
ABSTRACT
Glioblastoma is the most prevalent and aggressive primary brain tumour for which total tumour lysate-pulsed dendritic cell vaccination is currently under clinical evaluation. Glioblastoma extracellular vesicles (EVs) may represent an enriched cell-free source of tumour-associated (neo-) antigens to pulse dendritic cells (DCs) for the initiation of an anti-tumour immune response. Capture and uptake of EVs by DCs could occur in a receptor-mediated and presumably glycan-dependent way, yet the glycan composition of glioblastoma EVs is unknown. Here, we set out to characterize the glycocalyx composition of glioblastoma EVs by lectin-binding ELISA and comprehensive immunogold transmission electron microscopy (immuno-TEM). The surface glycan profile of human glioblastoma cell line-derived EVs (50–200 nm) was dominated by α-2,3- and α-2,6 linked sialic acid-capped complex N-glycans and bi-antennary N-glycans. Since sialic acids can trigger immune inhibitory sialic acid–binding Ig-like lectin (Siglec) receptors, we screened for Siglec ligands on the EVs. Glioblastoma EVs showed significant binding to Siglec-9, which is highly expressed on DCs. Surprisingly, however, glioblastoma EVs lack glycans that could bind Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN, CD209), a receptor that mediates uptake and induction of CD4+ and CD8+ T cell activation. Therefore, we explored whether modification of the EV glycan surface could reduce immune inhibitory Siglec binding, while enhancing EV internalization by DCs in a DC-SIGN dependent manner. Desialylation with a pan-sialic acid hydrolase led to reduction of sialic acid expression on EVs. Moreover, insertion of a high-affinity ligand (LewisY) for DC-SIGN resulted in a four-fold increase of uptake by monocyte-derived DCs. In conclusion, we show that the glycocalyx composition of EVs is a key factor of efficient DC targeting and that modification of the EV glycocalyx potentiates EVs as anti-cancer vaccine.
Introduction
EVs are membranous particles consisting of a lipid bilayer containing a cargo of functional proteins, RNA molecules and metabolites[Citation1]. All cells including glioblastoma cells produce EVs, thereby releasing small packages of information and delivering them to target cells [Citation1–Citation3]. Although capture and uptake of EVs by recipient cells could occur through membrane fusion or endocytosis, several studies have reported receptor-mediated EV uptake in a glycan-dependent way [Citation4–Citation6]. The glycan profile of EVs from different sources (T-cells, carcinoma and melanoma cell lines and human breast milk) has been studied by lectin microarray and mass spectrometry showing a relatively conserved glycan profile with enrichment of polylactosamine and α2-6 linked sialic acid residues, complex type N-glycans and high mannose structures [Citation7–Citation9]. Although the RNA and protein contents of glioblastoma-derived EVs are widely studied [Citation2,Citation3,Citation10–Citation14], their surface glycosylation has not been described.
Glioblastoma is the most prevalent and aggressive primary brain tumour with a lack of curative therapeutic interventions and a median survival of 14.6 months under the current standard of care [Citation15]. Although the central nervous system (CNS) has been considered an immune-privileged site, this dogma has now shifted towards a paradigm where there is room for two crucial aspects in immunotherapy: CNS infiltration of antigen-specific T cells, and a draining lymphatic system [Citation16]. Thanks to this paradigm shift, potentiating T cells to infiltrate and kill glioblastomas has become an appealing immunotherapeutic option [Citation17–Citation26]. Different types of immunotherapy, including peptide-based vaccines [Citation27], autologous vaccines, checkpoint inhibition and adoptive dendritic cells (DCs) and T cells have emerged with some showing encouraging results [Citation19–Citation25]. Current approaches for vaccine-based immunotherapy in glioblastoma include mono-antigenic vaccines targeting a single tumour-specific neo-antigen [Citation27] and predefined or personalized multi-antigenic vaccines [Citation18,Citation28]. Multi-antigenic vaccines can also comprise an undefined mix of tumour-associated antigens and neo-antigens in an autologous tumour lysate to stimulate dendritic cells [Citation21,Citation25,Citation29]. An interim analysis of a current Phase III clinical trial with an autologous tumour lysate-pulsed dendritic cell vaccine (DCVax®-L) for newly diagnosed glioblastoma shows a median overall survival of the total intention to treat study population (treatment arm plus control arm) of 23.1 months [Citation25], which suggests that the patients in the treatment arm of this trial live significantly longer than the expected 14.6 months based on the standard of care [Citation15].
DCs are antigen-presenting cells (APCs) with the ability to induce antigen-specific adaptive immune responses against pathogens and cancer [Citation30,Citation31]. In homeostatic conditions, immature DCs reside in the lymph nodes and peripheral tissues and sample the environment. After recognition of antigens in combination with pattern recognition receptor stimulation DCs mature and antigens are processed for presentation by major histocompatibility complex (MHC) class I and II molecules to CD8+ and CD4+ T cells, respectively, [Citation32–Citation34]. Glioblastoma-derived EVs are relatively enriched with plasma membrane proteins of the originating cell [Citation3], and thus could function as an autologous cell-free alternative to pulse DCs [Citation35]. Tumour EVs are more stable upon freezing and thawing than cells or tissues, and compared to tumour lysates, vesicles can likely be modified and thereby specifically targeted to an uptake receptor. Tumour-derived EVs, loaded onto DCs, are routed into the endo-lysosomal pathway [Citation36], and transferred tumour-specific antigens allow for specific activation of CD4+ and CD8+ T cells in different mouse tumours in vivo [Citation36,Citation37]. Specific delivery of tumour EVs to DCs by targeting an uptake receptor for endocytosis could enhance their effective uptake by DCs and intracellular routing for antigen (cross-) presentation [Citation30,Citation38]. DC-SIGN is a C-type lectin receptor (CLR) with known glycan specificity, and mediates antigen internalization, processing and presentation of antigens to CD4+ and cross-presentation to CD8+ T cells [Citation39–Citation42]. DC-SIGN recognizes its natural ligands, Lewis antigens [Citation43], through its carbohydrate recognition domain and traffics to the lysosomes upon internalization [Citation44].
We hypothesized that by characterization of the surface glycosylation of EVs and modification of their glycocalyx, we could enhance their internalization by DCs. The aim of this study was to identify the main groups of glycans in the glycocalyx of EVs that could provide ligands for DC-specific receptors by ELISA-based lectin-binding assays and immunogold transmission electron microscopy (TEM). We used a lectin panel including lectins recognizing sialic acids (α-2,3- and α-2,6, N-linked and O-linked), terminal GalNAc, different mannose configurations, bi-antennary glycans and fucose, revealing a dominance of sialic acid-capped N-glycans and bi-antennary glycans and a lack of DC-SIGN ligands. Therefore, we set out to enzymatically remove the immune inhibitory Siglec stimulating sialic acids and incorporate a high-affinity DC-SIGN ligand (Lewis Y) for enhanced uptake by DCs by conjugating it to a palmitic acid tail. The success of the modifications was verified by lectin-binding ELISA, immunogold TEM and EV internalization by DCs.
