
ABSTRACT
Extracellular vesicles (EVs) are secreted membrane vesicles, which play complex physiological and pathological functions in intercellular communication. Recently, we isolated natural killer (NK) cell-derived EVs (NK-EVs) from ex vivo expansion of NK cell cultures. The isolated NK-EVs contained cytotoxic proteins and several activated caspases, and they induced apoptosis in target cells. In this report, the protein levels of cytotoxic proteins from NK-EV isolates were analysed by ELISA. The mean values of perforin (PFN, 550 ng/mL), granzyme A (GzmA, 185 ng/mL), granzyme B (GzmB, 23.4 ng/mL), granulysin (GNLY, 56 ng/mL), and FasL (2.5 ng/mL) were obtained from >60 isolations using dot plots. The correlation between cytotoxicity and cytotoxic protein levels was examined by linear regression. PFN, GzmA, GzmB, GNLY all had a positive, moderate correlation with cytotoxicity, suggesting that there is not a single cytotoxic protein dominantly involved in killing and that all of these proteins may contribute to cytotoxicity. To further explore the possible killing mechanisms, cells were treated with NK-EVs, proteins extracted and lysates assessed by Western blotting. The levels of Gzm A substrates, SET and HMG2, were diminished in targeted cells, indicating that GzmA may induce a caspase-independent death pathway. Also, cytochrome C was released from mitochondria, a central hallmark of caspase-dependent death pathways. In addition, several ER-associated proteins were altered, suggesting that NK-EVs may induce ER stress resulting in cell death. Our results indicate that multiple killing mechanisms are activated by NK-derived EVs, including caspase-independent and -dependent cell death pathways, which can mediate cytotoxicity against cancer cells.
Abbreviations: NK: natural killer cells; aNK: activated NK cells; EV: extracellular vesicles; ER: endoplasmic reticulum; ALL: acute lymphoblastic leukaemia; FBS: foetal bovine serum. GzmA: granzyme A; GzmB: granzyme B; GNLY: granulysin; PFN: perforin
Introduction
Extracellular vesicles (EVs) are biological nanovesicles that act as natural delivery vessels, shuttling active molecules (e.g. mRNA, miRNA, proteins) to recipient cells and influencing cellular functions [Citation1,Citation2]. EVs represent an important mode of intercellular communication and play key roles in many physiological and pathological processes [Citation3–Citation5]. Accumulating data indicate that the contents, size and membrane composition of EVs are highly heterogeneous and dynamic, and depend on the cellular source, physiological status and environmental conditions [Citation6–Citation8]. In general, EVs can be broadly classified into three main classes: 1) microvesicles that are produced by outward budding and fission of the plasma membrane; 2) exosomes that are formed within the endosomal network and are released upon fusion of multivesicular bodies with the plasma membrane; and 3) apoptotic bodies that are released as blebs from cells undergoing apoptosis. Lower organism, such as pathogenic yeast Cryptococcus neoformans, secretes microvesicles which can activate brain endothelial cells to facilitate its traversal across the blood-brain barrier and thus promote its brain invasion [Citation9]. Bacteria and parasites are also able to secrete EVs, which are formed by outward bulging of the outer membrane. Thus, EVs are universal intercellular communicators, existing in higher and lower organisms.
Proteomic studies of EVs released by cultures of primary cells, cell lines, and tissues, or of EVs isolated from biofluids have yielded extensive catalogues of the protein abundance in different types of EVs. Usually, EVs contain high levels of cytoskeletal (e.g. actin, fibronectin), cytosolic (e.g. GAPDH, LDH, 14–3-3 protein), heat shock (e.g. HSP90, HSP70), antigen presentation (i.e. MHC-I, -II), and plasma membrane proteins (e.g. tetraspanins CD9, CD63, CD81), as well as proteins involved in vesicle trafficking (e.g. ESCRT, Tsg101, Alix) [Citation10]. These proteins may represent basic constituents commonly observed in all EVs. On the other hand, EVs derived from different cell types may exhibit a unique protein profile, which may be required for specific biological functions. For example, EVs from tumour cells contain tumour antigens, exosomes from dendritic cells express MHC-II-peptide complexes, and platelet-derived exosomes contain coagulation factors [Citation11]. In addition, EVs contain a repertoire of mRNA, miRNA or other RNA species distinct from those of their cell of origin [Citation12,Citation13]. As such, EVs can shuttle functional nucleic acids between cells and regulate the recipient cell at a post-transcriptional level [Citation13]. This process represents a novel mode of gene regulation.
One feature of EVs is that their composition varies as determined by different physiological or pathological conditions. The same cell type may secrete different subgroups of vesicles (e.g. exosomes or microvesicles) depending on the cell topography (e.g. from the apical or basolateral sides) [Citation14], environmental factors (e.g. oxygen tension) or activating stimulus (e.g. apoptosis or autophagy) [Citation15]. In addition, the protein contents of the same EV subgroups are regulated by external stimuli [Citation16]. Some studies have reported changes in the glycosylation pattern of EVs in pathological conditions such as ovarian cancer [Citation17]. Thus, the complexity and dynamic compositions of EVs point towards the unique properties that are required for their versatile roles in biological systems.
Human natural killer (NK) cells can secrete EVs that contain typical NK markers (e.g. CD56) and killer proteins (e.g. perforin, granzyme A & B, granulysin and FasL) [Citation18,Citation19]. We have recently developed a protocol to isolate large quantities of NK-derived EVs from ex vivo expansion cultures of activated NK cells, and the isolated NK-EV fractions were cytotoxic against several cancer types [Citation19]. Interestingly, the isolated NK-EVs contained the cytotoxic proteins perforin (PFN), granzyme A (GzmA), granzyme B (GzmB) and granulysin (GNLY), which originated from the NK cells. It is well known that activated human NK cells and cytotoxic T lymphocytes release these cytotoxic proteins as the major mechanism for their cytotoxicity [Citation20–Citation23]. As expected, we showed that activation of caspase −3, −7 and −9 was detected in cancer cells incubated with NK-EVs, and caspase inhibitors (−2, −3, −6, −8, −9, −10, −12) blocked NK-EV-induced cytotoxicity, suggesting that NK-EVs activate caspase pathways in target cells [Citation19]. The ability to isolate highly cytotoxic NK-EVs on a large scale may lead to new clinical applications [Citation24,Citation25]. In this scenario, one needs to determine whether the yield of EVs and levels of the cytotoxic proteins in separate isolations vary depending on sources and environmental factors. In addition, as NK-EVs contain various cytotoxic proteins that are involved in several cell death pathways, it is important to determine the killing mechanisms used by NK-EVs among this array of cytotoxic proteins. Our results indicate that multiple killing mechanisms are triggered through these cytotoxic proteins, resulting in cytotoxicity of target cells.
