
ABSTRACT
Highly accumulated hyaluronan (HA) not only provides a physiological barrier but also supports an immune-suppressive tumour microenvironment. High-molecular-weight (HMW)-HA inhibits the activation of immune cells and their access into tumour tissues, whereas, low-molecular-weight oligo-HA is known to potentially activate dendritic cells (DCs). In this paper, we investigated whether small extracellular vesicle (EVs)-PH20 hyaluronidase induces tumour HA degradation, which, in turn, activates DCs to promote anti-cancer immune responses. Informed by our previous work, we used a small EV carrying GPI-anchored PH20 hyaluronidase (Exo-PH20) that could deeply penetrate into tumour foci via HA degradation. We found that Exo-PH20-treatment successfully activates the maturation and migration of DCs in vivo, particularly CD103+ DCs leading to the activation of tumour-specific CD8+ T cells, which work together to inhibit tumour growth. Moreover, combination with anti-PD-L1 antibody provided potent tumour-specific CD8+ T cell immune responses as well as elicited prominent tumour growth inhibition both in syngenic and spontaneous breast cancer models, and this anti-tumour immunity was durable. Together, these results present new insights for HA degradation by Exo-PH20, providing a better understanding of oligo HA-triggered immune responses to cancer.
Introduction
During the early anti-tumour immune response, pattern recognition receptors, such as Toll-like receptors (TLRs), recognize components of damaged cells that are called damage-associated molecular patterns (DAMPs) [Citation1,Citation2]. The DAMPs contain high-mobility group box 1 proteins, heat shock proteins, and ATP [Citation3,Citation4], in addition to molecules released by the degradation of extracellular matrix (ECM) [Citation5]. The hyaluronan (HA), has been the focus of research interest because it is particularly abundant in tumour tissues as a main component of the ECM in the tumour microenvironment (TME) [Citation6]. HA, which is typically very large in its nascent form (mass >1,000 kDa), has been associated with an extraordinarily wide variety of biological functions [Citation7,Citation8]. In addition, high-molecular-weight (HMW)-HA blocks access of immune cells, induces dysfunction of immune cells, and polarizes immune cells towards an immunosuppressive phenotype [Citation9,Citation10]. The HMW-HA accumulation has been connected with more aggressive malignant tumours in preclinical models and patients [Citation11,Citation12].
HMW-HA is broken down by an enzymatic reaction into small fragments [Citation7] that can interact with endothelial cells, dendritic cells (DCs) and macrophages. The small fragments of HA (oligo-HA) have been shown to have a pro-inflammatory influence on DCs [Citation13–Citation15]; however, the DAMP-relevant physiological functions of oligo-HA is still controversial [Citation16–Citation18]. In the present work, therefore, we sought to specifically investigate a direct rationale for the potential use of HAse as an immune-stimulatory molecule for the cancer immunotherapy.
PH20 hyaluronidase is a GPI-anchored membrane-associated protein that plays functional roles in the degradation of HMW-HA polymers to oligo-HA. In a previous study, we have developed engineered small extracellular vesicles (EVs) harbouring GPI-anchored PH20 hyaluronidase (Exo-PH20). Remarkably, our PH20 on small EV surface exhibited a 3-fold higher enzymatic activity than truncated form of recombinant PH20 proteins [Citation19], corresponding with that EVs have a unique ability to transfer their biological information, especially membrane proteins [Citation20].
