
ABSTRACT
Extracellular vesicles have the capacity to transfer lipids, proteins, and nucleic acids between cells, thereby influencing the recipient cell’s phenotype. While the role of RNAs in EVs has been extensively studied, the function of DNA remains elusive. Here, we distinguished novel heterogeneous subpopulations of small extracellular vesicles (sEVs) based on their DNA content and topology. Low- and high-density sEV subsets from a human mast cell line (HMC-1) and an erythroleukemic cell line (TF-1) were separated using high-resolution iodixanol density gradients to discriminate the nature of the DNA cargo of the sEVs. Paired comparisons of the sEV-associated DNA and RNA molecules showed that RNA was more abundant than DNA and that most of the DNA was present in the high-density fractions, demonstrating that sEV subpopulations have different DNA content. DNA was predominately localised on the outside or surface of sEVs, with only a small portion being protected from enzymatic degradation. Whole-genome sequencing identified DNA fragments spanning all chromosomes and mitochondrial DNA when sEVs were analysed in bulk. Our work contributes to the understanding of how DNA is associated with sEVs and thus provides direction for distinguishing subtypes of EVs based on their DNA cargo and topology.
Introduction
Extracellular vesicles (EVs) are membrane-enclosed particles released by most cell types from both prokaryotic and eukaryotic organisms in an evolutionarily conserved and regulated manner [Citation1]. EVs generated during physiological and pathological conditions can be classified and named based on their biogenesis, cellular origin, and properties [Citation2,Citation3]. EVs can be divided into different subgroups: (i) exosomes, which are small EVs (sEVs) secreted as a result of the fusion of multivesicular bodies to the plasma membrane, (ii) microvesicles or microparticles, which are large EVs released by shedding or budding from the plasma membrane, (iii) and apoptotic bodies, which are cell membrane fragments generated during programmed cell death [Citation1–Citation3]. However, it has recently been suggested that EVs are more complex entities than previously perceived and that they can be further divided into subpopulations [Citation4–Citation6], with one potentially new EV subgroup based on their nucleic acid content.
The release of cellular DNA in many different structural forms such as apoptotic blebs, histone/DNA complexes or nucleosomes, DNA/RNA-lipoprotein complexes or virtosomes, DNA traps, etc., has been well documented [Citation7–Citation10]. Such extracellular structures, classified in umbrella terms such as circulating DNA or cell-free DNA, largely serve to protect the DNA from nucleases that are present in, for example, the circulation and to reduce the likelihood of DNA being seen as a danger signal by the immune system [Citation11]. As nucleases are essential enzymes that control DNA repair and therefore, genomic stability, their defects or absence are associated with diseases in which the sensing of self-nucleic acids is critical [Citation12]. For instance, the knockout of the DNase I and II family members are linked to severe autoimmune and metabolic diseases [Citation13].
The ability of EVs to transfer their cellular cargo and effectively deliver it to recipient cells has been primarily demonstrated for the transfer of functional RNA species and proteins [Citation14–Citation16]; however, the presence and role of DNA in EVs have not been characterized in detail. To date, only a limited number of reports have described the presence of DNA species, including single-stranded (ss)DNA, double-stranded (ds)DNA, and mitochondrial (mt)DNA, in EV subpopulations such as microvesicles and exosomes [Citation17–Citation29].
Over the last few years, several studies have shown that many different cell types, particularly cancer cells, actively release cell-free DNA that can be found in free form or associated with various EV subpopulations [Citation11]. Interestingly, cancer-derived EVs have been shown to contain DNase-resistant genomic DNA (gDNA) fragments of the entire genome spanning all chromosomes and reflecting the mutational signature of the original tumours [Citation19–Citation21,Citation28]. These studies have exclusively focused on DNA resistant to nucleases as DNase was applied to an EV pellet before vesicle lysis. However, it was not addressed how and to what degree the DNA is associated with EVs. Moreover, the DNA analyses were performed on pelleted vesicles, lacking extra purification steps to avoid the co-isolation of EVs with cell-free circulating DNA or other contaminants. We and others have recently shown that this extracellular DNA is sensitive to enzymatic digestion and is associated with the surface of EVs where it plays a role in EV aggregation as well as internalization, and it is responsible for the increased zeta-potential of EVs [Citation22,Citation30,Citation31]. Nonetheless, systematic analyses of extracellular DNA regarding its origin, organization, and association with vesicles are missing.
Here, we investigated the DNA content and topology of sEVs separated using high-resolution iodixanol density gradients. sEVs were characterized in terms of their protein and nucleic acid content (ss/dsDNA and total RNA). We found that most of the DNA associated with sEVs was in the high-density (HD) fractions, corresponding to non-canonical sEVs and/or non-vesicular material, and only small amounts of DNA were detected in the low-density (LD) fractions. The latter were sEVs with preserved shape, morphology, and canonical protein vesicle markers. In addition, most of the DNA content detected in both LD and HD fractions was surface-associated and was degraded upon enzymatic treatment without affecting the integrity of the vesicles. Taken together, the knowledge about the distribution of DNA cargo among density fractions as well as the topology of the DNA associated with sEVs provides a much-needed baseline for comparative studies of the nucleic acid cargo of sEVs and contributes to further understand the roles and implications of EVs in diseases.
Material and methods
Cell culture
The human mast cell line HMC-1 (a gift from Dr Joseph Butterfield, Mayo Clinic, Rochester, MN, USA) was cultured in IMDM (HyClone, Logan, UT) with 1.2 mM α-thioglycerol (Sigma-Aldrich, St Louis, MO). The erythroleukemic cell line TF-1 (ATCC: CRL-2003) was grown in RPMI 1640 medium supplemented with 5 ng/mL granulocyte-macrophage colony-stimulating factor (HyClone). All media were supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), 100 units/mL penicillin (HyClone), 100 μg/mL streptomycin (HyClone), and 2 mM L-glutamine (Sigma-Aldrich). The FBS was EV-depleted by ultracentrifugation at 120,000 × gavg for 18 h at 4°C (Type 45 Ti, Beckman Coulter, Brea, CA) and filtered through a 0.22 µm filter before added to the media. All cells were grown at 37°C in 5% CO2 humidified incubator. Cells were passaged every 2–3 days at 70%–80% confluence.
EV isolation and iodixanol density gradient separation
Cell-conditioned media (600–1,200 mL) from 80% confluent HMC-1 and TF-1 cells grown in T175 cell culture flasks (2 × 106 HMC-1 cells/mL) and (1 × 106 TF-1 cells/mL) was used for EV isolation. Cell viability at the time of EV harvesting was on average 99% for HMC-1 and 96% for TF-1 cells. For the DNA-capture experiments, EVs were isolated from cell-conditioned media from HMC-1 cells treated with 10 µM 5-Bromo-2´-Deoxyuridine (BrdU) (Thermo Fisher Scientific, Waltham, MA, USA). Cells were first removed by centrifugation at 300 × g for 10 min, and cell debris and larger EVs were removed by centrifugation at 2,000 × g for 20 min and then at 16,500 × gavg for 20 min. An exosome-enriched fraction, here called sEVs, was collected at 118,500 × gavg (Type 45 Ti, k-factor 217.6, Beckman Coulter) for 2.5 h. The sEV-enriched pellet was further fractionated by flotation on iodixanol density gradients (Sigma-Aldrich) (isopycnic centrifugation). A total of 1 mL of PBS-sEV sample was mixed with 3 mL of 60% iodixanol and laid at the bottom of the tube, and 1 mL layers of 35%, 30%, 28%, 26%, 24%, 22%, 22%, and 20% iodixanol were subsequently overlaid forming a discontinuous gradient. Samples were ultracentrifuged at 180,000 × gavg (SW 41 Ti, k-factor 143.9, Beckman Coulter) for 16 h. Fractions of 1 mL were collected from the top to bottom, and the density of each iodixanol fraction was measured by absorbance at 340 nm (Varioskan, Thermo Fisher Scientific). Two white layers were observed in the interphase of F1–F2 and F4–F5 in all gradients. Their distribution between those fractions could slightly vary upon collection. Next, samples were transferred to new tubes, diluted and washed in PBS (up to 94 mL) and ultracentrifuged at 118,500 × gavg for 3.5 h (Type 70 Ti or 45Ti, k-factor 133.7 or k-factor 217.6 respectively, Beckman Coulter). All centrifugations were done at 4°C. The sEV pellets were resuspended in PBS and were freshly processed or stored at −80°C.
