant of different FCS dilutions (prepared as described in (c), and RNA isolated from non-depleted medium (“medium”) were used for RT-qPCR analysis of the indicated RNAs. Depletion efficiencies were calculated from the raw Cq values shown in (e) using the ΔΔCq method [Citation33] relative to non-depleted medium. Indicated are mean log2fold change values ± SD of n = 3 experiments. Statistical differences were determined by ANOVA with Tukey HSD post-hoc test. * p < 0.05. No statistical differences were found between the depletion efficiencies in different FCS dilutions. (e) Raw Cq values of the RT-qPCR data shown in (d). Low Cq values mean high abundance. Indicated are mean values ± SD of n = 3 experiments. Statistical differences were determined by ANOVA with Tukey HSD post-hoc test. * p < 0.05 No statistical differences were observed between different FCS dilutions. (f) Suspensions of 30% FCS were depleted from EV by overnight ultracentrifugation (15–18h) at 100,000× g. EV-depleted supernatants were recovered from ultracentrifugation tubes by decanting (“sup decant”) or pipetting (“sup pipette”). RNA was isolated from EV-depleted supernatants collected by decanting and pipetting (equilibrated to 10% FCS) and from non-depleted medium (containing 10% FCS). Equal volumes of RNA were used for RT-qPCR analysis of the indicated RNAs. Log2fold change levels relative to non-depleted medium were calculated as in (d). Indicated are mean values ± SD of n = 3 experiments. Statistical differences were determined by ANOVA with Tukey HSD post-hoc test, * p < 0.05.
![Figure 1. (a) Schematic overview of the experimental setup. Critical methodological parameters that are compared are indicated in red. (I) Suspensions of undiluted, 3.3× and 10× diluted FCS (100%, 30%, 10% FCS) were depleted from EV by overnight ultracentrifugation (15–18 h) at 100,000× g. EV-depleted supernatant was recovered from ultracentrifugation tubes by pipetting. EV-depleted supernatant fractions were diluted 3× or 10× before RNA isolation to correct for differences in FCS concentration. RNA was additionally isolated from pellet fractions and non-depleted medium (containing 10% FCS). (II) A suspension of diluted FCS (30%) was depleted from EV by overnight ultracentrifugation (15–18 h) at 100,000× g. EV depleted medium was recovered from ultracentrifugation tubes by pipetting or decanting and diluted three times before RNA isolation. RNA was isolated from EV-depleted supernatant and from non-depleted medium (10% FCS). (III) EV-depleted supernatant containing 30% FCS was used to prepare EV-depleted medium in which HEK293T and A20 cells were cultured 20 h for EV production. Identical volumes of cell conditioned EV-depleted medium or non-conditioned EV-depleted medium were subjected to differential (ultra)centrifugation and density gradient centrifugation to purify cell-derived and/or residual bovine EV. RNA was isolated from EV-containing density fractions (1.12–1.18 g/mL). (b) Representative Bioanalyser Pico chip electropherograms of RNA isolated from non-depleted medium (I), supernatant (II), and pellet (III) fractions of EV-depleted medium. (c) Suspensions of 100%, 30%, or 10% FCS were depleted from EV by overnight ultracentrifugation (15–18 h) at 100,000× g. EV-depleted supernatant was recovered from ultracentrifugation tubes by pipetting. Supernatant fractions were diluted 3x and 10x before RNA isolation to correct for differences in FCS concentrations. The concentrations of RNA in supernatants and pellets were assessed on Bioanalyser. Total RNA quantity in the entire ultracentrifuge tube was calculated by adding up the total RNA quantities in pellet and supernatant fractions, corrected for the dilution factor before RNA isolation. Indicated is the distribution of RNA over supernatant and pellet fractions for each of the FCS dilutions. Mean ± SD of n = 3 experiments, * p < 0.05 ANOVA with Dunnett’s one-sided post-hoc test (control condition: 100% EV-depleted supernatant). (d) Equal volumes of RNA isolated from EV-depleted supernatant of different FCS dilutions (prepared as described in (c), and RNA isolated from non-depleted medium (“medium”) were used for RT-qPCR analysis of the indicated RNAs. Depletion efficiencies were calculated from the raw Cq values shown in (e) using the ΔΔCq method [Citation33] relative to non-depleted medium. Indicated are mean log2fold change values ± SD of n = 3 experiments. Statistical differences were determined by ANOVA with Tukey HSD post-hoc