There has been increasing interest in developing therapeutics aimed at interfering tumour growth by disrupting the ECM such as PEGylated recombinant human hyaluronidase (PEGPH20) has developed growing rapidly [Citation21]. However, challenges remain in our efforts to investigate the immunotherapeutic functions of molecules degraded from HMW-HA [Citation22,Citation23]. Here, we investigated whether PH20 hyaluronidase expressed on the small EV surface, which was previously shown to induce in situ HA degradation [Citation19], can activate DCs to promote anti-tumour immune responses. Indeed, our results revealed that Exo-PH20 produced HA fragments, and that this could induce DC activation via TLR-4 signalling. Our results also revealed that in vivo HA breakdown is an initiator of CD103+ DCs migration from the tumour site towards lymph nodes, and affects the capacity for T cell cross-priming. As a consequence of elevated production of IFN-γ, PD-L1 expression on tumour cells was also induced in vivo, that provides the rationale for combination therapy of Exo-PH20 with immune checkpoint blockade (ICB). Combination with the anti-PD-L1 antibody yielded improved tumour-specific CD8+ T cell immune responses and protected mice against tumour re-challenge. Collectively, these findings support the idea that Exo-PH20 could be useful for cancer immunotherapy, as it degrades HA in situ.
Materials and methods
Animals
Six- to seven-week-old male C57BL/6, female BALB/c and male nude mice were obtained from Orient Bio. OT-I mice (Tg(TcraTcrb)1100Mjb) and Batf3−/- mutant mice (B6.129S(C)-Batf3tm1.1Kmm/J)in the C5BL/6 background and FVB/N female mice (Tg(MMTVPyVT) 634Mul/J) were purchased from Jackson Laboratory. All animals were maintained in pathogen-free specific facilities and were used in accordance with protocols approved by the Korea Institute Science and Technology (KIST).
Reagents
HMW HA extracted from rooster comb was purchased from Sigma. Anti-PD-L1 (BE0061) antibodies and the isotype-rat IgG2b control (BE0090) were obtained from BioXcell. Anti-phospho MAPK, anti-MAPK and anti-NF-ĸB antibodies were purchased from Cell Signalling. APC-anti-CD11c (clone N418, 117310), PE-anti-CD40 (clone GL-1, 105008), PE-anti-CD86 (clone 3–23, 124610), PE-anti-CD80 (clone 16-10A1, 104707), PE-anti-H-2Kb bound to SIINFEKL (clone 25-D1.16, 141604), FITC-anti-CD103 (clone 2E7, 121419), PE/Cy7-anti-CCR7 (clone 4B12, 120124), PerCP/Cy5.5-anti-CD45.2 (clone 104, 109827), APC-anti-CD3 (clone 17A2, 100235), PE-anti-CD25 (clone 3C7, 101903), FITC-anti-CD4 (clone GK1.5, 100405), PE-anti-CD11b (clone M1/70, 101207), FITC-anti-Gr1 (clone RB6-8C5, 108405), FITC-anti-CD8α (clone 53–6.7, 100705), APC-anti-F4/80 (clone BM8, 123116), FITC-anti-MHCⅡ (clone M5/114.15.2, 107605) and PE-anti-CD206 (clone C068C2, 141705) antibodies were obtained from BioLegend. APC-Armenian hamster IgG (clone HTK888, 400912), APC-rat IgG2b (κ-isotype, clone RTK4530, 400605), PE-rat IgG2a (κ-isotype, clone RTK2758, 400508), PE-Armenian hamster IgG (cloneHTK888, 400907), FITC-Armenian hamster IgG (cloneHTK888, 400905), FITC-rat IgG2a (κ-isotype, clone RTK2758, 400505) PE/Cy7-rat IgG2b (κ-isotype, clone RTK2758, 400522), and PerCP/Cy5.5 Mouse IgG2a, (κ-isotype, MOPC-173, 400149) controls were purchased from BioLegend. Anti-CD8α antibodies (clone 53–6.7, 550281) and anti-CD16/CD32 (Fc blocker, clone 2.4G2, 553142) were purchased from BD Pharmingen. Anti-HABP2 (ab181837), anti-Tsg101 (ab83), anti-PH20 (ab196596) and anti-pan cytokeratin (ab86734) antibodies were obtained from Abcam. Anti-Alix (sc-9,010) and anti-CD81 (sc-166029) antibodies were purchased from Santa Cruz. Recombinant murine GM-CSF and IL-4 were obtained from Peprotech. Anti-PD-L1 (AF1019), anti-GAPDH (MAB5718) antibodies and the mouse IFN-gamma Quantikine ELISA Kit were purchased from R&D Systems. The CD11c, CD8a+ T cell and Naïve CD8a T cell isolation kit and the tumour dissociation kit were acquired from Miltenyi Biotech.