Materials and methods
Reagents and materials
Polyethylene glycol-8000 was purchased from Sigma-Aldrich Chem. Co (Saint Louis, MO). Interleukin-2 was obtained from PeproTech (Rocky Hill, NJ). Protein concentration was determined by the Bradford assay (Bio-Rad Laboratories, Inc., Hercules, CA). The G-Rex cell culture device was purchased from Wilson Wolf Manufacturing Corporation (New Brighton, MN). MitoTracker® Green FM (#9074S) was purchased from Cell Signalling Technology.
Isolation of activated NK-EVs from ex vivo NK cell culture
Here, 30 mL of blood was drawn from healthy donors under a protocol approved by the IRB at Children’s Hospital Los Angeles (Los Angeles, CA). Peripheral blood mononuclear cells (PBMC) were isolated by density separation using Histopaque-1077 (Sigma, cat.#10771), and T-cells were then absorbed using a human CD3 positive selection kit (STEMCELLTM, cat.#18051). K562 Clone 9.mbIL21 cells (clinical-grade master cell bank designated CJLCKT64.86.41BBL.CD19. mbIL21) were used as artificial antigen presenting cells (aAPC) for NK cell propagation and activation. These aAPCs express a membrane-bound variant of IL-21 [Citation26]. The aAPCs were γ-irradiated (100 Gy) and then suspended in RPMI-1640 and 10% foetal bovine serum (FBS) supplemented with 50 IU/mL recombinant human IL-2 (PeproTech). On day 0, PBMC from normal donors were incubated with the γ-irradiated-aAPC at a 1:1 ratio and cell concentration of 1 × 106 in the G-Rex culture device (Wilson Wolf Corp. New Brighton, NM). After 7 days of co-culture, cells were counted, and new γ-irradiated aAPC were added (total cell:aAPC ratio 1:0.5). Cells were grown for a total of 14 days, at which time they were phenotyped by flow cytometry, demonstrating that >95% of the cells in the cultures were NK cells (CD56+/CD16+/CD3ˉ). At day 19, the culture medium was replaced with exosome-free FBS and the culture supernatant was harvested 48 hr later and filtered with 0.8-μm pore size membranes (cat. # A080A047A; Advantec, Inc). An equal volume of 50% sterile PEG8000 was added to the filtrate to precipitate the EVs derived from the activated NK cells at 4°C overnight, followed by centrifugation at 10,000 xg for 20 min to pellet the vesicles and overnight dialysis at 4°C with PBS-5% glycerol and a 100 KDa cut-off dialysis bag (Spectrum Labs, Com; part #131420).
ELISA
ELISA assays were performed with commercial kits according to the individual manufacturer’s instructions. The following human ELISA kits were used: perforin (U-CyTech Biosciences, CT391A), granzyme-A (Boster Biosciences, EK1162), granzyme-B (U-CyTech Biosciences, CT211A), granulysin (Biolegend, 438007), and human FasL (RayBiotech, ELH-FAS1-1). Aliquots of NK-EV isolates were added to the reaction mixture into the 96-well strips from the kits. After 2 hr, the reactions were stopped, developed and read at 450 nm by spectrophotometry. All kits provide standard proteins. The amount of cytotoxic proteins was determined based on the standard curve from each kit.
Western blotting analysis
The CHLA255 (neuroblastoma), SubB15 (acute lymphoblastic leukemia, ATCC CRL-1929) and NALM6 (a B cell precursor leukemia cell line, ATCC CRL-3273) cells were grown in RPMI 1640 medium supplemented with 10% heat-inactivated foetal bovine serum, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin until log phase. Cell lysates of neuroblastoma (CHLA255), leukaemia cells (SupB15 or NAML-6) were treated with different amounts of NK-EVs as indicated in the figure legend. For each treatment, 30 µg of cell lysate proteins were loaded per lane and separated by SDS-PAGE, and proteins were electro-transferred to PVDF membranes (Millipore). The blots were probed with antibodies to HMG2 (Cell Signalling Technology, #14163, 1:500) or SET (Santa Cruz Biotech, sc-133138, 1:500) followed by incubation with anti-mouse-HRP secondary antibody conjugates (1:5,000 dilution). Proteins were detected with a chemiluminescence substrate (cat. #34080) from ThermoFisher Scientific, Inc. (Waltham, MA). ER Stress Antibody Sampler Kit was purchased from Cell Signalling Technology (#9956S). Protein size markers were purchased from Bioland Scientific, LLC. (cat. # PM01-01).
Cytotoxicity of NK-EVs
The cytotoxicity assay was performed using the Lunar Cell Counter (Logos Biosystems, Inc., Annandale, VA). SupB15 or CHLA255 (104 cells) were seeded into Costar 96-well plates (Corning) in 0.1 mL RPMI-1640 with 50 IU/mL IL-2 and 2% FBS, and 40 μg of isolated NK-EVs were added to triplicates. After 24 hr, the acridine orange/propidium iodide (AO/PI) dual fluorescence staining method was used to examine total (green, AO) and dead (red, PI) cells. Total, live and dead cell number, concentration, percentage of viability/death, cell size and distribution were all displayed by the Lunar Cell Counter. The built-in software provided data analyses, histograms and image storage. The ratio of dead cells to total cells was calculated. PBS was used in place of the NK-EVs as a negative control.