Materials and methods
Cell culture
U87 [Citation45] and GBM8 [Citation46] glioblastoma cell lines were cultured in a humidified incubator under standardized conditions with 5% CO2 and 37°C. U87 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher) supplemented with 100 U/ml penicillin/streptomycin (Lonza), 2 mM L-glutamine (Lonza) and 10% Fetal Calf Serum (FCS) (Biowest). GBM8 cells were cultured in Neurobasal medium (Invitrogen) supplemented with 100 U/ml penicillin/streptomycin (Lonza), 2 mM L-glutamine (Lonza), 1 × B27 supplement (Life Technologies), 0.5 × N2 supplement (Life Technologies), 20 nM recombinant human fibroblast growth factor (FGF, Pepro Tech), 20 nM recombinant human epidermal growth factor (EGF, Pepro Tech) and 20 μg/mL heparin [Citation46].
Human monocyte-derived dendritic cells (MoDC)
Monocytes were isolated from multiple healthy donor-derived buffy coats (Sanquin) by sequential Lymphoprep (Axis-Shield)/Percoll (Amersham) gradient centrifugation as previously described [Citation47]. Isolated monocytes were cultured in RPMI (Invitrogen) supplemented with 100 U/ml penicillin/streptomycin (Lonza), 10% Fetal Calf Serum (Biowest), 500 U/ml IL-4 and 800 U/ml granulocyte-macrophage colony-stimulating factor (GM-CSF, ImmunoTools) for 5–6 days. Dendritic cell differentiation and activation status were confirmed by flow cytometric analysis of the presence of DC-SIGN (AZN-D1 [Citation48], and secondary FITC-labelled polyclonal goat anti-mouse antibody, Jackson), CD80 (clone L307.4, BD Biosciences), and CD86 (clone 2331 (FUN-1), BD Bioscience) with and without lipopolysaccharide (LPS) stimulation.
Isolation of EVs
We used differential (ultra-)centrifugation [Citation49] and size-exclusion chromatography (SEC) [Citation50] for enrichment of EVs. EVs were isolated from conventional U87 and primary GBM8 cell cultures growing between 40% and 90% confluency. Unlike the medium for GBM8 cells, the medium for U87 cultures contained exosome depleted FCS, which was obtained by overnight (15 h) centrifugation at 70.000 x g at 4°C [Citation51]. EVs were isolated, as described previously [Citation52–Citation54], by sequential centrifugation of 240 mL cell culture medium; two times 500 × g at 4°C for 10 min, two times at 2000 × g at 4°C for 15 min and two times at 10,000 × g at 4°C for 30 min. The supernatant was then transferred to endotoxin-free ultracentrifuge tubes (Ultra-Clear) and centrifuged at 70,000 × g at 4°C for 1 h without a brake in an SW32Ti rotor (Beckman Coulter). Based on our previous work [Citation53,Citation54] we choose the 70,000 x g protocol to reduce protein contamination. The EV containing pellet was then resuspended, washed (2x) in PBS and used for further experiments or modification after resuspending the washed pellet in 400 μL PBS. EV preparations were characterized and stored at −80°C. The size distribution of EV preparations was analysed by transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA, Nanosight) after calibrating the system with Silicon Oxide Size Standards beads (105.2 nm, Microspheres-nanospheres).
Palmitoyl-Lewisy synthesis
LeY-glycolipid (LeY-hexadecanehydrazide) was prepared from LeY pentasaccharide (Elicityl) and palmitic anhydride (Sigma-Aldrich), the latter undergoing two subsequent chemical transformations, first to tert-butyl N-(hexadecanoylamino) carbamate, then to palmitic hydrazide through common reactivity. Palmitic hydrazide was coupled to LeY through a reductive amination reaction. Briefly, palmitic hydrazide (2 eq., Sigma-Aldrich) and picoline borane (10 eq., Sigma-Aldrich) were dissolved in DMSO/AcOH/CHCl3 (8:2:1, 200 μl). The mixture was added to LeY (1 eq.) and the reaction was stirred for 2.5 h at 65°C. Addition of CHCl3/MeOH/H2O at 8:1:8 v/v ml ratio allowed the extraction of LeY-glycolipid as white slurry at the interphase. The mixture was centrifuged at 4600 rpm for 20 min, then the aqueous and organic layers were carefully removed, and the washing step was repeated once more. The slurry was freeze-dried (methanol/water) to remove residual solvent. Glycan derivatization was confirmed by ESI-MS (LCQ-Deca XP Ion trap mass spectrometer in positive mode; Thermo Scientific) using nanospray capillary needle. LeY-glycolipid was post-inserted into the EVs by adding 1 ml of EV suspension to 0.75 mg of glycolipid, previously dissolved in 15 μl of methanol. After 15 min of vigorous stirring and overnight at 4°C, the EVs were and purified by SEC.
Modification of EV surface glycosylation
After ultracentrifugation at 70.000 x g, the containing pellets were carefully resuspended and pooled together in a total end volume of 400 μL. Next, the EV preparation was incubated with 0.5 μM of the fluorescent lipophilic fluorescent reporter DiD (1′- dioctadecyl-3,3,3′,3′-tetramethyl indodicarbocyanine, Life Technologies) at 37°C for 15 min. Half of the EV preparation was then treated with a pan-sialic acid hydrolase Neuraminidase (Roche) for 30 min at 37°C. Next, half of the unmodified and half of the Neuraminidase treated aliquots were incubated with palmitoyl-LewisY while vortexing for 10 min. All four preparations (of equal volume) were then diluted to a final volume of 1 mL and EVs were purified by SEC according to a previously described protocol [Citation50]. In order to maximize microvesicle concentration and purity fraction 8–11 were collected for further experiments.
Lectin-binding enzyme-linked immunosorbent assay (ELISA)
EVs were coated onto Nunc maxisorb 96-well plates (Nunc, Denmark) in 0.2M NaHCO3 buffer (pH 9.2) for 18 h at 22°C. Plates were then washed with TSM buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM CaCl2, 1mM MgCl2) twice, followed by blocking for 30 min at 37°C with TSM supplemented with 1% bovine serum albumin (BSA). Anti-CD63 (clone NKI/C3, a kind gift of prof. J. Neefjes, Dutch Cancer Institute) and lectins used for primary incubation were incubated for 120 min, mildly shaking at room temperature (RT) followed by detection with peroxidase-conjugated goat anti-mouse antibody or streptavidin. Glycan-specific lectins were used to identify glycan groups: biotinylated Concanavalin A (ConA, specific for high mannose, terminal mannose, bi-antennary glycans, α-linked mannose, Vector Laboratories, B-1005–5), biotinylated Galanthus nivalis (GNA, specific for α1-3 mannose), biotinylated Helix pomatia agglutinin (HPA, specific for αGalNAc (Tn antigen) and type A erythrocytes, Sigma Aldrich, L6512), biotinylated Lotus Tetragonolobus (LTA, specific for α1-6 polymannose, Vector Laboratories, B-1325–2), biotinylated Maackia Amurensis I (MAL-1, specific for O-linked α-2,3 sialic acids, Vector laboratories, B-1315), biotinylated Maackia Amurensis II (MAL-II, specific for N-linked α-2,3 sialic acids, Vector laboratories, B-1265), biotinylated Sambucus nigra (SNA, Specific for N-linked α-2,6 sialic acids, Vector laboratories, B-1305) (). Binding was visualized using a substrate buffer containing 0,1M Sodium Acetate (Sigma), 0,1M Citric acid (Sigma), pH4, 0,006% H2O2, and 0,1 mg/mL tetramethylbenzidine (TMB, Sigma) and the reaction was blocked with a 0.2 M H2SO4 stop buffer. Signal was measured using a microplate absorbance spectrophotometer at 450 nm (Bio-Rad, USA).