Cell culture
HEK293T cells and murine B16F10-Ova melanoma cells were maintained in Dulbecco’s modified Eagle’s medium (Hyclone) with 10% foetal bovine serum and 1% antibiotic-antimycotic. Murine 4T1 breast cancer cells were grown in RPMI-1640 midium (Hyclone) with 10% foetal bovine serum and 1% antibiotic-antimycotic. All cell lines were cultured at 37°C in 5% CO2.
To generate bone marrow-derived dendritic cells (BMDCs), bone marrow cells extracted from leg bones of C57BL/6 mouse. The cells were maintained in RPMI-1640 medium (Welgene) with 10% foetal bovine serum (Gibco) and 1% antibiotic-antimycotic. For differentiation into BMDCs, the bone marrow cells were treated with 20 ng/ml GM-CSF and 20 ng/ml IL-4 for 7 days.
Preparation and characterization of small EVs
HEK293T cells were cultured at 6.5 × 106 per 150 mm dish. After the medium was changed to serum-free DMEM containing Glutamax (Gibco), the cells were transfected with 20 μg of PH20 plasmid DNA (OriGene) using Lipofectamine 3000 (Invitrogen) and incubated for 48 h. For removal of debris and microvesicles, the cell culture supernatants were centrifuged with altered RCF (300 g for 10 min, at 2,000 g for 10 min and at 10,000 g for 30 min). The supernatants were concentrated using Amicon Ultra centrifugal Filters 10K (Merck) and subjected to ultracentrifugation at 150,000 g for 3 h in a 45 Ti rotor (Beckman Instruments). The small EVs were re-suspended in sterilized PBS with a protease inhibitor cocktail (Roche), and stored at 4°C. Western blot analysis, dynamic light scattering (DLS) and transmission electron microscopy (TEM) were used to verify the characteristic of the isolated small EVs and identify the expression of PH20.
Immunoblotting: To identify the expression of PH20, the small EVs were resuspended in RIPA buffer (Cell Signalling) and the concentration of protein of small EVs was quantified using a BCA Protein assay kit (Bio-Rad). The equal amounts (20 μg) of proteins were separated by gel electrophoresis and transferred to nitrocellulose membranes. The membranes were incubated overnight at 4°C with primary antibodies against PH20 (ab196596, Abcam), Alix (sc-99010, Santa Cruz), Tsg101 (ab83, Abcam), or CD81 (sc-166029, Santa Cruz) and then for 1 h at room temperature with anti-mouse peroxidase (A4416, Sigma) or anti-rabbit peroxidase (A0545, Sigma) secondary antibodies. The membranes were treated with an ECL substrate (Bio-Rad) and the results were visualized by chemiluminescence.
Dynamic light scattering (DLS): The size distribution of the small EVs was analysed by DLS, which was performed using a Zetasizer Nano ZS (Malvern Instruments) at fixed angles of 173° at 25°C. The data were processed using the provided software.
Transmission electron microscopy (TEM): Tecnai transmission electron microscopy (Tecnai TEM) was used to analyse the small EVs for size and shape. A Formvar/Carbon grid was loaded with small EVs for 5 min at room temperature, washed by being floated on PBS three times, and then incubated with 1% glutaraldehyde (Sigma) for 5 min. After being washed with sterile distilled water, the grid was stained with uranyl acetate solution for 1 min and analysed with the Tecnai TEM.