Immunofluorescence microscopy
Samples for immunofluorescence microscopy were prepared as follows. Neuroblastoma CHLA255 cells were plated onto glass coverslips (22 mm, square) that had been previously coated with type I collagen from rat tails (Upstate, 5–10 μg/cm2) and placed in an 8-well square culture system (Nalgene Nunc). Approximately 5 × 104 cells were seeded onto each coverslip 24 hr prior to the experiment. The mounted cells were prewashed four times with PBS, then fixed with 2% formaldehyde/PBS (v:v) for 30 min at room temperature. After three additional washes with PBS, non-specific binding was blocked with 5% milk/PBS for 30 min and cells were then incubated overnight with an antibody to cytochrome C (7H8.2C12; Biolegend; cat.#: 612503) and the MitoTracker FM dye at 4°C. The coverslips were then washed four times with PBS followed by 1% BSA/PBS. Anti-mouse IgG Rhodamine conjugate (1:100 dilution) was added into each well for 1 hr at 4°C. Another three washes were applied then a drop of Vectashield mounting solution containing DAPI (Vector Laboratories; H-120) was used to seal the coverslips onto slides. Samples were examined under a fluorescence microscope at the Congressman Dixon Cellular Imaging Core Facility of Children’s Hospital Los Angeles.
Cytochrome C release assay
Cytochrome C was examined by the Cytochrome C Releasing Apoptosis Assay Kit (Abcam, cat #65,311). Briefly, ALL SupB15 cells were grown to 1 × 106 cells/mL (viability ~95%). Then, 50 mL culture was used as a unit per experimental condition, as suggested by the manufacturer. The cultures were treated with (2 mg NK-EV/106 cells) for 18 hrs. After treatments (viability ~50%), cells were collected by centrifugation at 600 × g for 5 min at 4°C and washed for twice. In parallel, no NK-EV treatment was used as the control. Cells were homogenized and isolated as cytosolic and mitochondrial extraction by employing the reagents from the kit. A standard Western blot procedure was performed and probed with monoclonal mouse anti-cytochrome C antibody. Cytochrome C signals from Western blots (n = 3) were quantified by ImageJ program and subjected to statistical analysis.
Statistical analysis
Data from ELISA assays or Image J quantitation were entered into Microsoft Excel spreadsheets and converted automatically into statistical packages. Scatter plots were used to compare the protein concentrations with the cytotoxicity from CHLA255 and SupB15 cells, and R-squared values were calculated to determine linear regressions. Dot plots were analysed by the software GraphPad version 6.0 (). Standard derivation was calculated for Image J quantitation.
Figure 1. Expression of cytotoxic proteins from different batches of NK-EV isolates.
NK-EVs were isolated from PBMC using the standard protocol described in the Materials and Methods, prepared from >30 health donors. The levels of cytotoxic proteins were assayed using commercial ELISA kits for human perforin (U-CyTech Biosciences, CT391A), human granzyme-A (Boster Biosciences, EK1162), human granzyme-B (U-CyTech Biosciences, CT211A), granulysin (Biolegend, 438,007), and human FasL (RayBiotech, ELH-FAS1-1). The buffer contains non-ionic detergent to lyse the EVs. Thus, the quantitative measurements were the total proteins in our isolated NK EVs. All concentrations were calculated based on the standard curves from the individual kits. Dot Plots showed the concentration of NK-EVs versus EVs isolated from control cell lines, which included CHLA255 (neuroblastoma), SupB15 (acute lymphoblastic leukemia), D54MG (glioma), MCF7 (breast carcinoma), DU145 (prostate), Hela (cervical cancer), and HepG2 (hepatoma), were used as the controls. These non-NK-EV preparations have been described previously [Citation18]. Red circles indicate the protein level of the cytotoxic proteins derived from the EV preparation of a NK cell line, NK92. Y-axis indicates ng of a given cytotoxic protein per mL of NK-EV preparation. Note that the protein concentration scales on the Y-axis significantly differ among the cytotoxic proteins.
![Figure 1. Expression of cytotoxic proteins from different batches of NK-EV isolates.NK-EVs were isolated from PBMC using the standard protocol described in the Materials and Methods, prepared from >30 health donors. The levels of cytotoxic proteins were assayed using commercial ELISA kits for human perforin (U-CyTech Biosciences, CT391A), human granzyme-A (Boster Biosciences, EK1162), human granzyme-B (U-CyTech Biosciences, CT211A), granulysin (Biolegend, 438,007), and human FasL (RayBiotech, ELH-FAS1-1). The buffer contains non-ionic detergent to lyse the EVs. Thus, the quantitative measurements were the total proteins in our isolated NK EVs. All concentrations were calculated based on the standard curves from the individual kits. Dot Plots showed the concentration of NK-EVs versus EVs isolated from control cell lines, which included CHLA255 (neuroblastoma), SupB15 (acute lymphoblastic leukemia), D54MG (glioma), MCF7 (breast carcinoma), DU145 (prostate), Hela (cervical cancer), and HepG2 (hepatoma), were used as the controls. These non-NK-EV preparations have been described previously [Citation18]. Red circles indicate the protein level of the cytotoxic proteins derived from the EV preparation of a NK cell line, NK92. Y-axis indicates ng of a given cytotoxic protein per mL of NK-EV preparation. Note that the protein concentration scales on the Y-axis significantly differ among the cytotoxic proteins.](/cms/asset/ee4779d4-7ec0-4182-950f-04ce4f567f29/zjev_a_1588538_f0001_oc.jpg)
Results
Content of cytotoxic proteins from different NK-EV isolates
Large-scale isolation of NK-EVs from ex vivo expansion of NK cell cultures were produced using our previously developed methods [Citation19]. Protein levels were quantified by ELISA in >60 preparations. Non-NK cell lines or inactivated NK cell cultures (day-14, or no IL-2) were used as controls. The mean values for each preparation and each cytotoxic protein are shown in as dot plots. The EV preparations had very high PFN levels with a mean value of ~550 ng/mL and reached more than 1000 ng/mL in some NK-EV isolates. The protein levels of GzmA were also high with a mean value of ~185 ng/mL. In contrast, the levels of GzmB and GNLY were only moderate (~23.4 ng/mL and ~56 ng/mL, respectively). The amount of FasL in EV preparations was the lowest among the cytotoxic proteins (2.48 ng/mL). EVs were also prepared from the NK92 cell line, the only currently available commercial NK cell line. As shown in (red circle), the protein levels of the cytotoxic proteins in NK92-derived EVs were mostly lower than those from EVs obtained from ex vivo expansion culture, with the exception of GzmA (230 ng/mL) and FasL (6.4 ng/mL). Thus, the levels of the cytotoxic proteins from NK-EVs differ from each other and from batch-to-batch.