test. * p < 0.05. No statistical differences were found between the depletion efficiencies in different FCS dilutions. (e) Raw Cq values of the RT-qPCR data shown in (d). Low Cq values mean high abundance. Indicated are mean values ± SD of n = 3 experiments. Statistical differences were determined by ANOVA with Tukey HSD post-hoc test. * p < 0.05 No statistical differences were observed between different FCS dilutions. (f) Suspensions of 30% FCS were depleted from EV by overnight ultracentrifugation (15–18h) at 100,000× g. EV-depleted supernatants were recovered from ultracentrifugation tubes by decanting (“sup decant”) or pipetting (“sup pipette”). RNA was isolated from EV-depleted supernatants collected by decanting and pipetting (equilibrated to 10% FCS) and from non-depleted medium (containing 10% FCS). Equal volumes of RNA were used for RT-qPCR analysis of the indicated RNAs. Log2fold change levels relative to non-depleted medium were calculated as in (d). Indicated are mean values ± SD of n = 3 experiments. Statistical differences were determined by ANOVA with Tukey HSD post-hoc test, * p < 0.05.](/cms/asset/46a523ce-2244-416a-bebe-5e7cd62b7e6b/zjev_a_1552059_f0001_oc.jpg)
For preparation of EV-depleted medium, EV-depleted supernatant (30% FCS in IMDM) was collected with a pipette (as described above), subsequently diluted to 10% FCS, and supplemented with 2 mM Ultraglutamine (Lonza, Verviers, Belgium), 100 IU/mL penicillin and 100 µg/mL streptomycin (Gibco, Paisley, UK). Where indicated, EV-depleted supernatant (30% FCS in IMDM) was not recovered by pipetting (as described above), but instead by decanting the fluid (), panel II). Non-depleted medium was supplemented with 10% non-depleted FCS and the above mentioned concentrations of ultraglutamine, penicillin, and streptomycin.
Cell culture
Human Embryonic Kidney (HEK293T) cells (adherent) and murine B-lymphoblast (A20) cells (non-adherent) were grown in IMDM supplemented with 10% FCS, 2 mM Ultraglutamine, 100 IU/mL penicillin and 100 µg/mL streptomycin. HEK293T cells were passaged twice a week using 0.05% trypsin/EDTA for cell detachment (Gibco, Paisley, UK). A20 cells were passaged twice a week by dilution in medium. For EV-production, HEK293T cells were seeded in complete medium (36–45 million cells per batch, divided over 6 T175 flasks), and incubated for 24 h to attach to the flask surface. Subsequently, the monolayer was washed in 1x PBS (Gibco, Paisley, UK), after which cells were cultured in 210 mL EV-depleted medium per batch (prepared as described above) for 20 h. A20 cells (56–79 million cells per batch, divided over two T75 flasks placed upright) were spun down, medium was removed from the pellet and cells were cultured in 70 mL EV-depleted medium for 20 h. Confluency of HEK293T cells was more than 80% at harvest. Cell viability at harvest of HEK293T and A20 cells was determined by Trypan blue exclusion, which was above 90% for all cultures. All cells were maintained at 37°C and 5% CO2 in a humidified incubator.
Isolation and fluorescent labelling of EV
EV were isolated from identical volumes of cell conditioned medium and non-conditioned control medium (), panel III) via differential centrifugation and density gradient ultracentrifugation as described previously [Citation17]. In brief, supernatants were centrifuged 2 × 200× g for 10 min, 2 × 500× g for 10 min, 1 × 10,000× g for 30 min. Subsequently, EV were pelleted by ultracentrifugation for 65 min at 100,000× g in a SW28 rotor (k-factor 334.2) (Beckman Coulter, Brea, CA). EV pellets were resuspended in 50 µl PBS + 0.2% BSA (depleted from protein aggregates by overnight ultracentrifugation at 100,000× g), mixed with 1.5 mL 2.5 M sucrose, after which a linear sucrose gradient (2.0 M–0.4 M sucrose in PBS) was layered on top of the sample. Gradients were centrifuged 15–18 h at 192,000× g in a SW40 rotor (k-factor 144.5) (Beckman Coulter, Brea, CA). EV-containing gradient fractions with densities of 1.12–1.18 g/mL were diluted 6 times in PBS + 0.2% EV-depleted BSA after which EV were pelleted at 192,000× g for 65 min in a SW40 rotor (k-factor 144.5). For EV quantification by high-resolution flow cytometry, 100,000× g EV-pellets were resuspended in 20 µl PBS + 0.2% BSA and labelled with 1.5 µl PKH67 (Sigma, St. Louis, MO) in 180 µl Diluent C per pellet. Labelled EVs were purified by sucrose density gradient centrifugation as described above prior to analysis by high-resolution flow cytometric analysis (see below).