Preparation of the HA fragments
HMW HA extracted from rooster comb (Sigma) was dissolved to 0.5% in 150 mM sodium chloride and 100 mM phosphate buffer (pH 5.3), mixed with PBS, Exo-Con or Exo-PH20 and incubated for 24 h at 37°C. To collect the oligo HA, the sample was centrifuged using an Amicon® Ultra-0.5 mL Centrifugal Filter MWCO 10 kDa (Millipore). The size of the oligo HA fragments in each ultracentrifugation filtrate was determined by fluorophore-assisted carbohydrate electrophoresis (FACE) analysis. Briefly, the buffer was changed using a PD Minitrap™ G-25 (GE Healthcare), and the sample was freeze-dried, incubated with 10 μl 0.2 M ANTS and 10 μl 1 M NaCNBH4 for 16 h at 37°C, resuspended in 20 μl 40% glycerine solution, and analysed by 36% acrylamide gel electrophoresis. The results were visualized using a gel document system (AlphaImager HP). The size distribution of HA molecules larger than 10 kDa was analysed by electrophoresis. Briefly, 10 μl of each HA sample was mixed with 2 μl of 2M sucrose/TAE and 2 μl of 0.02% bromophenol blue/TAE and resolved by 0.5% agarose gel electrophoresis. The gel was stained overnight with 0.005% Stains-All/50% ethanol and then de-stained with distilled water, and the results were detected using a gel document system (AlphaImager HP).
In vitro enzymatic activity assay
To confirm the hyaluronidase activity of Exo-PH20, 4 × 104 B16F10-Ova or 4T1 cells were seeded to a 4-well chamber. After 24 h, the medium was changed to serum-free medium and the wells were treated with PBS or 20 μg of Exo-Con or Exo-PH20 for 2 h at 37°C in 5% CO2. The cells were fixed in 4% formaldehyde for 7 min and treated with anti-HABP2 antibody (ab181837, Abcam) for 30 min at 25°C followed by anti-rabbit Alexa Fluor 488-conjugated antibody (711-545-152, Jackson ImmunoResearch) for 30 min at 25°C. The samples were treated with Hoechst 33342 (H3570, Thermo Fisher Scientific) for 5 min and mounted on slides using Fluoromount-G® (SouthernBiotech). Immunofluorescent imaging was performed were using a Leica fluorescence microscope.
In vitro measurement of DC activation
For immune-phenotype analysis, BMDCs were treated with PBS, HMW HA or Exo-PH20-decomposed HA fragments (< 10 kDa) for 24 h. For blockade of TLR-4, BMDCs were incubated with anti-TLR-4 antibody for 30 min prior to stimulation with HA fragments. The cells were stained with fluorescent dye-conjugated mAbs for mouse CD11c, CD40, CD86 or CCR7, and detected using an AccuriTM C6 Flow Cytometer (BD Biosciences).
To identify the status of TLR-4 signalling on BMDCs exposed to oligo HA, BMDCs were treated with PBS, HMW HA or Exo-PH20-decompoased HA fragments (< 10 kDa) for 6 h. Nuclear proteins were extracted using a commercially available kit (Abcam) and 20 μg of cellular and nuclear protein lysates were separated by gel electrophoresis. After being transferred to nitrocellulose membranes, the membranes were incubated overnight at 4°C with primary antibodies against the following: p38 (8690S), phospho-p38 (4511S), p44/42 (4695S), phospho-p44/42 (4370S), NF-kB p65 (8242S, all from Cell Signalling) and GAPDH (MAB5718, R&D Systems). Each membrane was then incubated with anti-mouse peroxidase secondary antibody (A4416, Sigma) or anti-rabbit peroxidase secondary antibody (A0545, Sigma) for 1 h at 25°C . Finally, the membranes were treated with ECL substrate (Bio-Rad) and visualized for chemiluminescence.