Correlation between expression level of cytotoxic proteins from different NK-EV isolates
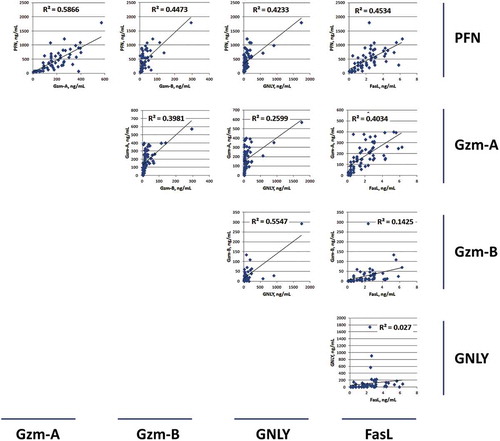
To understand the relationship between the individual content levels among these cytotoxic proteins, scatter plots were performed to compare the levels of PFN, GzmA, GzmB, GNLY and FasL between each other (). The highest R-square value was 0.5866 between PFN and GzmA, suggesting a possible coordinated function between these two proteins. The lowest R-square value was observed between GNLY and FasL (R2 = 0.027). Thus, there appears to be no functional relationship between these two proteins.
Figure 2. Correlation between levels of cytotoxic proteins from different NK-EV isolates.
The expression levels of individual cytotoxic proteins were cross-compared to each other to determine their R-square values.
Cytotoxic effects of NK-EVs on tumour cells
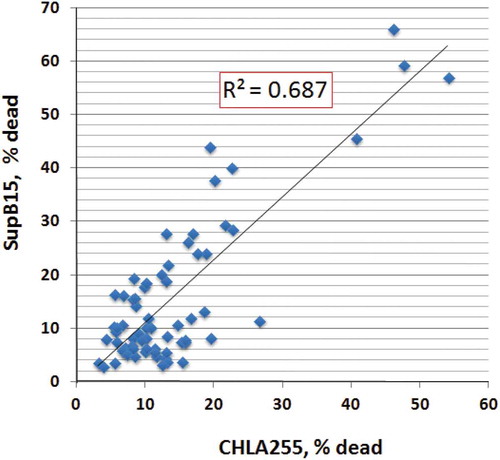
NK-EVs induce cytotoxicity in cancer cells of hematologic origin and in solid tumours [Citation19]. In this study, we selected two cancer cell lines, SupB15 (ALL, acute lymphoblastic leukemia) and CHLA255 (neuroblastoma), to determine if the isolated NK-EVs were functional. SupB15 cells are in suspension while CHLA255 cells are adherent to the culture plate. After 24 h incubation with NK-EVs, the percentage of dead cells from SupB15 or CHLA255 was counted. Scatter plots were used to compare the cytotoxicity of NK-EVs towards CHLA255 and SupB15 (). Linear regression showed that the R-square value was 0.687, suggesting that NK-EVs have similar cytotoxic activity towards these two target cells.
Figure 3. Cytotoxic activities of NK-EVs against solid tumors or tumor cell suspensions.
Cytotoxicity was determined based on the Acridine orange/Propidium iodide fluorescence assay: 104 cells (SupB15 or CHLA255) from log phase cell culture were transferred to 96-well plates in the presence of isolated EVs (40 μg) in a final volume of 100 μL. The suspensions were incubated at 37°C for 24 hr, and the percentage of dead cells was calculated by the LUNA cell counter. Linear regression line of cytotoxicity between CHLA255 and SupB15 was determined by the scatter plot.
Relation between cytotoxic proteins and cytotoxicity on neuroblastoma CHLA255 and ALL supB15
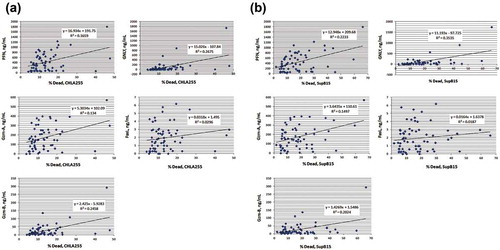
To determine the relation between cytotoxicity and individual cytotoxic proteins, scatter plots were used to determine the R-square values for each protein (). While there was a positive correlation between each cytotoxic protein and cytotoxicity, the R-square values were moderate, ranging from 0.15 to 0.35. These results suggest that there is not one single cytotoxic protein that is responsible for cytotoxicity, and that PFN, GzmA, GzmB and GNLY may all contribute to the cytotoxic function of NK-EVs towards these targets. In addition, the R-square value of FasL was very low for the CHLA255 and SupB15 cell lines, suggesting that FasL may play little, if any, role in the cytotoxicity to those cancer cells. The R-square values are summarized in .
Table 1. Summary of R-squared values for cytotoxic proteins against solid tumors and tumor cell suspensions.
Figure 4. Correlation between cytotoxic proteins and cytotoxicity against neuroblastoma CHLA255 and ALL SupB15.
The levels of the cytotoxic proteins and cytotoxicity of EV isolates were compared using scatter plots. R-squared values were determined using Excel software. The individual cytotoxic proteins versus cytotoxicity of CHLA255 (A) and SupB15 (B) from each EV isolate were plotted.