RNA isolation
For RNA isolation directly from solutions (non-depleted medium, EV-depleted supernatants and pellet fractions from 100%/30%/10% FCS dilutions), the Qiagen miRNeasy micro Serum/Plasma protocol (Qiagen, Hilden, Germany) was used. Prior to RNA isolation, supernatant samples of 30% and 100% serum conditions were diluted 3x or 10x, respectively, to prevent potential interference of serum proteins and/or lipids with RNA isolation efficiency. Input volumes were 1.2 mL for non-depleted medium and EV-depleted FCS supernatant (with FCS concentrations equalised to 10%), and 0.2 mL for pellet fractions. Volumes of reagents (Qiazol, chloroform and 100% ethanol) that are used before binding of RNA to the miRNeasy spin columns, were adjusted to sample volumes as recommended by the manufacturer. Each sample was loaded onto one miRNeasy spin column, after which regular volumes of wash buffers were used. Elution of each sample was done in 15 µl RNAse free water. To isolate RNA from density gradient purified EV pellets, the miRNeasy micro kit was used according to the manufacturer’s instructions without modifications (Qiagen, Hilden, Germany). RNA yields and size profile were assessed using the Agilent 2100 Bioanalyzer with Pico 6000 RNA chips (Agilent Technologies, Waldbronn, Germany).
RT-qPCR and cDNA synthesis
Equal volumes of isolated RNA were used to quantify differences in specific RNA transcripts between samples. cDNA was prepared using the miScript RT2 kit (Qiagen, Hilden, Germany). Per qPCR reaction, 2 μl of 10 times diluted cDNA template was used with 100 nM primers (IDT, Leuven, Belgium) and 4 µL SYBR Green Sensimix (Bioline Reagents Ltd., UK) in an 8 µl reaction. No-RT-controls confirmed the absence of genomic DNA and non-specific amplification. Cycling conditions were 95°C for 10 min followed by 50 cycles of 95°C for 10 s, 57°C for 30 s and 72°C for 20 s. Subsequently, a melting curve analysis was performed.
All PCR reactions were performed on the Bio-Rad iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Quantification cycle (Cq) values were determined using Bio-Rad CFX software using automatic baseline settings. Thresholds were set in the linear phase of the amplification curve.
List of DNA oligo probes
The following primers were used in RT-qPCR, primers 1–7 and 16–20 were used in combination with the miScript universal reverse primer, primers 8–15 were used as pairs of forward (F) and reverse (R) primers.
High resolution flow cytometric analysis of EV
PKH67-labelled EV were quantified by high resolution flow cytometric analysis using a BD Influx flow cytometer (BD Biosciences, San Jose, CA) with an optimised configuration, as previously described [Citation17,Citation18]. In brief, we applied threshold triggering on fluorescent signals from PKH67-labelled EV. Forward scatter (FSC) was detected with a collection angle of 15–25° (reduced wide-angle (rw) FSC). Fluorescence and rw-FCS settings were calibrated using fluorescent 100- and 200-nm polystyrene beads (FluoSpheres, Invitrogen, Carlsbad, CA). Sucrose gradient fractions containing PKH67-labelled EV were diluted 142× (for A20-derived EVs) or 20× (for all other EV measurements) in PBS and vortexed just before measurement. This dilution factor was sufficient to avoid “coincidence” (multiple EV arriving at the measuring spot at the same time), thereby enabling accurate quantification of EV numbers in different conditions. Moreover, samples were measured below 10,000 events per second, which is far below the limit of the electronic pulse processing speed of the BD Influx [Citation19].
EV-TRACK
Written details on experimental procedures have been submitted to the EV-TRACK knowledgebase (EV-TRACK ID: EV180039) [Citation20].