Protein extraction and Western blotting
For protein extraction, HMC-1 and TF-1 cells were collected, washed twice in ice-cold PBS, and solubilised in RIPA lysis and extraction buffer supplemented with Halt Protease Inhibitor Cocktail immediately before use (Thermo Fisher Scientific). Lysed samples were incubated on ice for 15 min, followed by three sonications of 5 min each with vortexing of samples in between. Total protein content was determined using the Qubit protein assay kit (Thermo Fisher Scientific) following the manufacturer´s protocol.
For Western blotting, sEV samples and lysed cells were prepared in NuPAGE lithium dodecyl sulfate sample buffer (4x) with NuPAGE Sample Reducing Agent (10x) (Thermo Fisher Scientific) except for samples blotted with CD63 and CD81 antibodies that were run under non-reducing conditions. Samples were heated to 70°C for 10 min and loaded on gels. Proteins were separated on 4%-12% SDS-PAGE Bis-Tris gels (Life Technologies) and transferred using Trans-Blot Turbo Mini or Midi polyvinylidene fluoride transfer packs (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked with Odyssey TBS Blocking Buffer (LI-COR Biosciences Inc, Lincoln, NE) for 1 h at room temperature (RT) and then incubated with the following primary antibodies diluted in TBS Odyssey blocking buffer at 4°C overnight: anti-CD81 (M38, 1:1,000 dilution, Abcam, Cambridge, UK, catalogue no. ab79559), anti-CD9 (1:1,000 dilution, Abcam, catalogue no. ab97999), anti-CD63 (TS63, 1:1,000 dilution, Abcam, catalogue no. ab59479), anti-Alix (3A9, 1:500 dilution, Abcam, catalogue no. ab117600), anti-Flotillin-1 (clone 18, 1:500 dilution, BD Biosciences, San Jose CA, catalogue no. 610820), anti-TSG101 (1:1,000 dilution, Abcam, catalogue no. ab30871), anti-β-Actin (AC-15, 1:4,000 dilution, Sigma-Aldrich, catalogue no. A1978), anti-Calnexin (1:1,000 dilution, Abcam, catalogue no. ab22595), anti-Histone H2A (1:1,000 dilution, Abcam, catalogue no. ab18255), and anti-Histone H3 (EPR17785, 1:1,000 dilution, Abcam, catalogue no. ab201456). The membranes were washed three times with 0.1% TBS-Tween and then incubated for 1 h at RT with the following secondary antibodies diluted 1:20,000 in 0.1% TBS-Tween: IRDye 680RD goat anti-mouse IgG (H + L) catalogue no. 925–68070, IRDye 680RD goat anti-rabbit IgG catalogue no. 925–68071, IRDye 800CW goat anti-mouse IgG catalogue no. 925–32210, or IRDye 800CW goat anti-rabbit IgG catalogue no. 926–32211 (all from LI-COR). The membranes were then washed three times for 5 min with TBS-Tween, visualized with the Odyssey CLx imaging system (LI-COR), and analysed with the Image Studio v.4.0.
Particle size and concentration
The sizes and particle concentrations of the sEVs present in each gradient fraction were measured using a ZetaView PMX 110 (Particle Metrix). Samples were diluted in 0.22 µm filtered PBS before the measurements. Triplicate measurements were obtained from each sample analysing two stationary layers with five measurements per layer. Camera sensitivity was kept at 70 (arb.unit) in all measurements. Data were analysed using the ZetaView analysis software (v 8.2.30.1) with a minimum size of 5 nm, a maximum size of 1,000 nm, and minimum brightness of 20. Settings were kept constant between measurements.
Transmission electron microscopy
Five microliters or three micrograms of sEVs were used for the electron microscopy (EM) characterization of all fractions and the DNase experiments, respectively. sEVs were loaded onto glow-discharged formvar/carbon-coated copper grids (Ted Pella, Inc., Redding, CA, USA). The grids were incubated with the samples for 15 min, washed three times in PBS, fixed in 2% paraformaldehyde and 2.5% glutaraldehyde with three PBS washing steps in between, and contrasted in 2% uranyl acetate at RT. The preparations were examined using a LEO 912AB Omega electron microscope with a 2k x 2k VELETA Olympus CCD camera (Carl Zeiss NTS, Jena, Germany).
Proteomic analysis
The sEV samples were lysed by the addition of SDS to a final concentration of 2%. Fifty micrograms of sEVs were digested with trypsin using the filter-aided sample preparation method, as previously described [Citation32]. Peptide samples were desalted with PepClean C18 spin columns (Thermo Fisher Scientific) according to the manufacturer’s guidelines prior to analysis on a Q Exactive mass spectrometer (Thermo Fisher Scientific) interfaced with an Easy nLC 1200 liquid chromatography system. Peptides were separated using an in-house constructed C18 analytical column (200 mm × 0.075 mm I.D., 3 μm pore size, Dr Maisch, Germany) and a gradient from 7% to 40% acetonitrile in 0.2% formic acid over 75 min. MS/MS analysis was performed in a data-dependent mode, where the most intense precursor ions at charge states 2 to 7 were selected for fragmentation. Dynamic exclusion was set to 30 s. Raw files were searched, and exclusion lists were generated for a second MS/MS analysis of each sample. Data analysis was performed using Proteome Discoverer v.1.4 (Thermo Fisher Scientific) against Human Swiss-Prot Database v. Nov. 2014 (Swiss Institute of Bioinformatics, Switzerland). The two raw files for each sample were merged in the searches. Mascot 2.3 (Matrix Science) was used as a search engine with a precursor mass tolerance of 5 ppm and a fragment mass tolerance of 100 mmu. Tryptic peptides were accepted with one missed cleavage, and methionine oxidation and cysteine alkylation were set as variable and static modifications. The detected peptide threshold in the software was set to Mascot99 by searching against a reversed database. Identified proteins were grouped by sharing the same sequences to minimize redundancy.
DNase treatment
Resuspended sEV pellets were divided into two aliquots (paired samples). One sample was subjected to DNase I treatment prior to further experiments such as DNA isolation, EM, flow cytometry, or enzyme-linked immunosorbent assay (ELISA). The Turbo DNA-free kit (Thermo Fisher Scientific) was used to completely digest the DNA present on the outside of the sEVs. The concentration of DNase I used for the treatment of sEVs was optimized using cellular DNA. One unit of Turbo DNase was found to digest 1 µg of total cellular DNA and therefore was chosen for the sEV experiments. A hundred microliters of each sEV sample (variable protein and particle count depending on the fraction processed) were mixed with 1U of Turbo DNase in 10x Turbo DNase buffer followed by incubation at 37°C for 30 min following the kit´s recommendations. For the DNA isolation experiments, 5 µL of resuspended DNase Inactivation Reagent was mixed with the samples to remove the Turbo DNase enzyme and divalent cations completely. The samples were then centrifuged at 10,000 × g for 2 min, and the supernatant containing the sEVs was transferred to a fresh tube and directly subjected to DNA isolation. For the flow cytometry and EM experiments, samples were vortexed twice for 30 sec, which is expected to reduce the DNase activity. The enzyme was not removed from the preparation as the addition of the DNase Inactivation Reagent was found to interfere with the downstream analysis of the sEV samples.