NK-EVs induce degradation of the SET and HMG2 proteins in target cells
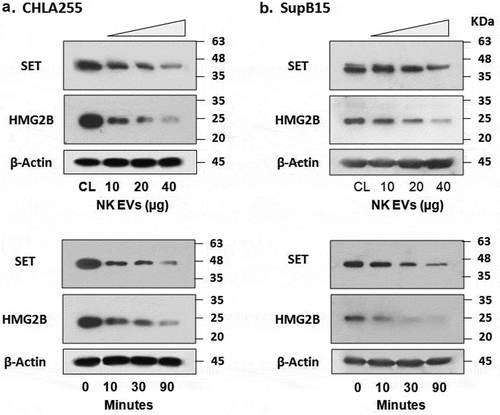
The SET and HMG2 proteins are GzmA substrates during programmed cell death [Citation27]. To test the role of GzmA contained in isolated NK-EVs, we used Western blots to detect these proteins (HMG2 and SET) in target cells (). The immunoblots were probed with an antibody to HMG2 first, and the same blots were stripped and re-probed with an antibody to SET (also known as 12PP2A). In CHLA255 cell lysates ()), SET (~45 KDa) decreased during incubation with increasing amounts of NK-EVs (upper panel), an effect that was time-dependent (lower panel). A similar effect was observed in SupB15 cell lysates, except that the protein level of HMG2B in SupB15 was less abundant than that in CHLA255, and the time course of HMG2B degradation in the presence of NK-EVs was more rapid ()). The Western-blot signals were quantified by the ImageJ program (Supplement Figure S5). Thus, titration and time-point studies showed that in the presence of NK-EVs, the degradation of SET and HMG2B can be observed in both CHLA255 and SupB15 cells. These results indicate that NK-EVs, most likely via GzmA, can cleave SET and HMG2 to promote cell death in a caspase-independent death pathway.
Figure 5. NK-EVs cleave granzyme A substrates SET and HMG2 proteins in the target cells.
Neuroblastoma CHLA255 (A) or ALL SupB15 (B) cells were treated with different amounts of NK-EVs (upper panel) or harvested at different time-points after treatment (lower panel) as indicated. Untreated cells were used as controls (CL). After incubation, cells were harvested and lysed in SDS-PAGE sample buffer. Each lane contains 30-μg cell lysate proteins as determined by the Orbit® protein assay (Life Technologies). Antibodies against SET (Santa Cruz Biotech, sc-133,138) were used to probe the blots. The same blot was stripped and re-probed with anti-HMG2 antibody (Cell Signalling Technology, #14,163). β-Actin was used as the loading control. Western blot signals were qualified by the program ImageJ from at least three independent experiments (n ≥ 3). The plots with the standard derivation were shown in Supplementary Figure S5.
NK-EVs induce the release of cytochrome C from mitochondria in neuroblastoma cells
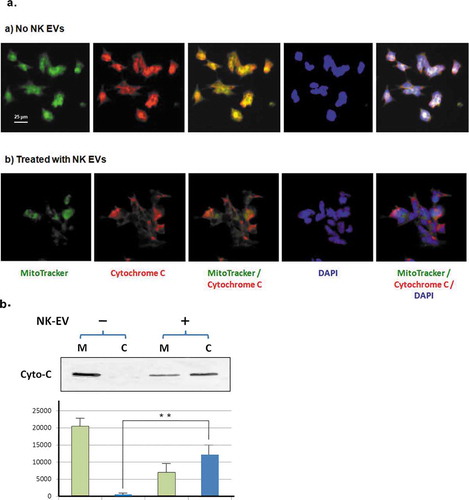
Release of cytochrome C from damaged mitochondria occurs in apoptotic cells and is often observed in GzmB-treated cells [Citation28]. To demonstrate the impact of NK-EVs on mitochondria, we first used immunofluorescence microscopy to examine the localization of cytochrome C with and without NK-EV treatment. Neuroblastoma cell CHLA255 was used, because they stick onto the slide well. As shown in ), the green MitoTracker dye was used to stain mitochondria and cytochrome C was localized by a rhodamine-conjugated antibody. The nucleus was visualized using DAPI nuclear DNA staining. In the absence of NK-EVs, the cytochrome C co-localized with the MitoTracker dye in neuroblastoma cells. With NK-EV treatment, however, the cytochrome C signals no longer co-localized with the MitoTracker dye, suggesting that damage to the mitochondria occurred in the treated neuroblastoma cells, presumably via apoptosis. In parallel, we used biochemical approach to extract mitochondria from ALL SupB15 cells, a suspension cell culture. In the absence of NK-EV treatment, cytochrome C was essentially detected in mitochondria fraction only. With the NK-EV treatment, a substantial amount of cytochrome C was released to the cytosolic fraction ()), consistent with the results of immunofluorescence images ()). Taken together, NK-EVs trigger the release of cytochrome C from the mitochondria into cytosol in the targeted cells.
Figure 6. NK-EVs induce release of cytochrome C from mitochondria in the target cells.
(a) CHLA255 cells were treated without (upper panel) or with NK-EVs (lower panel) for 12 hr and prepared for immunofluorescence microscopic studies, as described in the Methods and Material section. After extensive washes, the slides were probed with the MitoTracker dye (green), a Rhodamine conjugate of an antibody against cytochrome C (red). The slide was then sealed with mounting solution containing DAPI (blue).
(b)Upper panel: mitochondrial (M) and cytosolic (C) fractions were isolated by the Cytochrome C Release Apoptotic kit in the absence (-) or presence (+) of isolated NK-EVs, as described in Methods and Materials. Each sample of the mitochondrial or cytosolic fractions was derived from ~5x107 SupB15 cells and loaded on a 14% SDS-PAGE. The cytochrome C bands (Cyto-C) were shown on the Western blot. Lower panel: quantitative analysis of cytochrome C signals were measured by the program ImageJ from three independent experiments (n = 3). Mitochondrial fractions and cytosolic fractions were indicated as green bars and blue bars, respectively. The standard derivation was shown. ** P < 0.001.