Results and discussion
A range of different protocols for serum EV depletion are currently in use. To deplete EV from FCS, it was previously recommended to dilute serum prior to overnight ultracentrifugation. This reduces viscosity and increases EV depletion efficiency [Citation6,Citation21]. However, the effects of serum dilution on the depletion efficiency of different classes of (EV-associated) serum RNA have not been assessed in detail. We addressed this point by overnight ultracentrifugation of 100%, 30%, and 10% dilutions of FCS at 100,000× g, followed by RNA isolation from supernatants and pellets. For reference, RNA was isolated from medium prepared with FCS not depleted of EV (“non-depleted medium”) (), panel I). As expected, overnight centrifugation at 100,000× g led to accumulation of RNA in the pellets, while the amount of RNA in the EV-depleted supernatant was reduced compared to non-depleted medium ()). In line with earlier reports, we observed that not all FCS-derived RNA was depleted by overnight ultracentrifugation [Citation11]. We determined the distribution of FCS-RNA over EV-depleted supernatant and pellet fractions for each of the dilutions (with FCS concentrations equalised to 10% (v/v) before RNA isolation), and observed that FCS-derived RNA pelleted more efficiently in diluted FCS suspensions ()). This finding seems to contradict a previous study of Shelke et al., who reported that FCS dilution does not increase RNA depletion efficiency [Citation7]. However, Shelke et al. took a two-step approach by first preparing EV-depleted medium from FCS that was pre-diluted or not and then assessing the amount of residual RNA in pellets after a second centrifugation step. In our study, we directly quantified how much RNA was pelleted versus how much remained in the supernatant in the first centrifugation step (the actual EV depletion step). Data from these experiments indicated that dilution of serum increased the pelleting efficiency of RNA-containing structures.
Extracellular RNA in FCS can be associated with various macromolecular structures, such as EV, lipoprotein particles, and RNP [Citation12–Citation16,Citation22]. Alternatively, these RNAs may form stable extracellular RNA dimers [Citation23], which likely differ in sedimentation efficiency during ultracentrifugation. Moreover, the different RNA carriers in serum may be preferentially associated with different classes of small non-coding RNA. By using RT-qPCR analysis, we therefore assessed the efficiency with which various types of small non-coding RNA could be depleted from FCS. We selected example miRNAs (miR-451-5p, miR-122-5p, miR-148a-3p, miR-423-5p) that have been detected in multiple studies on EV-associated RNA, but which were also identified as potential FCS-derived contaminants by Wei et al [Citation11]. MiR-451-5p and miR-122-5p are mainly expressed in red blood cells and liver cells, respectively [Citation24], whereas miR-148a-3p (an immune-related miRNA [Citation25]) and miR-1246 (a fragment of snRNA U2 [Citation26]) are expressed in a broader range of cell types. Furthermore, we selected a number of other miRNAs which have been abundantly detected in serum and plasma (miR-16, miR-21, miR-93, and miR-142-3p) [Citation5,Citation27]. In addition to these miRNAs, we selected several cytoplasmic and nuclear RNAs (5S rRNA, U1 snRNA, Y-RNA, 7SL) that have been abundantly detected in multiple studies on EV-associated RNA [Citation28,Citation29]. The bovine and human sequences of these RNAs also show 100% homology. We compared the RNA content of EV-depleted supernatant versus non-depleted medium and observed striking differences in the efficiency with which each of the RNA types could be depleted from FCS ()). Various members of the Y-RNA family, 7SL, and miR-142 were efficiently depleted, with a 100-fold reduction compared to non-depleted medium. Intermediate depletion efficiencies (~30-fold reduction) were achieved for miR-16, 5S rRNA, U1 snRNA and miR-451. MiR-93 and miR-21 were depleted with low efficiency (~4-fold reduction). In contrast, no significant reduction in miR-1246, miR-148a, miR-122 and miR-423 was observed after EV depletion. We verified whether the calculated depletion efficiency was biased by differences in the abundance of transcripts before depletion. However, we found no correlation between the depletion efficiency depicted in ) and the abundance (Cq values) of transcripts before depletion ()). For example, 7SL was less abundant than 5S before depletion, but was depleted with higher efficiency than 5S. Additionally, miR-451 and miR-1246 were equally abundant before depletion, but miR-451 was depleted more efficiently than miR-1246. This suggests that the observed differences in depletion efficiencies are intrinsic characteristics of the studied example RNAs (and/or the macromolecular structures they are associated with), and do not depend on their abundance before depletion. These data illustrate that the FCS-RNA depletion by ultracentrifugation is variable between and even within different classes of non-coding RNAs. MiRNAs that could not be depleted by ultracentrifugation (e.g. miR-122 and miR-423, see also [Citation11]) may be predominantly associated with specific lipoprotein particles, or to subsets of RNP and EV that do not efficiently sediment during the centrifugation step. In contrast, the RNAs that efficiently sedimented (e.g. Y-RNA and 7SL) may be predominantly associated with larger EV and/or RNP.