DNA isolation and quantification
Small EV samples dissolved in 200 µL of PBS solution generated by iodixanol density gradient fractionation, underwent DNA extraction and analysis. DNA was isolated from all DNase-treated and non-treated sEV fractions using the QIAamp DNA Mini kit (Qiagen) following the manufacturer’s instructions. Small EV samples were treated with Proteinase K solution (40 mAU/mg protein) and RNase A (100 mg/mL) before DNA extraction. After DNA isolation, samples were eluted in 200 µL of DEPC water, and DNA size and concentrations were analysed using the Bioanalyzer 2100 instrument with High Sensitivity DNA and DNA 7500 kits (Agilent Technologies Inc., Palo Alto, CA, USA) according to the manufacturer´s protocols. The dsDNA High Sensitivity and ssDNA Assay Kits (Thermo Fisher Scientific) were also used to quantify ss/ds EV-associated DNA using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, California, CA, USA). All DNA samples were aliquoted and stored at −20°C.
Flow cytometry
Samples were incubated with anti-CD63-coated beads (Thermo Fisher Scientific, catalogue no. 10606D) overnight at 4°C with gentle agitation (10–15 µg protein/50,000 beads/antibody). The sEV-bead complexes were washed twice with 1% EV-depleted FBS in PBS, incubated with human IgG (Sigma-Aldrich, catalogue no. I4506) for 15 min at 4°C, washed twice, and incubated with phycoerythrin (PE)-labeled anti-CD63 or PE-isotype control (BD Bioscience, catalogue no. 556020 and 555749, 1:20 dilution) for 40 min at RT. The samples were washed twice, and 10,000 events were acquired on a FACSVerse (BD Bioscience) and analysed using FlowJo Software version 7.6.5 (Tri Star Inc., Ashland, OR, USA).
Whole-genome deep-sequencing and bioinformatics of sEV-associated DNA
After density floatation of TF-1 and HMC-1 sEVs, fractions 1 to 7 were pooled and washed in PBS by ultracentrifugation at 118,000 × gavg for 3.5 h using a Type 70 Ti rotor. The resulting pellet was divided into two samples, one that remained untreated and one that was treated with DNase prior to DNA extraction and sequencing. DNA sequencing libraries were fragmented with a Covaris E220 ultrasonicator to 350 bp insert sizes and prepared using the Truseq Nano DNA library kit on the NeoPrep instrument (Illumina, CA, USA) according to the manufacturer´s instructions. Input DNA (25 ng) was used for the non-DNase treated HMC-1 and TF-1 sEVs following Illumina´s recommendations. The amount of DNA for the TF-1 DNase-treated sEV sample was limited to a maximum of 9.5 ng, which was used for sequencing. Clustering was done by “cBot,” and samples were sequenced on a HiSeq2500 (HiSeq Control Software 2.2.58/RTA 1.18.64) with a 2 × 126 setup using “HiSeq SBS Kit v4” chemistry. The Bcl to FastQ conversion was performed using bcl2fastq_2.17 from the CASAVA software suite, and the Sanger/phred33/Illumina 1.8+ quality scale was used. The raw sequencing data were quality filtered with prinseq (v.0.20.3), and adapters were removed with cutadapt (v.1.9). The reads were mapped towards the human reference genome (hg19) using bowtie2 (v.2.2.9), and positions with fewer than five reads were removed. The remaining positions were binned within three ranges (5–10 x, 11–50 x, and 51 x or more). Parts per million (PPMs) were calculated and plotted with circus (v.0.67.7) using the ylgnbu-9-seq colour palette, with a max value of 0.05, except for where the max value was 4.
RNA isolation and quantification
RNA was isolated from all sEV fractions using the miRCURY RNA Isolation Cell and Plant kit (Exiqon A/S, Vedbaek, Denmark) with 1% 2-mercaptoethanol in the lysis buffer and following the manufacturer´s protocol. The optional on-column DNA removal protocol was applied to ensure the maximum removal of residual DNA from the RNA isolations. For that, 0.25 U of DNase I was added to each column and incubated at 37°C for 30 min according to the manufacturer´s recommendations. One microliter of the RNA from each sample was denatured at 72°C for 2 min, and the RNA concentration and size was measured with a Bioanalyzer 2100 using the RNA 6000 Nano and Pico total RNA kits (Agilent Technologies) according to the manufacturer´s protocol. RNA was also quantified using RNA BR and HS Assay kit using a Qubit 2.0 Fluorometer.
ELISA assays
For the indirect sandwich ELISA, an anti-BrdU antibody (BU1/75 (ICR1), 5 µg/mL, Abcam, catalogue no. ab6326) was coated on a black 96-well plate and incubated overnight at 4°C. Plates were blocked with 1% BSA in PBS for 1 h at RT. For each sEV sample, 100 µg were added to the plate and incubated for 2 h at RT. Next, anti-CD9 antibody (p24, 1 µg/mL, BD Biosciences, catalogue no. 555370) was added to the samples and incubated for 1 h at RT. After washing three times for 5 min with PBS, HRP-conjugated anti-mouse antibody was incubated with the samples for 1 h. Luminescent signal was measured with the BM Chemiluminescence ELISA Substrate (BD Biosciences) as recommended by the manufacturer. For the direct ELISA, 100 µg of sEVs were coated on 96-well plates overnight at 4°C. Plates were blocked with 1% BSA in PBS for 1 h and incubated with anti-CD9 antibody (BD Biosciences) for 2 h at RT. After washing three times for 5 min with PBS, an HRP-conjugated secondary antibody was incubated with the samples for 1 h at RT, and the luminescent signal was measured using a Varioskan instrument.
Data availability
The raw reads for the whole genome deep-sequencing were deposited to the Sequence Read Archive of the National Centre for Biotechnology Information under bioproject ID PRJNA52133. (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA521337). The proteomics data have been submitted to EVpedia [Citation33]. We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV190045) [Citation34].
Results
Identification of distinctive extracellular DNA and RNA profiles in sEVs
We have previously demonstrated that RNA cargo differs in subpopulations of EVs with different densities [Citation35]. To determine whether EV subpopulations also vary in their DNA content, sEVs were isolated from HMC-1 and TF-1 cell lines by differential centrifugation and bottom-up iodixanol density flotation gradient (Supplementary Figure 1(a)). From top to bottom, nine density layers were collected (F1–F9) () and total DNA and RNA were extracted and analysed for each fraction as paired samples. At this stage, no DNase or RNase were added to the samples and the nucleic acid content of the sEVs comprised both outer membrane-associated and intravesicular DNA and RNA molecules (). The percentages of DNA and RNA in the fractions showed that the nucleic acid distributions were similar for both cell lines across the gradients (). In general, DNA molecules were mainly found in the F4–F7 fractions, with most of the DNA being single-stranded for both HMC-1 and TF-1 fractions as determined by fluorometric quantification (Supplementary Figure 1(b)). Relatively little DNA was detected in F1–F3 and F8–F9 (). Large amounts of RNA were found in F1 as well as in F4–F7 (), and capillary electrophoretic analysis showed that the RNAs detected in F1–F3 had a narrow peak for short RNA species (25–200 nucleotides (nt)) and were enriched or co-sedimented with ribosomal RNA (rRNA), which was detected as distinct 18S and 28S peaks (). In contrast, the RNAs detected in F4–F7 had a broader peak for the RNAs (25–1,000 nt) than the narrow peak below 200 nt observed in F1–F3 for short RNA. Furthermore, no prominent rRNA peaks were observed in F4–F7. Regarding DNA, the detected fragments ranged from 500 to 10,000 bp in all density fractions (). Based on the DNA and RNA content and the nucleotide profiles in the different fractions, F1–F3 were grouped as they show similar features such as low DNA content and high RNA content with visible rRNA peaks. F1–F3 will hereafter be referred to as the low density (LD) fractions. Moreover, F4–F7 were grouped as they showed common characteristics such as high RNA and DNA content and no visible peaks for the rRNA subunits. F4–F7 will hereafter be referred to as high-density (HD) fractions. F8 and F9 were not included in these classifications due to their high density (1.22 to 1.28 g/mL) that indicated that these fractions could contain non-floated material.