Table 1. Lectin specificity.
Transmission electron microscopy
EVs were diluted 1:1 with PBS and coated for a minimum of 10 min on Formvar (EMS) coated copper grids (Veco). For the analysis of size distribution, grids were washed with PBS five times after coating, fixed in 1% glutaraldehyde (Millipore) and incubated in 0,5% uranyl acetate (SPI-Chem)/2% methylcellulose (Sigma). A total of nine images (three images per grid from three separate grids) were acquired using a transmission electron microscope (CM100 bio twin, company Philips/FEI), connected to a CCD camera (Morada G2, olympus). Diameters of randomly selected EVs found on these images were measured in iTEM (EMSIS) software, resulting in a total of 100 measurements. For immuno-gold stainings, grids were coated with EVs 1:1 diluted in PBS, washed five times with PBS supplemented with 1% BSAc (Aurion), blocked for 5 min in PBA and 10 min in blocking reagent (Aureon). Antibodies and lectins for primary stainings included biotinylated SNA, biotinylated MAL-1, biotinylated MAL-II, DC-SIGN/Fc (DC-SIGN/Fc consists of the extracellular portion of DC-SIGN (residues 64–404, fused at the C-terminus to a human IgG1/Fc fragment into the Sig-pIgG1-Fc vector; Fawcett et al. 1992)) and anti-CD63 (clone NKI/C3, a kind gift of prof. J. Neefjes, Dutch Cancer Institute). After a minimum of 30 min incubation with primary antibodies, grids were washed 5 times with PBA before incubation for 20 min with secondary reagents. Secondary reagents included Protein A-gold (Cell Microscopy Core, Utrecht), and goat-anti-biotin-Gold (Aureon). Lastly, grids were washed, fixed in 1% glutaraldehyde and incubated in 0,5% uranyl acetate/2% methylcellulose before imaging. Image acquisition was performed using a transmission electron microscope connected to a CCD camera and iTEM software.
Binding and uptake of EVs
EVs were isolated as described above. Per well, 500.000 MoDCs were incubated with DiD-labelled microvesicles for 45 min on ice to allow receptor binding. Subsequently, MoDCs were incubated at 37°C for 0, 30, 60 and 90 min prior to fixation with 4% PFA on ice for 20 min. Fluorescent DiD signal was measured by flow cytometry (Fortessa, BD Biosciences) and results were analysed using Cytobank [Citation55].
Internalization and intracellular routing of EVs
Internalization and intracellular routing of EVs in MoDCs was analysed by imaging flow cytometry following incubation with 1 million DC for 15 and 60 min at 37°C. After incubation, cells were immediately put on ice and washed in ice-cold PBS. Cells were fixed in ice-cold 4% paraformaldehyde in PBS for 20 min and permeabilized in 0.1% saponin in PBS for 30 min. Images were acquired on the ImageStream × (Amnis) imaging flow cytometer. A minimum of 15,000 cells was acquired per sample at a flow rate ranging between 50 and 100 cells/s at 60 × magnification. At least 2000 cells were acquired from single-stained samples to allow for compensation. The analysis was performed using the IDEAS v6.1 software (Amnis) as previously described [Citation56]. Cells were gated based on focus using the Gradient RMS (brightfield) feature, and on size using the features Area (brightfield) vs Aspect Ratio Intensity (brightfield). Internalization was addressed using the feature Internalization on a mask calculated by eroding eight pixels from the circumference of every cell. Co-localization was calculated using the feature Bright Detail Similarity R3.
Results
Glioblastoma-derived EV isolation and characterization for EV surface glycan analysis
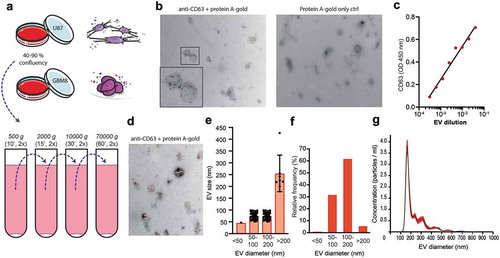
To study the glycan composition of glioblastoma-derived EV-membrane-associated glycoproteins and glycolipids, we isolated small EVs including exosomes and small membrane budded vesicles from cell culture media from two glioblastoma cell lines by ultracentrifugation [Citation49]. EVs were isolated from well-characterized glioblastoma cell lines, U87 [Citation45] and GBM8 [Citation46], following the protocol as described before [Citation52–Citation54] and schematically represented in ). U87 cells were maintained in a culturing medium with FCS pre-depleted from EVs by overnight ultracentrifugation [Citation51]. Because this depletion method results in incomplete (approximately 95%) elimination of EVs [Citation9,Citation51], we also included primary glioblastoma spheroids (GBM8 cells) [Citation46] in our experiments, cultured in serum-free neurobasal medium supplemented with growth factors. To confirm the quality of our EV preparations we imaged our preparations by immuno-TEM for CD63 ()). A subset of the EVs in the preparation showed a positive CD63 immune-gold staining. EV expression of CD63 was confirmed by CD63 ELISA on a titration of EVs adsorbed to a solid phase ()). TEM images also confirmed the typical morphology of EVs (,)). Size distribution profiles, quantified by measuring the diameter of 100 at random-selected EVs in eight TEM images (,,)) and by nanoparticle tracking analysis (NTA, )), were comparable to previously reported size of exosomes/small EVs (70–200 nm) isolated from glioblastoma culture media [Citation2,Citation4,Citation9,Citation57].
Figure 1. Glioblastoma cell line-derived EV isolation and characterization for EV surface glycan analysis. (a). EV isolation procedure by differential centrifugation from U87- and GBM8-conditioned medium. (b). Expression of CD63 on EVs by immuno-gold transmission electron microscopy (TEM) with a magnification of 24,500 x. Primary staining with anti-CD63 followed by protein A coupled to 15 nm gold nanoparticles. (c). CD63 ELISA on a titration of EVs adsorbed to a solid phase. (d). Measurement of EV diameter, determined on TEM micrographs with a magnification of 24,500 x (CD63 immuno-gold staining, N = 100 EVs measured at random). (e). Quantification of the diameter of 100 randomly selected EVs in nm per size category, measured in eight TEM images. (f). Quantification of the relative distribution of EV diameter in percentage per size category, determined on TEM in D. (g). Relative size distribution measured by nanoparticle tracking analysis (NTA).