Although FCS dilution showed a clear effect on pelleting efficiency of total FCS-RNA ()), the depletion efficiencies of the example RNAs, as tested by RT-qPCR, were similar for all dilutions ()). This suggests that serum contains additional types of complexed RNAs with sedimentation properties that are yet different from those observed in our data.
During ultracentrifugation, FCS-containing medium will concentrate into a protein-rich, high density fluid in which EV may become trapped. We therefore tested whether the method used for recovery of EV-depleted supernatant after ultracentrifugation affected the amount of contaminant FCS-RNA carried over to the FCS-supplemented culture medium. Although exact steps in EV depletion protocols are often not reported in publications, a widely used method to collect EV-depleted supernatant is to pour off supernatant after centrifugation (decanting method). We compared this method with a more careful approach in which supernatant was pipetted off (), panel II). Strikingly, the depletion efficiency of several of the example non-coding RNAs was strongly reduced when the decanting method was used instead of the pipetting method ()). Quantities of residual Y-RNA and 7SL in decanted supernatants even reversed to values similar to those observed in non-depleted medium ()). However, other non-coding RNAs, such as 5S, U1, miR-16, and miR-451, were depleted with similar efficiencies using the two methods. As expected, no differences were observed between the two recovery methods for miRNAs that could not be depleted by ultracentrifugation (miR-93, miR-1246, miR-148a, miR-122 and miR-423). These data indicate that small technical details in the serum EV depletion procedure strongly affect the efficiency with which different types of bovine RNAs can be depleted. Subsequently, this will have an effect on the scale and composition of the contaminating pool of bovine RNA in serum-supplemented culture medium. This urges the need to accurately describe these depletion procedures in publications to improve the reproducibility of EV-RNA studies.
The data above indicate that not all RNAs can be efficiently depleted from bovine serum. We aimed to assess the extent to which residual bovine RNAs affect the analysis of cell culture EV-associated RNA after high-grade EV purification by density gradient centrifugation. First, we analysed the number of bovine EV remaining in EV-depleted FCS after using the most optimal protocol for EV depletion (overnight ultracentrifugation of 30% FCS, collect EV-depleted supernatant by pipetting method). Ultracentrifuged FCS or non-depleted FCS was used to prepare culture medium, after which we applied differential centrifugation followed by density gradient ultracentrifugation to purify EV from both media (), panel III). Using high resolution flow cytometry [Citation17,Citation18], we observed that the EV depletion procedure led to 90% reduction in the number of bovine EV in culture medium ()). Next, we tested to what extent residual bovine (EV-associated) RNA in this EV-depleted medium influenced the analysis of EV-associated RNA released by cells cultured in this medium. We used two cell lines that differed in the quantity of released EV, because the number of EV per volume of cell culture medium will affect the relative contribution of FCS-derived RNA to the total amount of EV-RNA. A20 murine B cell lymphoma cells released six times more EV than the HEK293T human embryonic kidney cells, as determined by high-resolution flow-cytometry-based EV quantification ()). Cell culture EV fractions contained 2.5 times (HEK293T) and 112 times (A20) more RNA than residual bovine EV present in equal volumes of non-conditioned medium (,)). Next, we used RT-qPCR to assess whether specific EV-associated RNAs released by the cells in culture could be detected above background levels of these RNAs in EV fractions of non-conditioned medium. We selected RNAs from ,) with sequences that were identical in all tested species (human, mouse, bovine), allowing quantification with the same primer pairs. Due to low EV numbers in HEK293T cultures, three times larger volumes of HEK293T-conditioned medium compared to A20-condition medium were used to be able to detect the EV-associated RNAs in all conditions. Since equal volumes of control non-conditioned medium were analysed in parallel, this explains the lower Cq values for medium EV in HEK293T experiments ()) compared to A20 experiments ()). For A20 cell cultures, the quantified levels of all tested RNAs in cell-derived EV by far exceeded and significantly differed from the background levels in corresponding medium control samples ()). Although miR-451 was considered a red blood cell specific miRNA, our data indicate that