NK-EVs induce cell death in target cells via ER stress
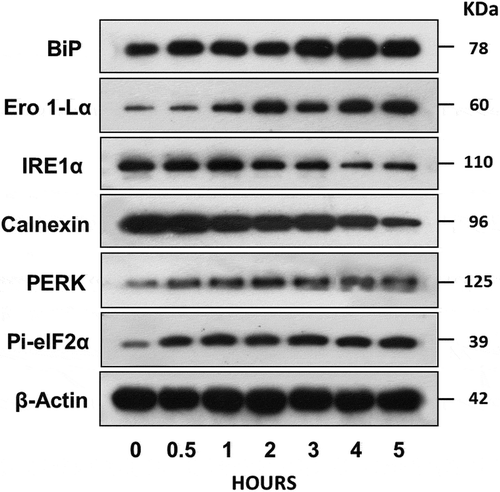
Cytotoxic proteins such as GNLY and GzmB are known to induce ER stress-mediated cell death. ER contains a pool of molecular chaperone proteins that counteract compromised protein folding, and alteration of chaperone protein levels may reflect the ER stress status. We tested the response of the target cells in the presence of active NK-EVs using an ER stress kit (). Indeed, incubation of NALM-6 cells with NK-EVs increased BiP levels, suggesting irregular protein folding within the ER [Citation29]. Incubation with NK-EVs also induced the accumulation of the unfolded protein chaperone, Ero1-Lα, a marker for disruption of ER homeostasis [Citation30]. Some ER components such as IRE1α and calnexin may be downregulated in apoptotic cells. IRE1α is a serine/threonine kinase that counteracts compromised synthesized glycoproteins [Citation28]. Both IRE1α and calnexin levels were reduced with prolonged incubation with NK-EVs. It is also well-known that ER stress increases PERK activity, which phosphorylates eIF2α to reduce protein translation [Citation31]. Phosphorylation of eIF2α, which has been known as a biomarker of immunogenic cell death [Citation32], was time-dependently increased after incubation with NK-EVs. The Western-blot signals were quantified by the ImageJ program (Supplement Figure S7). Thus, NK-EV treatment of ALL cells elicits ER stress that mediates cell death, most likely due to GzmB, GNLY [Citation33], and possibly other yet to be identified factors from NK-EVs.
Figure 7. NK-EVs induce ER stress in the target cells.
ALL NALM6 cells were treated with NK-EVs for different time points as indicated. Cells were then harvested and lysed in SDS-PAGE sample buffer. Each lane contains 30-μg cell lysate proteins as determined by the Orbit® protein assay (Life Technologies). Antibodies against BiP, Ero1-Lα, IRE1α, Calnexin, PERK and Pi-eIF2α from the ER Stress Antibody Sampler Kit (Cell Signalling technology, #9956S) were used to probe the blots according to the manufacturer’s protocol. β-Actin was used as the loading control. Abbreviations: BiP: pre-B Immunoglobulin heavy chain binding Protein; Ero 1-Lα: ER-residing protein endoplasmic oxidoreductin-1-like α-form; IRE1α: Inositol-requiring enzyme 1α; Calnexin: calcium-binding, endoplasmic reticulum (ER)-associated protein; PERK: Protein kinase-like endoplasmic reticulum kinase; Pi-elF2 α: phosphorylated elongation factor 2 α. Western blot signals were qualified by the program ImageJ from at least three independent experiments (n ≥ 3). The plots with the standard derivation were shown in Supplementary Figure S7.
Discussion
Cytotoxic lymphocytes such as NK and T cells release cytotoxic proteins, including PFN, GzmA, GzmB, GNLY and FasL that are major participants in the killing of target cells. These proteins have been studied extensively in their purified forms or as recombinant proteins. We recently demonstrated that EVs derived from NK cells exhibit cytotoxic activities and contained such proteins; however, the respective role of these proteins in isolated NK-EVs has not been previously investigated. We used quantitative ELISA to detect the presence of these cytotoxic proteins in NK-derived EVs. As with other EV isolates, the levels of PFN, GzmA, GzmB, GNLY and FasL varied from batch-to-batch, resulting in different protein profiles (). However, NK-EVs of individual batch have similar cytotoxic activity towards CHLA255 (neuroblastoma) and SupB15 (ALL) cells ().
PFN is required for pore formation [Citation33]. Among those tested cytotoxic proteins, the PFN had very high levels (~550 ng/mL mean value) relative to other cytotoxic proteins in the EV preparations. As the size of PFN (60 KDa), GzmA (60 KDa, dimer), GzmB (30 KDa) and GNLY (~12 KDa) are known, the molar ratio of these proteins can be stoichiometrically calculated with the approximate value of 10:3:1:4. PFN usually forms a polyperforin pore complex (PPC) with ~20 subunits [Citation34]. Therefore, the molar ratio of the PPC to other cytotoxic proteins becomes actually 0.5:3:1:4, e.g. more than one GzmA, GzmB, or GNLY molecules may pass through one PPC to enter target cells and activate cell death pathways. Among these cytotoxic proteins, the protein level of FasL in NK-EVs was negligible and its level did not correlate with cytotoxicity. Therefore, unlike in NK cells, FasL may not play a significant role in NK-derived EV-mediated cytotoxicity.
Despite the variations in cytotoxic protein concentrations, there was a good correlation between cytotoxic protein levels and cytotoxicity. Scatter plots and R-square values were used to determine the best-fitting regression line between individual levels of each cytotoxic protein (ELISA data) versus cytotoxicity (% dead cell) in >60 EV isolations (). The results did not identify any very high R2-value for any cytotoxic protein, suggesting that there is no single dominant killing factor. Instead, we observed that most cytotoxic proteins (PFN, GzmA, GzmB, and GNLY) fell into moderate R2 values (, and ), suggesting that they all contribute to cytotoxicity to some degrees. This supports our hypothesis that NK-EVs may use multiple killing mechanisms through these cytotoxic proteins to target cells and may activate various cell death pathways. Of note, our results do not rule out that other cytotoxic factors might be involved. Overall, the current results indicate that PFN, GzmA, GzmB and GNLY from NK-EVs together play prominent roles in inducing cytotoxicity.