The surface glycoconjugates of glioblastoma cell line-derived EVs are dominated by sialic acid-capped N-glycans and bi-antennary N-glycans
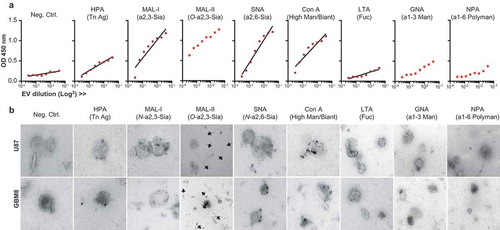
The glycocalyces of T cell-, melanoma-, colon cancer- and breast milk-derived EVs have been profiled using lectin microarray technology [Citation8,Citation9]. We selected a panel of lectins representing the main groups of glycans of our interest () and studied the lectin-binding profile of glioblastoma (U87 and GBM8) derived EVs. Lectin-binding assays on a titration of U87 EVs adsorbed to a solid phase (ELISA, )) revealed that glioblastoma EV glycosylation was mainly dominated by N-linked α-2,3 sialic acids (MAL-I), O-linked α-2,3 sialic acids (MAL-II) and N-linked α-2,6 sialic acids (SNA). They also express bi-antennary glycans (strong binding of ConA while GNA- and NPA binding is low) and some terminal GalNAc (HPA). A drawback of lectin microarrays or ELISAs in the characterization of the glycosylation of EVs is the impossibility to discard any contribution by contaminants in the EV preparation, such as soluble proteins or cell membrane fragments [Citation58]. To confirm the expression of these glycans on EVs we performed immuno-gold TEM, studying immunoreactivity of membrane vesicles derived from both cell lines with HPA, MAL-I, MAL-II, SNA, ConA, GNA and NPA (), Supplementary Figs. 1 and 2). In concert with the recently studied and described heterogeneity in size, and RNA, protein and lipid content of small EVs [Citation59] the single-vesicle resolution provided by TEM revealed also an expected heterogeneity in EV surface glycosylation. In addition, the images showed protein contamination of the EV preparations despite the intended reduction thereof by lowering the ultracentrifugation speed to 70,000 x g. The TEM images in (and Supplementary Figs. 1 and 2) showed the binding of MAL-II to free protein aggregates instead of EVs, showing the dominance of N-linked sialic acids over O-linked sialic acids on our EVs. Our EV preparations showed little to no binding to GNA or NPA indicating the absence of glycoconjugates containing α-1,3 mannoses or α-1,6 polymannoses. Our data, therefore, confirms the relatively conserved glycan signature for microvesicles [Citation9] but also indicates that we should be cautious interpreting glycan analyses performed on full preparations without a single-vesicle resolution such as TEM or FACS. As demonstrated here, detected glycans can be expressed both by EVs as well as free proteins in the preparation. As a consequence, we decided for future experiments to purify vesicle fractions by size exclusion chromatography (SEC) [Citation50] after the first 70.000 x g ultracentrifugation step, excluding fractions containing free proteins. Altogether, the glycocalyx of glioblastoma cell line-derived EVs showed a glycan profile dominated by α-2,3 and α-2,6 sialic acid-capped complex N-glycans and bi-antennary N-glycans.
Figure 2. The surface glycoconjugates of glioblastoma cell line-derived EVs are dominated by sialic acid-capped N-glycans and bi-antennary glycans. (a). Lectin-binding ELISA (HPA, MAL-I, MAL-II, SNA, ConA, LTA, GNA, NPA) on a titration of U87 EVs adsorbed to a solid phase (carbohydrate specificity in brackets and in ). (b). Immuno-gold TEM pictures of U87 and GBM8 EVs stained with the same biotinylated lectins and streptavidin conjugated to 15 nm gold nanoparticles.
Glioblastoma-derived EVs express ligands for Siglec-9, not DC-SIGN
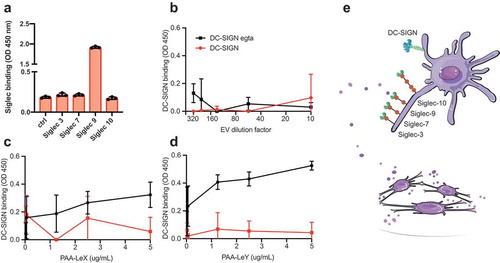
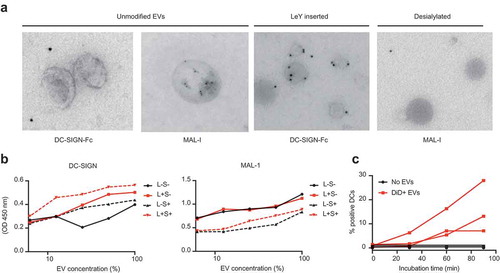
Next, we focused on two types of glycans that are of interest for receptor-mediated uptake of EVs by antigen-presenting cells in the context of vaccination, namely, sialic acids, fucosylated structures such as Lewis antigens. Sialic acids were detected on the surface of EVs and could represent ligands for the well-known immunosuppressive receptor family of Siglecs [Citation60]. Since DCs take up T cell-derived EVs in a Siglec-1 dependent way [Citation5], and splenic marginal zone macrophages take up B cell-derived EVs in a Siglec-1 dependent way [Citation6] we screened for Siglec ligands on our EV preparations. We immobilized EVs on a solid phase and performed Siglec binding ELISA assays with recombinant Siglec-Fc constructs. We tested the binding of the signalling Siglecs (Siglec-3, −7, −9 and −10) on moDCs [Citation61]. Upon INFα stimulation moDCs upregulate Siglec-1, but since this receptor is an uptake receptor without signalling properties we excluded it from the screen. U87-derived EVs expressed ligands for Siglec-9 (CD329), whereas no ligands for Siglec-3, −7 and −10 were detected ()). Siglec-9 is a CD33-related Siglec, which recognizes sialic acid in either α-2,3 or α-2,6 to galactose [Citation62]. Siglec-9 is mainly expressed on immune cells such as NK cells, T cells, neutrophils, monocytes and immature DCs, and has two intracellular immune receptor tyrosine-based inhibitory motifs (ITIMs) [Citation60–Citation63]. The dendritic cell-specific C-type lectin DC-SIGN binds high mannose glycans and polysaccharides containing fucose [Citation64]. DC-SIGN was previously successfully targeted with glyco-liposomes containing antigen for anti-cancer vaccination strategies [Citation65]. Although our EVs express some fucosylated glycans () and ConA reactive glycans, we could not detect GNA- or NPA reactive glycans or DC-SIGN ligands on their surface ()). DC-SIGN however strongly binds LewisX (LeX, )) and LewisY (LeY, )) in a calcium-dependent manner [Citation43,Citation66]. Thus, our EVs are likely to bind to Siglec-9 on immature dendritic cells but lack the glycans that serve as ligands for DC-SIGN ()).