Figure 1. Floatation on iodixanol gradients separates sEVs with distinct RNA and DNA profiles.
(a) Density gradient fractionation of HMC-1 and TF-1 small EVs (sEVs). Nine fractions of 1 mL each were collected from top to bottom from iodixanol density gradients and washed in 38-94 mL of PBS by ultracentrifugation. Their densities were analysed by measuring the absorbance at 340 nm ranging from 1.111 to 1.278 g/mL for HMC-1 (F1–F9) and from 1.103 to 1.284 g/mL for TF-1 (F1–F9). Data are plotted as the mean of three independent experiments ± SEM. F1–F3 were considered as low-density (LD) fractions and F4–F7 as high-density (HD) fractions. (b) Representative illustration of nucleic acids, including RNA species (blue) and DNA species (orange), associated with the inside and outside of the sEVs. (c–d) Percentage of DNA (orange) and RNA (blue) in the HMC-1 and TF-1 gradient fractions. DNA and RNA concentrations were quantified with High-Sensitivity DNA and DNA 7500 chips and RNA 6000 Nano and Pico total RNA kits. Bars represent the mean + SEM of three independent experiments for DNA and two independent experiments for RNA. (e) Representative RNA Bioanalyzer profiles (blue) of HMC-1 and TF-1 F1–F7 from Pico total RNA chips. (f) Representative DNA Bioanalyzer profiles (orange) from HMC-1 and TF-1 F1–F7 run in High-Sensitivity DNA chips. The y-axis of the electropherograms represents fluorescent units (FU) and the x-axis represents the nucleotide length (nt). Please note that the y-axes have different scales. (g-h) Bar graphs representing the total amount (ng) of DNA (orange) and RNA (blue) in LD and HD fractions from HMC-1 and TF-1 density gradients. Bars show the mean + SEM of three independent experiments for DNA and two independent experiments for RNA.
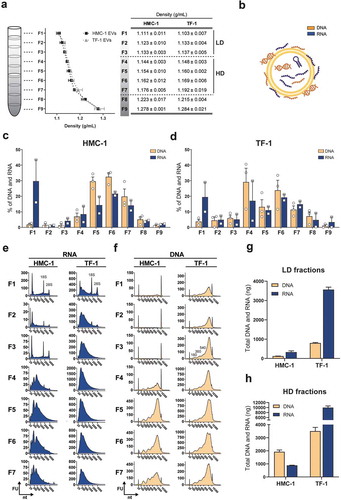
The total RNA and DNA quantifications from capillary electrophoretic measurements showed that the LD fractions were mostly enriched in RNAs compared to DNA, primarily due to the presence of rRNA in F1, with a DNA to RNA ratio of 1:2.6 for HMC-1 and 1:4.5 for TF-1 (). For the HD fractions, the total nucleic acid content differed between EV origins. HMC-1 fractions were enriched in DNA molecules with a DNA to RNA ratio of 2.2:1, while TF-1 fractions were mainly enriched in RNAs with a 1:2.9 DNA to RNA ratio (). Together this shows that the RNA/DNA cargo varied among the density fractions from different cell lines, with LD fractions having the most prominent rRNA peaks and little DNA and HD fractions having most of the DNA cargo and small RNAs with no visible peaks for rRNA.
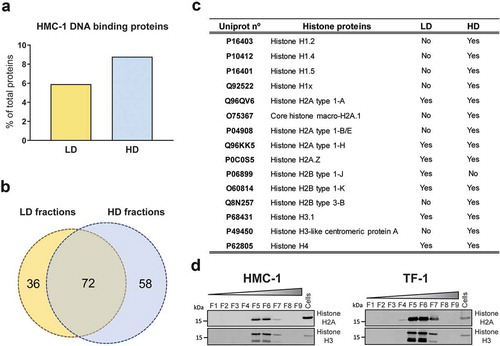
Identification of DNA binding proteins by proteomic analysis of LD and HD fractions
Notably, the DNA fragments identified in most of the gradient fractions had characteristic peaks around 180, 360, and 540 bp (most clearly visible in F5 from HMC-1 and F3 from TF-1, ) resembling those of nucleosomes (DNA wrapped around histone proteins). Interestingly, the proteomic analysis of HMC-1 fractions showed the presence of notably more DNA-binding proteins in the HD fractions (8.9% of the proteins) compared to the LD fractions (5.9% of the proteins) (). As shown in the Venn diagram (), 72 DNA-binding proteins were identified in both preparations, whereas 36 and 58 proteins were only detected in LD and HD HMC-1 fractions, respectively. Analysing the DNA binding proteins in more detail, the HD fractions were found to contain more DNA-binding histone proteins, including the core histones H2A, H2B, H3, and H4 that form the nucleosome and the linker histone H1 (). These results were validated by Western blotting, confirming that DNA-binding histones H2A and H3 were mainly associated with HD fractions rather than LD sEVs (). Overall, characteristic DNA profiles of nucleosomal DNA were detected in most of the fractions. Mass spectrometry and Western blotting confirmed the enrichment of DNA binding proteins, including core and linker histones, in the HD fractions that contained most of the DNA. However, the same proteins were limited in number or absent in the LD fractions, which contained less DNA.
Figure 2. Proteomics analysis of DNA-binding proteins in LD and HD fractions.
(a) Percentage of DNA-binding proteins from the total number of proteins identified in the analysis of LD (yellow) and HD (blue) HMC-1 floated fractions. (b) Venn diagram showing the number of unique and common DNA-binding proteins identified in LD and HD HMC-1 fractions. (c) Table displaying the Histone proteins identified in the LD and HD fractions. UniProt accession numbers and complete histone names are shown. (d) After density flotation and fractionation of HMC-1 and TF-1 small EVs in high-resolution iodixanol gradients, equal volume (9 µL) of each fraction (F1–F9) and 10 µg of total whole-cell lysates were loaded on SDS-PAGE gels. Membranes were blotted with Histone H2A and Histone H3 antibodies.
Characterization of sEVs from LD and HD fractions
Analysis of the presence of canonical EV proteins and non-vesicular proteins markers in all the density fractions from both cell lines confirmed the separation of LD and HD vesicles. LD sEV fractions (mostly visible in F1) were characterised by the enrichment of proteins like Alix, Flotillin-1, TSG101, CD63, CD81, and CD9 (except for CD9 in TF-1) compared to cell lysates. Moreover, LD sEVs were characterised by the absence of other non-vesicular proteins such as β-Actin and Calnexin. In contrast, HD fractions (mostly visible in F5 and F6) showed variable levels of luminal and transmembrane EV proteins, with detectable but lower levels of Calnexin compared to cell lysates and high levels of β-Actin ().
Figure 3. Characterization of sEVs in LD and HD fractions.