Granzyme A, a serine protease in the cytotoxic granules of NK cells and cytotoxic T lymphocytes, induces caspase-independent cell death when introduced into target cells [Citation35]. A special target of GzmA in the cell death pathway is an ER-associated complex, called the SET complex, which contains at least two GzmA substrates, the nucleosome assembly protein SET (or 12PP2A), and the DNA bending protein HMG2 (). GzmA cleavage of SET releases the inhibition on NM23-H1 and leads to single-stranded DNA damage [Citation23] (). The unique death pathway initiated by GzmA provides a parallel route for programmed cell death, which is important for targets that are insensitive to caspases. Our results in clearly demonstrate that NK-EVs are able to degrade both SET and HMG2 proteins, and thus, induce programmed cell death that is caspase-independent.
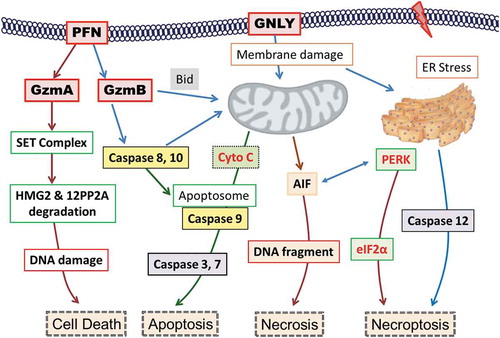
Figure 8. Summary of the potential killing mechanisms of NK-EVs exerting onto target cells.
Cell death may be induced by perforin-generated membrane holes for granzyme A and granzyme B entry, or by granulysin-damaged membrane, or by other un-defined factors. The cytotoxic proteins of NK-EVs may rely on these mechanisms to activate the caspase-dependent and -independent cascade, to cause ER stress, and subsequently to induce cell death.PFN, perforin; GzmA, granzyme A; GzmB, granzyme B; GNLY, granulysin; SET: an 270 ~ 420 KDa ER-associated complex; HMG: high mobility globulin; 12PP2A: 12 protein phosphatase 2A; CytoC, cytochrome C; AIF, apoptosis-inducing factor; PERK, Protein kinase-like endoplasmic reticulum kinase; elF2α, elongation factor 2α; Bid, BH3-interacting domain.
Granzyme B is the granzyme family member with the strongest apoptotic activity as a result of its caspase-like ability to cleave substrates at aspartic acid residues, thereby activating procaspases directly and cleaving downstream caspase substrates [Citation22]. GzmB prominently induces damage to mitochondria, a central hallmark of apoptosis [Citation36]. Cytochrome C is a well-conserved electron-transport protein and is part of the respiratory chain localized to mitochondrial intermembrane space [Citation37]. Upon apoptotic stimulation, cytochrome C released from mitochondria associates with procaspase-9 [Citation38]. The complex then processes caspase-9 from an inactive proenzyme to its active form. This event further triggers activation of caspase-3 and −7, and eventually leads to apoptosis (). We observed the release of cytochrome C () and the activation of caspase-3, −7, and −9 in NK-EV-treated cells [Citation19], suggesting damage to mitochondria and a role for GzmB (). The release of AIF (apoptosis inducing factor) may cause DNA damage and induce necrosis. It has also been reported that both GzmA and GNLY may damage mitochondria to induce cell death. Thus, multiple cytotoxic actions may centre at the mitochondrial death pathway.
It has been reported that GzmB and GNLY may induce ER stress-mediated apoptosis [Citation21]. Our results showed alterations of several ER markers during NK-EVs insults (). Importantly, the increase of PERK and phosphor-eIF2α indicates activation of apoptosis and/or necroptosis via ER stress [Citation32] (). ER stress may also activate caspase-7 and −12 pathways to induce cell death, which has previously been demonstrated [Citation19].
As a whole, our results indicate that multiple killing mechanisms are mediated by NK-derived EVs (–), including caspase-independent () and -dependent cell death pathways ( & ), resulting cytotoxicity of cancer cells. The potential killing mechanisms of NK-EVs are summarized in . In this scenario, NK cell-induced cytotoxicity may be executed in, at least in part, through NK-EVs’ actions. Finally, the protein levels of PFN, GzmA, GzmB and GNLY are related to the ability of NK-EVs to induce cytotoxicity. Consequently, understanding how they are packaged into NK-EVs during EV biogenesis may be critical to developing methods to improve their killing abilities, which in turn might enable clinical applications of NK-EVs.
Supplemental Material
Download MS Power Point (370.7 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Bobrie A, Colombo M, Raposo G, et al. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic. 2011;12(12):1659–13. Epub 2011/ 06/08. PubMed PMID: 21645191.
- Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. . PubMed PMID: 12154376
- Camussi G, Deregibus MC, Bruno S, et al. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010;78(9):838–848. Epub 2010/08/13. PubMed PMID: 20703216.
- Mittelbrunn M, Sanchez-Madrid F. Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol. 2012;13(5):328–335. Epub 2012/04/19. PubMed PMID: 22510790; PMCID: 3738855.
- Wen C, Seeger RC, Fabbri M, et al. Biological roles and potential applications of immune cell-derived extracellular vesicles. J Extracell Vesicles. 2017;6(1):1400370. Epub 2017/11/22. PubMed PMID: 29209467; PMCID: PMC5706476.
- Sódar BW, Kittel Á, Pálóczi K, et al. Low-density lipoprotein mimics blood plasma-derived exosomes and microvesicles during isolation and detection. Sci Rep. 2016;6:24316. Epub 2016/04/18. PubMed PMID: 27087061; PMCID: PMC4834552.
- Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):E968–77. Epub 2016/02/08. PubMed PMID: 26858453; PMCID: PMC4776515.
- Yanez-Mo M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066. PubMed PMID: 25979354; PMCID: PMC4433489.
- Huang SH, Wu CH, Chang YC, et al. Cryptococcus neoformans-derived microvesicles enhance the pathogenesis of fungal brain infection. PLoS One. 2012;7(11):e48570. Epub 2012/11/13. PubMed PMID: 23144903; PMCID: 3492498.
- Lötvall J, Hill AF, Hochberg F, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the international society for extracellular vesicles. J Extracell Vesicles. 2014;3:26913. PubMed PMID: 25536934; PMCID: PMC4275645
- Choi DS, Kim DK, Kim YK, et al. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics. 2013;13(10–11):1554–1571. . PubMed PMID: 23401200
- Challagundla KB, Wise PM, Neviani P, et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst. 2015;107(7). DOI:10.1093/jnci/djv135. PubMed PMID: 25972604; PMCID: PMC4651042
- Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. . PubMed PMID: 17486113
- Tauro BJ, Greening DW, Mathias RA, et al. Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol Cell Proteomics. 2013;12(3):587–598. . PubMed PMID: 23230278; PMCID: PMC3591653
- Pallet N, Sirois I, Bell C, et al. A comprehensive characterization of membrane vesicles released by autophagic human endothelial cells. Proteomics. 2013;13(7):1108–1120. . PubMed PMID: 23436686
- Aatonen MT, Ohman T, Nyman TA, et al. Isolation and characterization of platelet-derived extracellular vesicles. J Extracell Vesicles. 2014;3. DOI:10.3402/jev.v3.24692. PubMed PMID: 25147646; PMCID: PMC4125723
- Escrevente C, Keller S, Altevogt P, et al. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer. 2011;11:108. Epub 2011/03/29. PubMed PMID: 21439085; PMCID: 3072949.
- Lugini L, Cecchetti S, Huber V, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol. 2012;189(6):2833–2842. Epub 2012/08/21. PubMed PMID: 22904309.
- Jong AY, Wu CH, Li J, et al. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J Extracell Vesicles. 2017;6(1):1294368. Epub 2017/02/28. PubMed PMID: 28326171; PMCID: PMC5345580.
- Kaspar AA, Okada S, Kumar J, et al. A distinct pathway of cell-mediated apoptosis initiated by granulysin. J Immunol. 2001;167(1):350–356. PubMed PMID: 11418670
- Saini RV, Wilson C, Finn MW, et al. Granulysin delivered by cytotoxic cells damages endoplasmic reticulum and activates caspase-7 in target cells. J Immunol. 2011;186(6):3497–3504. Epub 2011/02/08. PubMed PMID: 21296981.
- Ewen CL, Kane KP, Bleackley RC. A quarter century of granzymes. Cell Death Differ. 2012;19(1):28–35. PubMed PMID: 22052191; PMCID: PMC3252830
- Lieberman J. Granzyme A activates another way to die. Immunol Rev. 2010;235(1):93–104. PubMed PMID: 20536557; PMCID: PMC2905780
- Zhu L, Kalimuthu S, Gangadaran P, et al. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics. 2017;7(10):2732–2745. Epub 2017/07/07. PubMed PMID: 28819459; PMCID: PMC5558565.
- Zhu L, Gangadaran P, Kalimuthu S, et al. Novel alternatives to extracellular vesicle-based immunotherapy - exosome mimetics derived from natural killer cells. Artif Cells Nanomed Biotechnol. 2018:1–14. Epub 2018/08/09. DOI:10.1080/21691401.2018.1489824. PubMed PMID: 30092165.
- Denman CJ, Senyukov VV, Somanchi SS, et al. Membrane-Bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PloS one. 2012;7(1):e30264. Epub 2012/01/27. PubMed PMID: 22279576; PMCID: 3261192.
- Zhou F. Expression of multiple granzymes by cytotoxic T lymphocyte implies that they activate diverse apoptotic pathways in target cells. Int Rev Immunol. 2010;29(1):38–55. . PubMed PMID: 20100081
- Williams DB. Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J Cell Sci. 2006;119(Pt 4):615–623. . PubMed PMID: 16467570
- Kaufman RJ, Scheuner D, Schröder M, et al. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol. 2002;3(6):411–421. . PubMed PMID: 12042763
- Cabibbo A, Pagani M, Fabbri M, et al. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J Biol Chem. 2000;275(7):4827–4833. PubMed PMID: 10671517
- Martins I, Kepp O, Schlemmer F, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30(10):1147–1158. Epub 2010/ 12/15. PubMed PMID: 21151176.
- Kepp O, Semeraro M, Bravo-San Pedro JM, et al. eIF2alpha phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol. 2015. DOI:10.1016/j.semcancer.2015.02.004 PubMed PMID: 25749194.
- Podack ER, Hengartner H, Lichtenheld MG. A central role of perforin in cytolysis? Annu Rev Immunol. 1991;9:129–157. . PubMed PMID: 1910674
- Law RH, Lukoyanova N, Voskoboinik I, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468(7322):447–451. . PubMed PMID: 21037563
- Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. 2010;17(4):616–623. . PubMed PMID: 20075940
- MacDonald G, Shi L, Vande Velde C, et al. Mitochondria-dependent and -independent regulation of Granzyme B-induced apoptosis. J Exp Med. 1999;189(1):131–144. PubMed PMID: 9874570; PMCID: PMC1887691
- Chavez-Galan L, Arenas-Del Angel MC, Zenteno E, et al. Cell death mechanisms induced by cytotoxic lymphocytes. Cell Mol Immunol. 2009;6(1):15–25. Epub 2009/03/04. PubMed PMID: 19254476.
- Slee EA, Harte MT, Kluck RM, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, −3, −6, −7, −8, and −10 in a caspase-9-dependent manner. J Cell Biol. 1999;144(2):281–292. PubMed PMID: 9922454; PMCID: PMC2132895