Figure 3. Glioblastoma-derived EVs bind Siglec-9, not DC-SIGN. (a). Siglec-binding ELISA on U87-derived EVs adsorbed to a solid phase. Screen for binding of recombinant Siglec-3, −7, −9 and −10 constructs (N = 3). (b). Absence of calcium-dependent DC-SIGN binding to ligands on U87-derived EVs by DC-SIGN binding ELISA on a titration of EVs adsorbed to a solid phase (N = 3). (c-d). DC-SIGN binding to polyacrylamide (PAA)-coupled LeX and -LeY glycoconjugates by DC-SIGN binding ELISA on titrations of PAA-LeX and PAA-LeY adsorbed to a solid phase (N = 3). (e). Schematic representation of possible uptake of glioblastoma EVs by DCs via Siglec 9, not DC-SIGN.
Glycan modification for receptor-mediated targeting to dendritic cells
We hypothesized that the elimination of Siglec ligands on the surface of EVs would prevent EVs from triggering Siglecs on DCs and, therefore, avoid immunosuppressive signalling. Since we could not observe the expression of DC-SIGN ligands on the surface of EVs, we aimed at both the removal of sialic acids from the surface of EVs and the incorporation of DC-SIGN ligands for receptor-mediated glycan-dependent targeting to DCs. To this end, we conjugated LeY to the reducing end of a palmitic acid tail ()) with a purity of over 95% (,)), and modified EV glycosylation after incorporating the fluorescent reporter DiD into the EVs ()). Sialic acids were removed by enzymatic desialylation with a pan-sialic acid hydrolase and DC-SIGN ligands were incorporated by insertion of palmitoyl-LeY, followed by an SEC step to eliminate free DiD, protein, enzyme and palmitoyl-LeY. Chemo-enzymatic modification of EVs resulted in enhancement of DC-SIGN binding in the palmitoyl-LeY modified EVs and a decrease in sialylation in the sialidase-treated EVs (), Supplementary Figure 3) by immuno-TEM analysis. Using ELISA we showed that chemo-enzymatic modification of EVs did not alter the expression of the EV marker CD63 (Supplementary Figure 4) and confirmed the altered glyco-phenotypes with decreased MAL-I binding after desialylation and increased DC-SIGN binding after palmitoyl-LeY insertion (), Supplementary Figure 4). Incubation of our UM DiD-labelled EVs with human monocyte-derived dendritic cells (moDCs) resulted in an incremental proportion of positive moDCs over time, indicating binding/uptake of DiD-labelled EVs by dendritic cells and effective labelling of EVs by DiD ()).
Figure 4. Chemo-enzymatic glycan modification for receptor-mediated targeting to dendritic cells. (a). Palmitoyl-LeY synthesis. (b). MS spectrum of palmitoyl-LeY. (c). MSMS spectrum of palmitoyl-LeY with peaks representing: 1074 = palmitoyl-LeY – H2O, 946 = palmitoyl-LeY – fucose, 658 = LeY, 511 = fucose(glcnac-gal). (d). Schematic representation of the fluorescent labelling and glycan modification of EV preparations by treatment with pan-sialic acid hydrolases and postinsertion of palmitoyl-LeY.
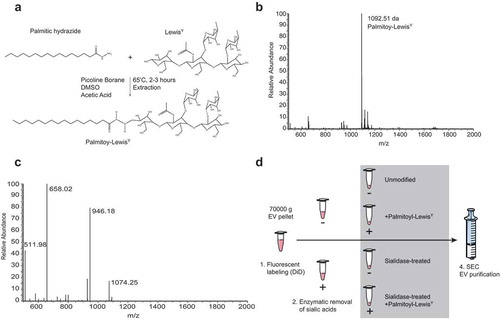
Figure 5. Glycan modification of EVs resulted in altered glyco-phenotypes. (a). Glycan modification resulted in enhanced recognition by DC-SIGN and less expression of sialic acids, measured by immuno-gold TEM. (b). Chemo-enzymatic modification of EVs resulted in enhancement of DC-SIGN binding in the palmitoyl-LeY incorporated EVs and a decrease in sialylation in the sialidase-treated EVs, measured by lectin-binding ELISA on EVs adsorbed to a solid phase (independent validations in Supplementary Figure 4). (c). Uptake of GBM-derived DiD-labelled EVs by MoDCs, measured as % DiD+ moDCs by FACS (three donors).
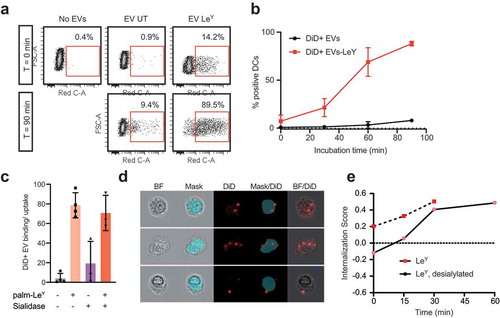
Glycan modification of EVs improved binding and internalization by DCs
We measured a four-fold increase of binding/uptake of DiD-labelled EVs by moDCs after LeY incorporation (,)). ) shows example dot plots of DiD signal due to binding of DiD+ EVs to moDCs at 4°C (T = 0) and DiD signal due to DiD+ EV-binding/uptake by moDCs after 90 min at 37°C, representative for three donors. In ) binding/uptake of DiD+ EVs in this pulse-chase experiment was plotted over time, comparing unmodified EVs and LeY+ EVs in three donors. To visualize binding versus internalization, we performed imaging flow cytometry analysis of moDCs of three additional donors, incubated with our four glycoforms of DiD-labelled EVs (unmodified, LeY+, sialidase-treated LeY- and sialidase-treated LeY+) in a pulse-chase experiment. This data also showed a four-fold increase of EV binding/uptake by moDCs after incorporation of palmitoyl-LeY ()). Binding/uptake of unmodified EVs and sialidase-treated EVs by moDCs was similar (10–20% DiD positive moDCs) after 60 min incubation at 37°C, whereas 80% of moDCs were DiD positive after incubation with our LeY+ EVs ()). Fluorescent DiD signal in DCs, however, does not prove internalization of EVs by the DCs. Therefore, we analysed internalization of both our LeY+ EV preparations, which showed efficient internalization by moDCs in an imaging flow cytometry experiment (,)). Internalization scores were measured by calculating co-localization of DiD signal with a mask covering the intracellular area as shown in ). At 4°C EVs remained at the surface of the moDCs indicated by the low internalization score (time point 0), whereas at 37°C the increasing internalization score represents rapid uptake of the LeY+ EVs.