EV from A20 B-cell lymphoma cells contained miR-451. Indeed, it was previously demonstrated that B-cell lymphomas [Citation30] and primary B-cells [Citation31] can express miR-451. The liver-specific miR-122 was not expressed by the A20 cells and was therefore not detected in A20-EV. In cultures of HEK293T cells, which produce low numbers of EV, the differences in Cq values in cell-derived EV versus medium-EV were substantially smaller ()). For 7SL, Y3, 5S, and U1, which are abundantly present in EV, as well as for the less abundant miR-16, miR-148a, miR-93 and miR-423, we observed significantly increased levels in cell culture EV compared to background levels in medium controls. Other EV-associated RNAs in the HEK293T cell cultures, such as Y1, miR-21, miR-142-3p, miR-1246, and miR-122 could not be reliably detected above background levels. Some of these RNAs (such as miR-122 and miR-451) were simply not expressed by the HEK293T cells. Detection of other RNAs (such as Y1) above background levels was hampered by the fact that bovine EV carrying the same RNA type contaminated the EV fraction of the cell-conditioned medium. In the experiments shown in , it cannot be excluded that part of the non-depleted bovine RNA is taken up by cultured cells [Citation11,Citation12]. However, little is known about the extent to which this may occur and on whether such RNAs are degraded or released back into the medium associated with cell-derived EV. More importantly, these data create awareness that the Cq values for each of the tested RNAs not only differ in the cell-derived EV but also in the medium control samples. This implies that the abundance of a specific RNA in cell culture EV can only be determined if measured relative to background levels of this RNA in medium control samples.
Figure 2. (a) EV-depleted medium was prepared using supernatant of ultracentrifuged solutions of 30% FCS. EV from equal volumes of EV-depleted and non-depleted medium were isolated by ultracentrifugation, fluorescently labelled with PKH67, and further purified by sucrose density gradient centrifugation. High-resolution flow cytometry was used to determine the number of PKH67-labeled EV. Indicated are the number of fluorescent EV detected in 30 s in pools of 1.12–1.18 g/mL sucrose fractions. (b) HEK293T and A20 cells were grown 20h in EV-depleted medium (prepared from 30% EV-depleted FCS). EV were isolated from equal volumes of HEK293T and A20 cell-conditioned supernatant, labelled with PKH67, and quantified using high-resolution flow cytometry. Indicated are the number of fluorescent EV in 30 s in pools of 1.12–1.18 g/mL sucrose fractions, normalised to the number of cultured cells to allow direct comparison of EV-release by both cell types. (c, d) HEK293T cells or A20 cells were cultured in EV-depleted medium (prepared from 30%-depleted FCS). EV were isolated from equal volumes of HEK293T (c) or A20 (d) conditioned medium and non-conditioned medium using differential centrifugation followed by sucrose density gradient centrifugation. RNA was isolated from EV pelleted from 1.12–1.18 g/mL sucrose fractions. Indicated are mean RNA concentrations ± SD of n = 2 (HEK293T) or n = 3 (A20) replicates. Statistical differences were determined by independent samples t-test, * p < 0.05. (E, F) EV-RNA isolated from A20 (e) and HEK293T (f) conditioned medium and equal volumes of non-conditioned EV-depleted medium (prepared as in d) were used for analysis of the indicated RNAs using RT-qPCR. Indicated are mean Cq values ± SD of n = 3 replicates. Statistical differences were determined by independent samples t-test, * p < 0.05.
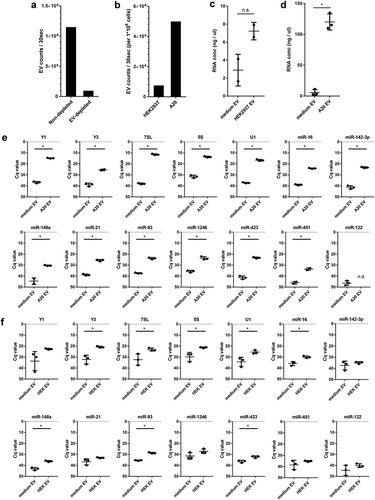
Combined, our data indicate that RNAs that can be depleted from FCS with high efficiency may still contaminate cell culture derived EV fractions after purification. For example, bovine Y1 is depleted with high efficiency ()), but residual Y1 can still be detected in purified EV fractions of medium control samples. This contaminating bovine Y1 can interfere with detection of human/murine Y1 derived from low-EV producing cell lines such as HEK293T. In contrast, miR-423 is inefficiently depleted from FCS ()), but is hardly detectable in purified EV fractions of medium control samples (,)). Parallel processing and analysis of medium control samples is therefore essential to accurately assess the RNA content of cell culture EV. Currently used sources of EV-depleted FCS include commercially available “exosome-depleted FBS” and FCS depleted from EV by individual researchers using different ultracentrifugation protocols or alternative methods [Citation10]. For each of these protocols, it is necessary to identify technical parameters affecting the depletion efficiency of RNA from FCS.