(a) After density flotation and fractionation of HMC-1 and TF-1 sEVs in high-resolution iodixanol gradients, equal volume (15 µL) of each fraction (F1–F9) and 10 µg of total whole-cell lysates were loaded on SDS-PAGE gels. Membranes were blotted with the following antibodies: Alix (~100 kDa), Flotillin-1 (~48 kDa), TSG101 (~47 kDa), CD63 (~30–60 kDa), CD81 (~22 kDa), CD9 (~20 kDa), β-Actin (~42 kDa) and Calnexin (~90 kDa). (b) Nanoparticle Tracking Analysis of the total number of particles per fraction from the HMC-1 and TF-1 gradients (ZetaView). Bars represent the mean ± SEM of two independent experiments run in triplicate. (c) Representative negative staining electron transmission microscopy images of HMC-1 and TF-1 high-resolution density fractions (F1, F2, F5, F6). Five microliters were loaded to the grids per each fraction. Scale bars = 200 nm.
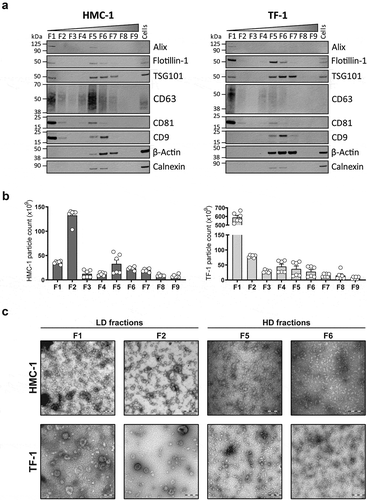
Regarding particle counts per fraction, most of the sEV material was isolated in F1 and F2 (). The particle counts for the remaining fractions were significantly lower, with a slight increase in particle number from F5–F7 for HMC-1 and F4–F6 for TF-1. Nanoparticle Tracking Analysis (NTA) of F1 and F2 showed a narrow size distribution centred around 120 nm for sEVs from both cell sources with similar size distribution but much lower particle count for HD fractions (Supplementary Figure 2(a)). The comparison of HMC-1 and TF-1 sEVs isolated from the same volume of supernatant revealed that TF-1 cells produce over 2.5-fold more particles than HMC-1 (). However, it is worth highlighting that the majority of the current NTA-based techniques cannot detect vesicles with a diameter below 70 nm [Citation36]. Therefore, the sEVs (<70 nm) present in the LD and HD fractions are not detected, which can affect the measurements on particle number and size distribution.
EM analysis of all fractions confirmed the separation of different sEV subpopulations based on their density (, Supplementary Figure 2(b)). Characteristic EV structures were identified from F1–F6 with different morphology, abundance, and purity of the preparations. Both F1 and F2 from HMC-1 and TF-1 cells contained most of the sEVs with preserved shape and high purity. From F3 to F6, the number of sEVs observed was significantly lower, and the preparations had co-precipitants. We could not visually identify sEVs in F8–F9 as the samples contained mostly non-vesicular material (Supplementary Figure 2(b)).
DNA in sEVs is associated with the vesicle surface
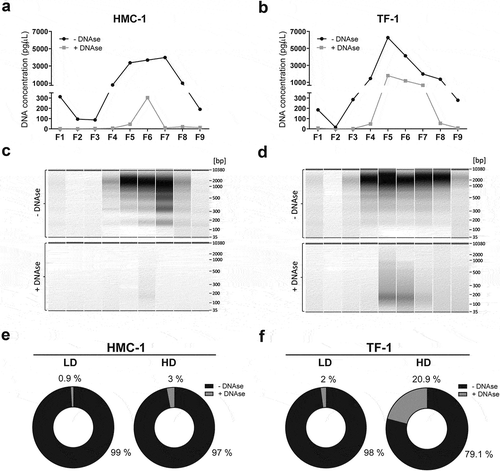
To investigate whether the DNA of sEVs is associated with the vesicle surface or is protected within the particles, HMC-1 and TF-1 fractions were divided into two samples. One sample was treated with DNase, and thus, any DNA on the outside of the sEVs would be degraded. The other sample was kept untreated and therefore would contain DNA from both the vesicle surface as well as internally protected DNA. Next, DNA was extracted from both samples following the same procedure including vesicle lysis. When the amount of DNA in the individual fractions was analysed before, and after DNase treatment, HMC-1 LD sEVs were shown to carry small quantities of DNA most of it degraded upon the enzymatic treatment, whereas HMC-1 HD fractions contained more DNA partly protected from enzymatic degradation (). When the DNA from TF-1 was analysed, a similar trend was observed (). TF-1 LD fractions carried small quantities of DNA that were mainly degraded after DNase treatment, whereas the HD fractions contained more substantial amounts of DNA that were partly protected from enzymatic degradation (). The electrophoretic DNA profiles of HMC-1 and TF-1 fractions prior to and after DNase treatment showed that only F5–F6 from HMC-1 gradients and F4–F8 from TF-1 gradients had remaining DNA resistant to DNase ( and Supplementary Figure 3). While many of the large DNA fragments (>0.7 kb) were removed after enzymatic treatment, smaller DNA fragments (<0.4 kb) remained in the fraction isolates, as seen in the TF-1 DNA electrophoretic profiles from F5–F7 ( and Supplementary Figure 3). Almost all the DNA found in the LD fractions for both cell types corresponded to DNA associated with the outside of the sEVs as over 97% was degraded by DNase treatment (). Most of the DNA found in HD fractions was also associated with the vesicle surface. However, 3% of the DNA in HMC-1 and 20.9% of the DNA in TF-1 fractions was protected from enzymatic degradation, suggesting either its localisation on the inside of the EV membrane or its protection by other non-vesicular material (). Together, these results indicate that a substantial portion of the LD and HD EV-associated DNA is not protected by the phospholipid bilayer membrane of the sEVs and is thus, present on the outside of the vesicles where it is susceptible to degradation by DNase.
Figure 4. Most of the vesicle-associated DNA is not protected against DNase treatment and thus is localised on the vesicle surface.
Each collected gradient fraction was divided into two. One half was DNase treated (grey) while the other half served as untreated control (black). (a-b) The DNA concentration before and after Turbo DNase I digestion of HMC-1 fractions and TF-1 fractions was determined by capillary electrophoresis using high-sensitivity DNA bioanalyzer chips. (c-d) Representative capillary electrophoretic gels of HMC-1 and TF-1 fractions (F1–F9) prior to DNase addition (represented as −) and after DNase treatment (represented as +). (e-f) Pie charts representing the percentage of total DNA of LD and HD HMC-1 and TF-1 density fractions before (−DNase, black) and after DNase treatment (+DNase, grey).
To verify that the loss of DNA signal was not a consequence of the sEVs being disrupted after the DNase, the integrity of the sEVs was analysed by EM and flow cytometry (). EM analysis of HMC-1 and TF-1 LD and HD fractions revealed that the sEVs retained their integrity and morphology after the enzymatic treatment (). These results were confirmed by flow cytometry, where anti-CD63 beads were used to capture sEVs after DNase treatment. As shown in the histograms (), non-treated and DNase-treated sEVs had a corresponding signal, suggesting that the integrity of the sEVs was preserved.
Figure 5. sEVs are intact after DNase treatment.