Figure 6. Glycan modification of EVs improved internalization by DCs. (a). Dot plots showing binding/uptake of unmodified or LeY-incorporated DiD+ EVs by moDCs after 30 min incubation at 4°C (T = 0) or after 90 min incubation at 37°C (T = 90) measured by FACS, representative of three donors. (b). Percentage DiD+ moDCs after incubation with unmodified or LeY-incorporated DiD+ EVs for 30 min at 4°C (T = 0). MoDCs were fixed after 0, 30, 60, or 90 min incubation at 37°C and DiD signal was measured by imaging flow cytometry (three donors). (c). Binding/uptake of EVs by moDCs (three donors), measured by imaging flow cytometry. (d). Representative imaging flow cytometry brightfield (BF) pictures of moDCs with DiD positive spots on the outside and inside of cells. Blue area masks inside area of cells, and colocalization of DiD signal with the mask was used to calculate internalization scores. (e). Internalization of EVs by moDCs in a pulse-chase experiment with cells fixed in PFA after 15, 30, 45 and 60 min incubation at 37°C visualized in one donor by imaging flow cytometry.
Discussion
Immunization of patients with a newly diagnosed glioblastoma by highly personalized synthetic multi-peptide- or autologous tumour lysate-pulsed DC vaccination has recently emerged as a promising approach to boost CD4+ and CD8+ T cell responses [Citation18,Citation25,Citation28]. To circumvent the need for personalized neo-antigen identification and peptide synthesis, we proposed that EVs may represent a more comprehensive, cell-free, autologous source of neo-antigens and tumour-associated antigens, which could be targeted for efficient DC loading [Citation3,Citation35,Citation67]. Since EVs can be internalized by recipient cells via glycan-specific receptors [Citation4–Citation6], we profiled the surface glycosylation of glioblastoma cell line-derived EVs and modified their glycosylation for enhanced internalization by DCs. By measuring lectin-binding properties of EVs in both ELISA-based assays and immunogold-TEM, we showed that the surface glycoconjugates of our EV preparations were dominated by immune inhibitory sialic acid-capped N-glycans and complex bi-antennary glycans. Desialylation with a pan-sialic acid hydrolase reduced EV recognition by sialic acid-specific lectins (MAL-I and SNA) and insertion of palmitoyl-LeY enhanced their capacity to be captured by DC-SIGN and internalized by moDCs.
The glycocalyx of T cell-derived EVs was previously characterized by lectin-microarray analysis showing a glycan profile that differed from that of the parent cell plasma membrane [Citation8]. In a later study, comparison of the EV glycocalyces of different cell lines (T cells, melanoma and colon cancer) and breast milk-derived EVs revealed enrichment in high mannose, polylactosamine, α-2,6 sialic acid and complex N-linked glycans while blood group antigens were excluded [Citation9]. These results are in accordance with the high expression of α-2,3- and α-2,6-sialic acids (binding of MAL-1 and SNA, respectively) and the presence of bi-antennary N-glycans (Con A-binding) in our glioblastoma-derived EV preparations. We, however, did not find high mannose glycans in our glioblastoma EV preparations, indicated by the absence of NPA binding or DC-SIGN ligands. In this and other studies, purification steps and techniques for glycan detection could have influenced the specificity of lectin binding to EVs. Here, we demonstrated a discrepancy between lectin-binding assays based on ELISA versus immunogold TEM. Therefore, detected glycans in EV preparations should always be considered to be due to contaminating proteins rather than the EVs themselves, unless lectin binding has been assessed at a single EV level with TEM or FACS. Moreover, we demonstrate heterogeneity in lectin binding to EVs, suggesting selective glycan expression on subsets of EVs.
Surface glycans may be important for the uptake of EVs by recipient cells [Citation9,Citation68,Citation69]. B cell-derived EVs, for example, are enriched with α-2,3-linked sialic acid allowing their capture by Siglec-1 (CD169) on macrophages in both spleen and lymph nodes [Citation6]. The inhibitory effect of heparin on EV uptake indicates an important role for heparin sulphate proteoglycans in EV uptake [Citation70,Citation71], and interference of D-mannose with EV uptake by DCs suggests an EV uptake mechanism via C-type lectin receptors [Citation72]. Glycoengineering has been used previously to alter the stability and pharmacokinetics of protein biopharmaceuticals [Citation73]. In this study, we enzymatically removed immune inhibitory sialic acids using a pan-sialic acid hydrolase. These sialidases have also been used to effectively remove sialic acids from EVs as a control for lectin binding to EV-associated sialic acids [Citation74]. We have previously demonstrated efficient DC targeting via DC-SIGN with glycopeptides and glyco-liposomes for antigen presentation and CD4+ and CD8+ T cell induction [Citation41,Citation65,Citation75]. Here we incorporated the high-affinity DC-SIGN ligand LeY into glioblastoma EVs for targeting of DCs.
Although anti-cancer vaccination with tumour-derived EVs has been evaluated in several studies [Citation37,Citation76,Citation77], concerns about the immunosuppressive properties of tumour EVs on immune effector cells and their role in metastatic niche formation led to caution in the use of these EVs for vaccination [Citation78,Citation79]. Combining tumour-derived EVs with the right immune-stimulating adjuvant such as a Toll-like receptor-3 agonist [Citation80], granulocyte-macrophage colony-stimulating factor (GM-CSF) [Citation81] or alpha-galactosylceramide [Citation76] has overcome this risk of immune inhibition by tumour-derived EVs. In patients with colorectal cancer, the feasibility and safety of immunization with ascites-derived EVs plus GM-CSF has been demonstrated with a few patients benefitting from this treatment [Citation81]. We previously showed that simultaneous triggering of TLR4 and DC-SIGN improves intracellular routing for cross-presentation on MHC-I for CD8+ T cell activation [Citation42], and that the incorporation of the TLR4 ligand MPLA into liposomes favours T cell responses after targeting antigen-containing liposomes to DCs [Citation65]. Thus, in future studies, different TLR stimulating adjuvants in combination with targeted tumour-derived EVs need to be compared for optimal DC activation and T cell induction.
Multi-antigenic immunization of patients with glioblastoma raised high hopes [Citation18,Citation25,Citation28]. Moreover, multi-antigenic immunization using EVs in mice with other types of tumours induced CD8+ T cell cross-priming and tumour rejection [Citation37]. Therefore, we hypothesize that multi-antigenic vaccination using highly stable EVs, glyco-modified for enhanced DC priming, could induce a potent anti-glioblastoma immune response. Thus, in future studies, EV immunization in a glioblastoma mouse model is indispensable. Aiming at personalized, multi-antigenic vaccination, subsequent studies should also focus on the identification of neoantigens in patient-derived exosomes and their capacity to induce in vitro autologous T cell activation and tumour killing. It is unknown whether desialylation of neo-antigens on the surface of EVs would affect their processing by dendritic cells, subsequent presentation of the antigens to lymphocytes and tumour recognition by induced lymphocytes. Therefore, future studies should also compare the induction of autologous T cell activation by glioblastoma-derived EVs before and after glyco-modification. To enable clinical application, the source of a sufficient yield of autologous tumour-derived EVs needs to be explored. A commercial hemopurifier for the extracorporeal capture of circulating EVs from blood [Citation82] has recently been developed. However, the yield and purity of tumour-derived EVs has yet to be demonstrated. Alternatively, the yield of tumour-derived EVs from cerebrospinal fluid or from conditioned culture medium with resected tumour tissue could be explored. Also, the timing of vaccination in the context of standard of care with dexamethasone, temozolomide and radiotherapy will be pivotal. T cell activation is hampered by the use of dexamethasone, a potent corticosteroid often used to treat intracranial oedema in patients with glioblastoma [Citation18]. Temozolomide, the alkylating chemotherapeutic agent used in the first line of treatment of glioblastoma and radiotherapy, could potentially increase the mutational load and immunogenicity of the tumour and its EVs, but continuous administration of low-dose TMZ is also associated with a high incidence of lymphocytopenia [Citation83]. Radiotherapy is also associated with lymphocytopenia but could on the other hand aid in turning the usually immune desert glioblastoma microenvironment into an inflamed tumour attracting immune effector cells needed in a vaccination strategy. Although immune stimulation using PD-1 blockade alone does not seem to result in a marked delay or prevention of disease relapse following surgery in patients, anti-PD-1 enhances the local and systemic immune response [Citation22] and might therefore accelerate a vaccine-induced immune response [Citation23].