Conclusion and recommendations
We show that optimisation of FCS-EV depletion protocols and high-grade EV purification can reduce the levels of contaminating bovine RNA in in vitro cell culture experiments. To deplete RNA from FCS, we recommend to dilute the FCS to 30% and to ultracentrifuge for at least 15h at 100,000 × g. EV-depleted medium should then be carefully removed by pipetting from the top of the tube downward, leaving approximately 10% of the supernatant volume on top of the pellet. During purification of EV from cells cultured in EV-depleted medium, it is advisable to process an equal volume of EV-depleted medium in parallel to conditioned medium so that background levels of RNA in RT-qPCR can be assessed. Overall, the reliability with which conserved RNAs in cell culture EV can be detected over background levels in culture medium depends on several different factors: 1) the quantity of EV released by the cultured cells; 2) the abundance of this RNA in these EV; and 3) the amount of this RNA that remains present in FCS-EV-depleted culture medium. Our data further underscore the importance of developing and implementing guidelines [Citation32] for dealing with FCS-derived background RNA in EV-RNA research, and urge for accurate reporting of technical details of depletion procedures to increase experimental reproducibility [Citation20].
Acknowledgments
We thank Dr. G. J. A. Arkesteijn for assistance on the high-resolution flow cytometric measurements.
Disclosure statement
The authors reported no potential conflict of interest.
Additional information
Funding
Related Research Data
References
- van der Grein SG, Nolte-’t Hoen ENM. “Small Talk” in the innate immune system via RNA-containing extracellular vesicles. Front Immunol. [Internet]. 2014 [cited 2014 Nov 11];5:1–10. Available from: http://journal.frontiersin.org/journal/10.3389/fimmu.2014.00542/full
- Redzic JS, Balaj L, van der Vos KE, et al. Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol. 2014;28:14–23. [Internet].
- Cheng L, Doecke JD, Sharples RA, et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol Psychiatry. [Internet]. 2014 [cited 2015 May 8];1–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25349172
- Martinez BV, Dhahbi JM, Nunez Lopez YO, et al. Circulating small non coding RNA signature in head and neck squamous cell carcinoma. Oncotarget. 2015;6:19246–19263. [Internet]. Available from: http://www.oncotarget.com/fulltext/4266
- Buschmann D, Kirchner B, Hermann S, et al. Evaluation of serum extracellular vesicle isolation methods for profiling miRNAs by next-generation sequencing. J Extracell Vesicles. 2018;7. [Internet]. doi: 10.1080/20013078.2018.1481321.
- Théry C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants. Curr Protoc Cell Biol. 2006;3:1–29. [Internet]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18228490
- Shelke GV, Lässer C, Gho YS, et al. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J Extracell Vesicles. 2014;3:1–8.
- Eitan E, Zhang S, Witwer KW, et al. Extracellular vesicle-depleted fetal bovine and human sera have reduced capacity to support cell growth. J Extracell Vesicles. 2015;4:1–10.
- Mateescu B, Kowal EJK, van Balkom BWM, et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA – an ISEV position paper. J Extracell Vesicles. 2017;6:1286095. [Internet]. Available from: https://www.tandfonline.com/doi/full/10.1080/20013078.2017.1286095
- Kornilov R, Puhka M, Mannerström B, et al. Efficient ultrafiltration-based protocol to deplete extracellular vesicles from fetal bovine serum. J Extracell Vesicles. [Internet]. 2018;7:1422674. Available from: https://www.tandfonline.com/doi/full/10.1080/20013078.2017.1422674.
- Wei Z, Batagov AO, Carter DRF, et al. Fetal bovine serum RNA interferes with the cell culture derived extracellular RNA. Sci Rep. [Internet]. 2016;6:31175. Available from: http://www.nature.com/articles/srep31175.