The resulting 1 mL fractions from the HMC-1 and TF-1 high-resolution iodixanol density fractionation were pooled based on low-density (LD) (F1–F3) or high-density (HD) F4–F7, washed with PBS, and re-pelleted by ultracentrifugation at 118,500 × gavg for 3.5 h. sEV samples were divided into two aliquots, one of which was DNase-treated (+DNase), and the other was kept untreated and used as a control (Ctrl). (a) Representative electron microscopy images of +DNase and Ctrl sEVs are shown. Three micrograms of the sample were loaded per each grid. Scale bars are 500 nm in the full pictures and 100 nm in the magnifications. (b) The DNase-treated and non-treated sEVs were bound to anti-CD63 beads, labelled with anti-CD63-Phycoerythrin (PE), and evaluated using flow cytometry. Histograms of non-treated (NT) sEVsCD63+ (blue), DNase-treated (+DNase) sEVs CD63+ (red), and isotype control (grey) are shown. Count (y-axis) vs. PE signal (x-axis) is shown. (c) HMC-1 cells were treated with 10 µM 5-Bromo-2´-Deoxyuridine (BrdU) to label newly synthesized DNA. Next, cell-conditioned media from the BrdU-treated cells was collected and used to isolate sEVs using differential centrifugation followed by density fractionations as described in material and methods. Next, LD and HD fractions were run in an in-house generated enzyme-linked immunosorbent assay (ELISA). The schematic representation of the sandwich ELISA system designed to measure and capture BrdU+/CD9+ sEV is shown. LD and HD samples were NT or DNase+, and the graph represents the mean ± SEM of three independent experiments per group. (d) Direct ELISA-based detection of CD9+ signal on pre-captured DNase+ sEV. The graph represents the mean ± SEM of three independent experiments per group. HRP = horseradish peroxidase, RLU = relative luminescence units.
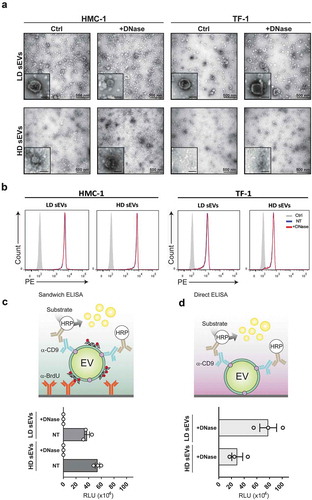
After demonstrating that most of the EV-associated DNAs were present on the outside of the sEVs, we asked, whether the surface DNA could be used to capture sEV subpopulations. For this, sEVs from BrdU-treated cells were isolated following differential centrifugation and density gradient floatation as described above. A sandwich ELISA detection system was developed with anti-BrdU as the capturing antibody and anti-CD9 antibody as the detection antibody (). The BrdU/CD9 antibody combination allowed the specific capture and detection of CD9+ EVs by their surface-associated DNA in LD and HD HMC-1 fractions (). To confirm that the assay specifically bound surface DNA, DNase treated sEVs were analysed as a control. A total reduction of the luminescence signal was observed when DNase-treated EVs were incubated with the BrdU antibody, indicating that the binding was specific for surface-associated DNA. A higher signal was also detected in the HD fractions compared to LD fractions, despite the lower particle number in HD fractions, indicating that HD fractions contained more DNA on the sEV surface or more DNA associated with protein complexes in comparison to LD fractions. To validate that the lack of signal was not due to the disruption of vesicles by the enzymatic treatment, the EV integrity was investigated using a direct ELISA for the detection of CD9+ sEVs after DNase treatment (). In both LD and HD HMC-1 fractions, the CD9 signal was retained indicating that after DNase treatment the samples contained intact CD9+ sEVs, thus complementing the EM and flow cytometry results.
The entire human genome is represented both on the inside and outside of the sEVs
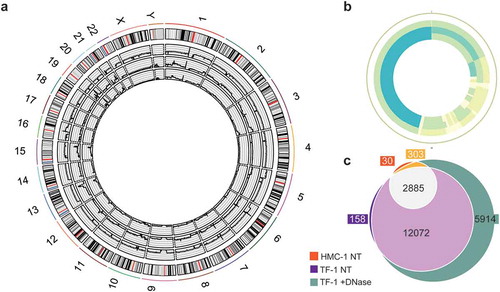
The DNA from HMC-1 and TF-1 fractions was subjected to whole-genome sequencing (WGS). sEVs from HMC-1 and TF-1 cells were isolated and floated according to Supplementary Figure 1(a). Then F1–F7 were pooled, pelleted, and divided into two aliquots, one that was DNase-treated (protected DNA) and one that was kept untreated (total DNA) prior to DNA isolation. Next, the protected DNA from TF-1 fractions, the total DNA from TF-1 sEVs fractions, and the total DNA from HMC-1 fractions were analysed by WGS. Only 3.9% of the DNA of the HMC-1 fractions remained after DNase treatment (). The remaining amount of DNA was too small for sequencing analysis, and consequently, this sample was excluded.
The sequencing analysis revealed broadly distributed regions of the entire human genome in both DNase-treated and non-treated samples, except for the Y chromosome in the HMC-1 sample (, Supplementary Figure 4). In the WGS analysis, 113, 176, and 203 million reads were identified in the total DNA from HMC-1 fractions, total DNA from TF-1 fractions, and DNase-treated TF-1 fractions, resulting in a mean depth of 9.25 X, 14.47 X, and 15.88 X, respectively. All samples had over 88% of bases with a quality score above 30, and of all the reads 95.8%, 95.4%, and 94.3% were aligned in the HMC-1 total DNA, TF-1 total DNA, and TF-1 protected DNA, respectively. DNA fragments of various lengths spanning all chromosomes were detected, including the mitochondrial chromosome, which was highly covered in all samples (, Supplementary Figure 4), demonstrating that both gDNA and mtDNA were resistant to DNase and thus, protected from degradation by sEVs or other non-vesicular material. mtDNA genes coding for subunits of OXPHOS complex I (ND1–ND6 and ND4L), cytochrome C oxidase of complex IV (COI-COIII), cytochrome B oxidase of complex III (CytB) and complex V (ATPase 6 and 8), 22 tRNAs, 2 rRNAs (12S and 16S), and the D-loop containing sequences for initiation, replication, and transcription were identified. Because the areas around the centrosomes are highly repetitive, these areas are commonly hard to map, which was also seen in our data set (Supplementary Figure 4). Several regions of the genome were highly covered with >0.05 RPMs (represented in dark blue, Supplementary Figure 4). The genes located in these regions were listed and compared, showing that the DNA isolated from TF-1 fractions had more genes mapped with coverage of over 0.05 RPM compared to the DNA isolated from HMC-1 fractions (). Furthermore, the DNase-treated TF-1 fractions that only contained DNA protected from degradation had more genes with high coverage than the non-DNase-treated samples that contained total DNA, including DNA on the outside of the vesicles and/or DNA inside the sEVs or associated to protein complexes (). Together, this demonstrates that gDNA spanning all chromosomes and mtDNA fragments from sEVs are protected from enzymatic treatment and therefore, existing as a luminal EV cargo or as a non-vesicular DNA-protein complex. The DNase-treated samples had the highest coverage despite the low amount of DNA detected after DNase treatment. Interestingly, we did not identify DNA fragments exclusively present on the sEV surface.
Figure 6. Whole-genome coverage on the sEV-associated DNA.
F1–F7 from HMC-1 and TF-1 gradients were pooled, pelleted, and divided into two aliquots, one that was DNase-treated (protected DNA) and one that was left untreated (total DNA) prior to DNA isolation. Next, total DNA from TF-1 fractions, HMC-1 fractions, and DNase-treated TF-1 fractions was analysed by WGS. (a) Circos plot presenting the 46 chromosomes of the three sequenced DNA samples: HMC-1 non-treated (edge sample), TF-1 non-treated (middle sample), and TF-1 DNase treated (inner circle sample). (b) Circos plot representing the mitochondrial chromosome: HMC-1 non-treated (edge sample), TF-1 non-treated (middle sample), and TF-1 DNase treated (inner circle sample). Coverage colouring from low to high (max four parts per million) – white (lowest), light green, light blue, dark blue (highest). (c) Venn diagram comparing the genes in the three samples with coverage of more than 50 reads: orange (HMC-1 non-treated, NT), purple (TF-1 NT), and green (TF-1 +DNase-treated).