In conclusion, in this study, the glycocalyx of glioblastoma-derived EVs was found to be dominated by immunosuppressive sialic acid-capped N-glycans and complex bi-antennary glycans while lacking ligands for DC-SIGN, an effective receptor for DC targeting and antigen presentation to both CD4+ and CD8+ T cells. Glycan modification of glioblastoma-derived EVs strongly enhanced their capacity to be internalized by moDCs, making modified EVs an attractive cell-free source for multi-antigenic pulsing of DCs in the context of anti-glioblastoma vaccination.
Author contributions
SD, SK and NP performed experiments, HK and MA synthesized palmitoyl-LewisY, SD and JG analysed the data, RN, MP, TW, YK and JG supervised experiments. SD and JG drafted the manuscript. All authors revised the article and gave their approval for publication.
Disclosure of interest
The authors report no conflict of interest.
Supplemental Material
Download Zip (189.7 MB)Acknowledgments
We kindly acknowledge Jacques Neefjes for providing anti-CD63 antibody for ELISA and TEM experiments. We would like to thank Nicole van der Wel and Jan van Weering for advice for improvement of our TEM images. Rubina Baglio and Monique A. J. van Eijndhoven kindly advised us about EV isolation procedures for which we are very grateful. We thank Edwin van der Pol his assistance with NTA analysis. The authors thank Xandra O. Breakefield for critically reading the manuscript.
This work was financially supported by the Institute for Chemical Immunology (ICI0011), ERC Advanced Glycotreat 339977 and the Cancer Center Amsterdam (CCA2014-5-18).
Supplementary material
Supplemental data for the article can be accessed here.
Additional information
Funding
Related Research Data
References
- Yáñez-Mó M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:1–14.
- Skog J, Würdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476.
- de Vrij J, Maas SLN, Kwappenberg KMC, et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int J Cancer. 2015;137:1630–1642.
- Berenguer J, Lagerweij T, Zhao XW, et al. Glycosylated extracellular vesicles released by glioblastoma cells are decorated by CCL18 allowing for cellular uptake via chemokine receptor CCR8. J Extracell Vesicles. 2018;7:1–21.
- Izquierdo-Useros N, Lorizate M, Puertas MC et al. Siglec-1 is a novel dendritic cell receptor that mediates HIV-1 trans-infection through recognition of viral membrane gangliosides. PLoS Biol. 2012;10:1–13.
- Saunderson SC, Dunn AC, Crocker PR, et al. CD169 mediates the capture of exosomes in spleen and lymph node. Blood. 2014;123:208–216.
- Zaia J. Mass spectrometry and glycomics. Supramol Struct Funct. 2010;9(14):89–102.
- Krishnamoorthy L, Bess JW, Preston AB, et al. HIV-1 and microvesicles from T cells share a common glycome, arguing for a common origin. Nat Chem Biol. 2009;5:244–250.
- Batista BS, Eng WS, Pilobello KT, et al. Identification of a conserved glycan signature for microvesicles. J Proteome Res. 2011;10:4624–4633.
- van der Vos KE, Abels ER, Zhang X et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 2015;18:244.
- Mallawaaratchy DM, Hallal S, Russell B, et al. Comprehensive proteome profiling of glioblastoma-derived extracellular vesicles identifies markers for more aggressive disease. J Neurooncol. 2017;131:233–244.
- Van Der Vos KE, Abels ER, Zhang X, et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 2016;18:58–69.
- Wei Z, Batagov AO, Schinelli S, et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat Commun. 2017;8.
- Figueroa JM, Skog J, Akers J, et al. Detection of wild-Type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro Oncol. 2017;19:1494–1502.
- Stupp R, Mason WP, Van Den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996.
- Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341.
- Baratta MG. Glioblastoma is ‘hot’ for personalized vaccines. Nat Rev Cancer. 2019;245:41568.
- Keskin DB, Anandappa AJ, Sun J et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2018. DOI:10.1038/s41586-018-0792-9.
- Ampie L, Woolf EC, Dardis C. Immunotherapeutic advancements for glioblastoma. Front Oncol. 2015;5:1–8.
- Lim M, Xia Y, Bettegowda C, et al. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–442.
- Weller M, Roth P, Preusser M, et al. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat Rev Neurol. 2017;13:363–374.
- Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–486.
- Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma Letters. Nat Med. 2019;25:470–476.
- Zhao J, Chen AX, Gartrell RD, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25:462–469.
- Liau LM, Ashkan K, Tran DD et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16:1.
- Platten M, Bunse L, Wick W, et al. Concepts in glioma immunotherapy. Cancer Immunol Immunother. 2016;65:1269–1275.
- Weller M, Butowski N, Tran DD et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373–1385.
- Hilf N, Kuttruff-Coqui S, Frenzel K et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2018. DOI:10.1038/s41586-018-0810-y.
- Ridler C. Personalized vaccines use tumour fingerprint to target glioblastoma. Nat Rev Neurol. 2019;15:2019.
- Tacken PJ, Torensma R, Figdor CG. Targeting antigens to dendritic cells in vivo. Immunobiology. 2006;211:599–608.
- Caminschi I, Maraskovsky E, Heath WR. Targeting dendritic cells in vivo for cancer therapy. Front Immunol. 2012;3:1–13.
- Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258.
- Jensen PE. Recent advances in antigen processing and presentation. Nat Immunol. 2007;8:1041–1048.
- Neefjes J, Jongsma MLM, Paul P, et al. Towards a systems understanding of MHC class i and MHC class II antigen presentation. Nat Rev Immunol. 2011;11:823–836.
- Syn NL, Wang L, Chow EKH, et al. Exosomes in cancer nanomedicine and immunotherapy: prospects and challenges. Trends Biotechnol. 2017;35:665–676.
- Morelli AE, Larregina AT, Shufesky WJ et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 2004;104:3257–3266.
- Wolfers J, Lozier A, Raposo G et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;38:297–303.