- Tosar JP, Cayota A, Eitan E, et al. Ribonucleic artefacts: are some extracellular RNA discoveries driven by cell culture medium components? J Extracell Vesicles. [Internet]. 2017;6:1–10.
- Vickers KC, Palmisano BT, Shoucri BM, et al. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. [Internet]. 2011;13:423–433. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21423178%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3074610.
- Driedonks TAP, van der Grein SG, Ariyurek Y, et al. Immune stimuli shape the small non‑coding transcriptome of extracellular vesicles released by dendritic cells. Cell Mol Life Sci. 2018. [Internet]. doi: 10.1007/s00018-018-2842-8
- Arroyo JD, Chevillet JR, Kroh EM, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A. 2011;108:5003–5008. [Internet]. Available from: http://www.pnas.org/content/108/12/5003
- Turchinovich A, Weiz L, Langheinz A, et al. Characterization of extracellular circulating microRNA. Nucleic Acids Res. [Internet]. 2011 [cited 2014 Jul 13];39:7223–7233. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3167594&tool=pmcentrez&rendertype=abstract
- van der Vlist EJ, Nolte-'t Hoen ENM, Stoorvogel W, et al. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat Protoc. 2012;7:1311–1326. [Internet]. Available from: http://www.nature.com/doifinder/10.1038/nprot.2012.065
- Nolte-’t Hoen ENM, van der Vlist EJ, Aalberts M, et al. Quantitative and qualitative flow cytometric analysis of nanosized cell-derived membrane vesicles. Nanomedicine. [Internet]. 2012 [cited 2014 Nov 5];8:712–720. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22024193
- Groot Kormelink T, Arkesteijn GJA, Nauwelaers FA, et al. Prerequisites for the analysis and sorting of extracellular vesicle subpopulations by high-resolution flow cytometry. Cytom Part A. 2016;89:135–147.
- Van Deun J, Mestdagh P, Agostinis P, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14:228–232. [Internet]. Available from: http://www.nature.com/doifinder/10.1038/nmeth.4185
- Momen-Heravi F, Balaj L, Alian S, et al. Impact of biofluid viscosity on size and sedimentation efficiency of the isolated microvesicles. Front Physiol. 2012;3(MAY):1–6.
- Tosar JP, Gambaro F, Sanguinetti J, et al. Assessment of small RNA sorting into different extracellular fractions revealed by high-throughput sequencing of breast cell lines. Nucleic Acids Res. [Internet]. 2015;1–16. Available from: http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gkv432
- Tosar JP, Gámbaro F, Darré L, et al. Dimerization confers increased stability to nucleases in 5 halves from glycine and glutamic acid tRNAs. Nucleic Acids Res. 2018;1:1–13.
- Kent OA, McCall MN, Cornish TC, et al. Lessons from miR-143/145: the importance of cell-type localization of miRNAs. Nucleic Acids Res. 2014;42:7528–7538.
- Gonzalez-Martin A, Adams BD, Lai M, et al. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat Immunol. 2016;17:433–440.
- Baraniskin A, Nöpel-Dünnebacke S, Ahrens M, et al. Circulating U2 small nuclear RNA fragments as a novel diagnostic biomarker for pancreatic and colorectal adenocarcinoma. Int J Cancer. 2013;132:48–57.
- Williams Z, Ben-Dov IZ, Elias R, et al. Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc Natl Acad Sci. [Internet]. 2013 [cited 2015 Mar 17];110:4255–4260. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1214046110
- Nolte-'t Hoen ENM, Buermans HPJ, Waasdorp M, et al. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res. [Internet]. 2012 [cited 2014 Oct 21];40:9272–9285. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3467056&tool=pmcentrez&rendertype=abstract
- Bellingham SA, Coleman BM, Hill AF. Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res. [Internet]. 2012;40:10937–10949. Available from: http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gks832.
- Montes-Moreno S, Martinez N, Sanchez-Espiridión B, et al. MiRNA expression in diffuse large B-cell lymphoma treated with chemoimmunotherapy. Blood. 2011;118:1034–1040.
- Juzenas S, Venkatesh G, Hübenthal M, et al. A comprehensive, cell specific microRNA catalogue of human peripheral blood. Nucleic Acids Res. 2017;45:9290–9301.
- Lötvall J. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for extracellular vesicles. J Extracell Vesicles. Vol. 1. 2014. p. 1–6.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408.