Discussion
In contrast to extracellular RNA, EV-associated DNA represents a more unexplored source of information, possibly due to the lack of understanding of how the DNA might be associated or packed within sEVs and what its function in the extracellular space is. In this study, by using state-of-the-art high-resolution density gradients, we separated two distinct sEV subpopulations that differed in their nucleic acid content. Moreover, we compared in a paired fashion the total RNA and DNA cargo associated with LD and HD fractions, and we were able to characterise their distinct DNA content and topology from two different cell types.
Our analysis revealed a distinct separation of sEV subpopulations based on density. The LD fractions contained the majority of the sEV particles with characteristic vesicle morphology, size, and enrichment for canonical EV protein markers with an absence of non-vesicular proteins. In contrast, fewer sEVs were found in the HD fractions, and although they were positive to a lesser extent for canonical EV protein markers, they also contained histone proteins and other non-vesicular proteins. These results indicate that LD sEVs are most likely what we know as classical “exosomes” derived from intraluminal vesicles within the endosomal compartment, whereas HD sEVs might well represent non-canonical exosomes, sEVs released by different subcellular organelles like plasma membrane, or protein/nucleic acid complexes.
Our RNA/DNA analysis showed that LD sEV fractions carried minor quantities of DNA and contained relatively large amounts of RNA detectable as 18S and 28S rRNA peaks. In comparison, HD fractions were enriched in DNA, and the RNA profiles consisted mainly of small RNA and mRNAs with the absence of prominent rRNA peaks. These observations support previous findings from our group and others regarding the RNA content of EVs isolated using sucrose or iodixanol gradients in which full-length 18S and 28S rRNA peaks were detected in the LD fractions and absent or detected to a lesser extent in the HD fractions [Citation35,Citation37,Citation38]. However, this might not be a general feature of LD EVs, as other studies analysing sEVs from different origins do not detect or showed variable rRNA content in EVs [Citation39–Citation41]. Interestingly, this study demonstrates that HD fractions comprising the “non-canonical exosomes” and/or macromolecular complexes carry most of the DNA cargo that can be co-isolated with vesicle and non-vesicle like material, with only minor quantities of DNA detected in LD sEVs “canonical exosomes”. Both ssDNA and dsDNA fragments were identified in the LD and HD fractions from both cell types, although most of the fragments were ssDNA in nature. Similar results for the ss/ds DNA ratio have recently been reported for large EVs [Citation42], although the dsDNA cargo of vesicles has been investigated in more detail [Citation18–Citation21,Citation28]. Overall, the nature of this finding and whether the relative abundance of ssDNA is due to, for example, dsDNA denaturation or due to the inaccuracy of the current techniques to precisely differentiate between short ss/ds DNA fragments remains to be determined. It is worth mentioning that fluorometric quantification assays are not completely precise in the discrimination between ssDNA and dsDNA fragments as these assays can have a cross-detection of DNA species. Therefore, future studies should consider the use of specific ss/ds nucleases or atomic force microscopy for a proper determination of the ssDNA vs dsDNA ratio in EVs. A proper enzymatic quantification of ssDNA vs dsDNA fragments in all fractions would also require the isolation of LD sEVs containing enough DNA for all treatments, which was not possible in our study using sEVs from HMC-1 and TF-1 origin.
Interestingly, the characteristic profile of sEVs in which nucleosomal DNA is found together with DNA-binding proteins prompted us to examine whether the DNA detected in the high-resolution density fractions is associated with DNA histone proteins. We could indeed confirm that HD fractions contained larger DNA fragments up to 4 kb and a higher number of core and linker histones compared with LD fractions. These observations are not surprising because several previous proteomics studies have described the presence of histone proteins in EVs [Citation40,Citation43], and they also support that most of the gDNA detected in large EVs is chromatinized [Citation42]. The presence of histones H2A and H3 in HD fractions and absence in LD fractions was validated by Western blotting, in which histone positive fractions co-localized with HD sEVs. A recent study also showed that histones H2A, H3, and H4 could be found in HD fractions where most of the DNA was detected, in which even some EV markers were partly identified [Citation29]. Increasing evidence suggests that linker and core histones are actively mobile complexes that can cross the plasma membrane and can be localised in various cell compartments [Citation11]. Since histones associate with DNA, these complexes are likely capable of moving DNA across cells, which could potentially explain one of the mechanisms of DNA transfer.
Our observation that distinct subtypes of sEVs have different DNA contents led us to examine the nature of the DNA association to vesicles and the degree of protection of the DNA. DNase digestion showed that most of the sEV-associated DNA (79–99%) was not protected from enzymatic degradation and consequently, it was present on the vesicle surface of both LD and HD fractions. The smaller fraction of the DNA that was resistant to degradation suggested that this DNA was most likely localised within the EV lumen or it was partially protected by other non-vesicular material. These results confirm our previous work describing the association of DNA with the EV surface [Citation30] and support recent findings showing that the DNA content of sEVs is mainly surface-associated [Citation31,Citation44]. It is also worth mentioning that the first studies reporting the presence of DNA in EVs strongly suggested that the DNA was present inside EVs because it was protected against DNA nucleases [Citation18–Citation21,Citation28]. For example, Thakur et al. showed that after enzymatic treatment, DNA species greater than 2.5 kb in size were degraded while DNA fragments between 100 bp and 2.5 kb remained within the internal EV cargo [Citation21]. In contrast, others have suggested that long DNA fragments are mostly found within the EVs rather than outside [Citation18]. Although no further separation methods were applied at that time, we also found that a minor amount of DNase-resistant DNA can be detected in sEVs. Importantly, our study further revealed that the vesicle integrity was preserved after enzymatic treatment, which is crucial for downstream applications. Moreover, since most of the DNA is present on the vesicle surface in the sEVs, we developed a proof-of-concept system for capturing sEVs by their surface DNA, which together with the analysis of DNA content, might represent a unique opportunity to capture EVs for biomarker discovery.
Our WGS analysis of the density fractions further revealed a high coverage of genes from the entire human genome with a representation of all chromosomes. The coverage was higher in DNase-treated fractions despite the DNA concentration being significantly lower. We could not identify any specific genomic regions that were selectively included in EVs based on coverage, which agrees with previous findings [Citation19–Citation23,Citation25]. Notably, high coverage of the mitochondrial chromosome with extensive copies of mtDNA in both DNase-treated and non-treated fractions was observed. The presence of mtDNA in EVs has been ambiguously reported. While several groups have detected mtDNA in EVs derived from various sources, e.g. human plasma, vascular smooth muscle cells, glioblastoma cells, etc. [Citation17,Citation18,Citation23], most of the studies describing DNA in vesicles have not considered mtDNA [Citation19,Citation20] or have concluded that it is absent in EVs [Citation21,Citation44]. These discrepancies in the DNA content of EVs among studies could reflect, once again, the diversity and heterogeneity of EVs and/or the isolation methods, and this highlights the need to study vesicle subsets from various origins [Citation4,Citation24,Citation28,Citation40]. Previous reports have shown that cancer cell-derived EVs contain more DNA in comparison to benign cells [Citation21] and that the DNA content is increased in response to antibiotic and antitumor agents [Citation31,Citation45]. This heterogeneity is likely a consequence of the diverse mechanisms of DNA packing or association within sEVs, which might vary in cells of different origins. On the other hand, this could also be explained by various studies having enriched diverse EV subsets, highlighting the importance of dissecting the cargo based on the vesicle subtype. To date, the mechanisms behind DNA incorporation into the EV surface or within the vesicle lumen remain unknown. It has recently been suggested that the diversity of EV DNA content might also reflect DNA damage and possible chromosomal fragmentation within the cell [Citation42]. However, we and others have shown that EVs harbour DNA-binding proteins that might assist in the attachment of DNA to the EV surface [Citation31], which could contribute to the transfer of DNA across the cell membranes [Citation11].