- Fehres CM, Garcia-Vallejo JJ, Unger WWJ, et al. Skin-resident antigen-presenting cells: instruction manual for vaccine development. Front Immunol. 2013;4:157.
- Unger WWJ, Van Kooyk Y. ‘Dressed for success’ C-type lectin receptors for the delivery of glyco-vaccines to dendritic cells. Curr Opin Immunol. 2011;23:131–137.
- van Kooyk Y, Unger WWJ, Fehres CM, et al. Glycan-based DC-SIGN targeting vaccines to enhance antigen cross-presentation. Mol Immunol. 2013;55:143–145.
- Fehres CM, van Beelen AJ, Bruijns SCM, et al. In situ delivery of antigen to DC-SIGN + CD14 + dermal dendritic cells results in enhanced CD8 + T-cell responses. J Invest Dermatol. 2015;135:2228–2236.
- Horrevorts SK, Duinkerken S, Bloem K, et al. Toll-like receptor 4 triggering promotes cytosolic routing of DC-SIGN-targeted antigens for presentation on MHC class I. Front Immunol. 2018;9:1–13.
- Appelmelk BJ, van Die I, van Vliet SJ, et al. Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol. 2003;170:1635–1639.
- Engering A, Geijtenbeek TBH, van Vliet SJ, et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol. 2002;168:2118–2126.
- Pontén J, MACINTYRE E. Long term culture of normal and neoplastic human glia. Acta Pathol Microbiol Scand. 1968;74:465–486.
- Wakimoto H, Kesari S, Farrell CJ et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481.
- Bax M, García-Vallejo JJ, Jang-Lee J, et al. Dendritic cell maturation results in pronounced changes in glycan expression affecting recognition by siglecs and galectins. J Immunol. 2007;179:8216–8224.
- Geijtenbeek TBH, Torensma R, van Vliet SJ, et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses efficient T cell activation (Hernandez-Caselles et al., 1993). Indeed, antibody engagement of ICAM-3 results in elevation of intracellular ca. Cell. 2000;100:575–585.
- Thery C, Clayton A, Sebastian Amigorena GR. Isolation and characterization of exosomes from cell culture supernatants. Curr Protoc Cell Biol. 2006;supplement:1–29.
- Böing AN, Van Der Pol E, Grootemaat AE et al. Single-step isolation of extracellular vesicles from plasma by size-exclusion chromatography. Int Meet ISEV Rotterdam. 2014;3:118.
- Shelke GV, Lässer C, Gho YS, et al. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J Extracell Vesicles. 2014;3:1–8.
- Raposo G, Nijman HW, Stoorvogel W, et al. Lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161–1172.
- Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci. 2010;107:6328–6333.
- Baglio SR, van Eijndhoven MAJ, Koppers-Lalic D, et al. Sensing of latent EBV infection through exosomal transfer of 5′pppRNA. Proc Natl Acad Sci. 2016;113:E587–E596.
- Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom. 2010;53:10.17.1–10.17.24.
- Garcia-Vallejo JJ, Bloem K, Knippels LMJ, et al. The consequences of multiple simultaneous C-type lectin-ligand interactions: DCIR alters the endo-lysosomal routing of DC-SIGN. Front Immunol. 2015;6:1–12.
- Maas SLN, De VJ, Broekman MLD Quantification and size-profiling of extracellular vesicles using tunable resistive pulse sensing. 1–7 (2014). doi:10.3791/51623
- Lötvall J, Hill AF, Hochberg F, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the international society for extracellular vesicles. J Extracell Vesicles. 2014;3:26913.
- Roy S, Lin H-Y, Chou C-Y, et al. Navigating the landscape of tumor extracellular vesicle heterogeneity. Int J Mol Sci. 2019;20:1–22.
- Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266.
- Lübbers J, Rodríguez E, Kooyk YV. Modulation of immune tolerance via siglec-sialic acid interactions. Front Immunol. 2018;9:1–13.
- Zhang JQ, Nicoll G, Jones C, et al. Siglec-9, a novel sialic acid binding member of the immunoglobulin superfamily expressed broadly on human blood leukocytes.. J Biol Chem. 2000;275:22121–22126.
- Stanczak MA, Siddiqui SS, Trefny MP, et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. 2018;128:4912–4923.
- van Liempt E, Bank CMC, Mehta P, et al. Specificity of DC-SIGN for mannose- and fucose-containing glycans. FEBS Lett. 2006;580:6123–6131.
- Boks MA, Ambrosini M, Bruijns SC, et al. MPLA incorporation into DC-targeting glycoliposomes favours anti-tumour T cell responses. J Control Release. 2015;216:37–46.
- van Kooyk Y, Rabinovich G. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9:593–601.
- Lener T, Gimona M, Aigner L, et al. Applying extracellular vesicles based therapeutics in clinical trials - An ISEV position paper. J Extracell Vesicles. 2015;4.
- Escrevente C, Grammel N, Kandzia S, et al. Sialoglycoproteins and N-glycans from secreted exosomes of ovarian carcinoma cells. PLoS One. 2013;8:e78631.
- Barres C, Blanc L, Bette-Bobillo P et al. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood. 2010;115:696–706.
- Christianson HC, Svensson KJ, van Kuppevelt TH, et al. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci. 2013;110:17380–17385.
- Atai NA, Balaj L, van Veen H, et al. Heparin blocks transfer of extracellular vesicles between donor and recipient cells. J Neurooncol. 2013;115:343–351.
- Hao S, Bai O, Li F et al. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology. 2006;120:90–102.
- Sola RJ, Griebenow K. Glycosylation of therapeutic proteins an effective strategy to optimize efficacy. BioDrugs. 2010;24:9–21.
- Gomes J, Gomes-Alves P, Carvalho S, et al. Extracellular vesicles from ovarian carcinoma cells display specific glycosignatures. Biomolecules. 2015;5:1741–1761.
- García-vallejo JJ, Unger WWJ, Kalay H, et al. Glycan-based DC-SIGN targeting to enhance antigen cross-presentation in anticancer vaccines. 1–3 (2013).
- Liu H, Chen L, Liu J, et al. Co-delivery of tumor-derived exosomes with alpha-galactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett. 2017;411:182–190.
- Andre F, Schartz NE, Movassagh M et al. Mechanisms of disease Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002;360:295–305.
- Altevogt P, Bretz NP, Ridinger J, et al. Novel insights into exosome-induced, tumor-associated inflammation and immunomodulation. Semin Cancer Biol. 2014;28:51–57.
- Peinado H, Alečković M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891.
- Adams M, Navabi H, Croston D et al. The rationale for combined chemo/immunotherapy using a Toll-like receptor 3 (TLR3) agonist and tumour-derived exosomes in advanced ovarian cancer. Vaccine. 2005;23:2374–2378.
- Dai S, Wei D, Wu Z, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther. 2008;16:782–790.
- Marleau AM, Chen CS, Joyce JA, et al. Exosome removal as a therapeutic adjuvant in cancer. J Transl Med. 2012;10:1–12.
- Brock CS, Newlands ES, Wedge SR et al. Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res. 1998;58:4363–4367.