Our study presents a new and comprehensive process for systematically separating LD and HD sEVs with a different nucleic acid content using high-resolution iodixanol density gradients, and it offers a pair-wise comparison of the distribution of sEV-associated DNA and RNA molecules in HMC-1 and TF-1 fractions. The topology of the DNA found in EVs was also analysed in detail. Most of the sEV-harbored DNA was not protected from enzymatic degradation and thus, was localised to the EV surface. We also provide evidence that both protected and DNase-sensitive sEV-associated DNA covered the entire human genome with DNA fragments from both mitochondrial and genomic origin. Although our study answers many relevant questions related to DNA cargo, association, and topology in sEV subpopulations, more mechanistic studies are required to clarify the origin of the DNA in EVs and its specific functions.
Declaration of Interests
ELI is currently employed and holds equity in AstraZeneca R&D. SCJ is presently employed by Codiak BioSciences Inc., USA. JL holds equity in Codiak BioSciences, Inc. JL, ELI, CL, and SCJ are inventors of patents using EVs as either therapeutic or diagnostics tools.
Supplemental Material
Download Zip (5.5 MB)Acknowledgments
The authors acknowledge support from the National Genomics Infrastructure in Stockholm funded by the Science for Life Laboratory, the Knut and Alice Wallenberg Foundation, and the Swedish Research Council and acknowledge the SNIC/Uppsala Multidisciplinary Centre for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure. We want to thank Gunnar Nilsson at the Karolinska Institute, Stockholm, Sweden, for the kind gift of the human mast cell line HMC-1. The authors would like to acknowledge data analysis support from the Bioinformatics Core Facility at the Sahlgrenska Academy, University of Gothenburg, and microscopy support from the Centre for Cellular Imaging at the University of Gothenburg and the National Microscopy Infrastructure, NMI (VR-RFI 2016-00968). The authors acknowledge Lukas Badertscher for his assistance with graphic design. We thank the Proteomics Core Facility at Sahlgrenska Academy, University of Gothenburg University, for performing the proteomic analysis.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Yáñez-Mó M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066.
- van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213.
- Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–16.
- Lässer C, Jang SC, Lötvall J. Subpopulations of extracellular vesicles and their therapeutic potential. Mol Aspects Med. 2018;60:1–14.
- Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):E968–77.
- Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750.
- Thierry A, El Messaoudi S, Gahan PB, et al. Origins, structures, and functions of circulating DNA in oncology. Cancer Metast Rev. 2016;35(3):347–376.
- von Köckritz-Blickwede M, Goldmann O, Thulin P, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. 2008;111(6):3070–3080.
- Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241.
- Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61(4):1659–1665.
- Peters DL, Pretorius PJ. Origin, translocation and destination of extracellular occurring DNA—a new paradigm in genetic behaviour. Clin Chim Acta. 2011;412(11):806–811.
- Yang W. Nucleases: diversity of structure, function and mechanism. Q Rev Biophys. 2011;44(1):1–93.
- Keyel PA. Dnases in health and disease. Dev Biol. 2017;429(1):1–11.
- Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659.
- Ratajczak J, Miekus K, Kucia M, et al. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006;20(5):847–856.
- Deregibus MC, Cantaluppi V, Calogero R, et al. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood. 2007;110(7):2440–2448.
- Guescini M, Genedani S, Stocchi V, et al. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J Neural Transm. 2010;117(1):1–4.
- Cai J, Han Y, Ren H, et al. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J Mol Cell Biol. 2013;5(4):227–238.
- Kahlert C, Melo SA, Protopopov A, et al. Identification of Double stranded genomic DNA Spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;288(12):26888–26897.
- Lee TH, Chennakrishnaiah S, Audemard E, et al. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem Biophys Res Commun. 2014;451(2):295–301.
- Thakur BK, Zhang H, Becker A, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24(6):766–769.
- Fischer S, Cornils K, Speiseder T, et al. Indication of horizontal DNA gene transfer by extracellular vesicles. PLoS One. 2016;11(9):e0163665.
- Sisquella X, Ofir-Birin Y, Pimentel MA, et al. Malaria parasite DNA-harbouring vesicles activate cytosolic immune sensors. Nat Commun. 2017;8(1):1985.
- García-Romero N, Carrión-Navarro J, Esteban-Rubio S, et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget. 2016,8(1):1416.
- San Lucas FA, Allenson K, Bernard V, et al. Minimally invasive genomic and transcriptomic profiling of visceral cancers by next-generation sequencing of circulating exosomes. Ann Oncol. 2016;27(4):635–641.
- Yang S, Che SPY, Kurywchak P, et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol Ther. 2017;18(3):158–165.
- Allenson K, Castillo J, San Lucas FA, et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol. 2017;28(4):741–747.
- Lázaro‐Ibáñez E, Sanz-Garcia A, Visakorpi T, et al. Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: apoptotic bodies, microvesicles, and exosomes. Prostate. 2014;74(14):1379–1390.
- Jeppesen DK, Fenix AM, Franklin JL, et al. Reassessment of exosome composition. Cell. 2019;177(2):428–445. e18.
- Shelke GV, Jang SC, Yin Y, et al. Human mast cells release extracellular vesicle-associated DNA. Matters. 2016;2(2):e201602000034.
- Németh A, Orgovan N, Sódar BW, et al. Antibiotic-induced release of small extracellular vesicles (exosomes) with surface-associated DNA. Sci Rep. 2017;7(1):8202.
- Wiśniewski JR, Zougman A, Mann M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J Proteome Res. 2009;8(12):5674–5678.
- Kim D-K. EVpedia: a community web portal for extracellular vesicles research. Bioinformatics. 2014;31(6):933–939.
- Van Deun J, Mestdagh P, Agostinis P, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14(3):228.
- Lässer C, Shelke GV, Yeri A, et al. Two distinct extracellular RNA signatures released by a single cell type identified by microarray and next-generation sequencing. RNA Biol. 2017;14(1):58–72.
- Van der Pol E, Coumans FAW, Grootemaat AE, et al. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J Thromb Haemost. 2014;12(7):1182–1192.
- Willms E, Johansson HJ, Mäger I, et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci Rep. 2016;6:22519.
- Hong BS, Cho J-H, Kim H, et al. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics. 2009;10:556–2164-10–556.
- Lázaro-Ibáñez E, Lunavat TR, Jang SC, et al. Distinct prostate cancer-related mRNA cargo in extracellular vesicle subsets from prostate cell lines. BMC Cancer. 2017;17(1):92.
- Zhang H, Freitas D, Kim HS, et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat Cell Biol. 2018;20(3):332.
- Turchinovich A, Drapkina O, Tonevitsky A, et al. Transcriptome of extracellular vesicles: State-of-the-art. Front Immunol. 2019;10:202.
- Vagner T, Spinelli C, Minciacchi VR, et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J Extracell Vesicles. 2018;7(1):1505403.
- Kruger S, Elmageed ZYA, Hawke DH, et al. Molecular characterization of exosome-like vesicles from breast cancer cells. BMC Cancer. 2014;14:44.
- Takahashi A, Okada R, Nagao K, et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. 2017;8:15287.
- Kitai Y,Kawasaki T, Sueyoshi T, et al. DNA-containing exosomes derived from cancer cells treated with topotecan activate a STING-dependent pathway and reinforce antitumor immunity. J Immunol. 2017;198(4):1649–1659.