In vivo anti-tumour effects
Murine B16F10-Ova melanoma cells (1 x 106) were inoculated into the left hind legs of male C57BL/6 mice, Batf3−/- mice and BALB/c nu/nu mice. Female BALB/c mice were orthotopically inoculated with murine 4T1 breast cancer cells (1 x 106) into the mammary fat pad. Tumour volume (mm3) was calculated as (width)2 x (length) x 0.5. After the average tumour size reached 80–100 mm3, each subject was injected intratumorally with PBS or 50 μg of Exo-Con or Exo-PH20 every three days for three rounds. In TLR-4-blocking experiments, B16F10-Ova bearing mice were injected intraperitoneally with 200 μg/kg of anti-TLR-4 antibody (HTA125, Thermo Fisher Scientific) on days 0, 8, 11 and 14. To investigate the effects of the combination of Exo-PH20 and anti-PD-L1, 1 × 106 murine B16F10-Ova melanoma cells were transplanted into male C57BL/6 mice and 1 × 106 murine 4T1 breast cancer cells were orthotopically inoculated into female BALB/c mice. The mice were intraperitoneally treated with 2 mg/kg anti-PD-L1 the day after the initial intratumoral injection of Exo-PH20. The anti-PD-L1 injection was repeated three times at intervals of three days in B16F10-Ova tumour-bearing mice and five times at intervals of three days in the 4T1 orthotopic model. To assess the anti-tumour activity of Exo-PH20/anti-PD-L1 in the MMTV-PyMT spontaneous mouse model, age-matched (day 80–85) mice were used. When the average tumour size reached 50–110 mm3, the mice were intraperitoneally treated with 2 mg/kg anti-PD-L1 on the day following the first intravenous injection of Exo-PH20. The injection of anti-PD-L1 was repeated five times at intervals of three days.
Tumour-free mice generated by combination treatment with Exo-PH20 and anti-PD-L1 were re-challenged with 2 × 106 B16F10-Ova cells on the opposite flank. Age-matched C57BL/6 mice were used as controls. Tumour volumes were measured every three days.
Analysis of DC maturation, migration and cross-presentation
Three days after the last injection, tumour draining lymph nodes (TDLNs) were extracted and mechanically digested. Single cells were pre-incubated with 2 μg anti-mouse CD16/CD32 (BD Biosciences) for 5 min at 4°C. For DC maturation analysis, the cells were stained with APC-anti-CD11c (clone N418, 117310), PE-anti-CD40 (clone GL-1, 105008), PE-anti-CD86 (clone 3–23, 124610), PE-anti-CD80 (clone 16-10A1, 104707) or PE/Cy7-anti-CCR7 (clone 4B12, 120124). For quantification of the CD103+ DCs in TDLNs, the cells were stained with APC-anti-CD11c (clone N418, 117310) and FITC-anti-CD103 (clone 2E7, 121419). For cross-presentation analysis of DCs, the cells were stained with APC-anti-CD11c (clone N418, 117310), FITC-anti-CD103 (clone 2E7, 121419) and PE-anti-H-2Kb bound to SIINFEKL (clone 25-D1.16, 141604). Data were obtained using an AccuriTM C6 Flow Cytometer and analysed with the FlowJo-V10 software (BD Biosciences).
Ex vivo and in vivo cross-priming analysis
For analysis of ex vivo cross-priming, tumours and TDLNs were harvested three days after the last injection. Excised tumour tissues were digested with a tumour dissociation kit (Miltenyi Biotec) and dissociated to single cells using a gentleMACSTM Octo Dissociator (Miltenyi Biotec). After being filtered with a 40-μm strainer, single cell suspensions obtained from tumours or TDLNs were incubated with RBC lysis buffer for 5 min at 4°C. CD11c+ DCs were selected using CD11c microbeads (Miltenyi Biotec). Spleens extracted from OT-I mice were dissociated to single cells, strained (40 μm), and treated with RBC lysis buffer for 5 min at 4°C. Naïve CD8 T cell selection was performed utilizing a naïve CD8 T cell isolation kit (Miltenyi Biotec). After sorting of the CD11c+ DCs and naïve CD8+ T cells, a 1:3 ratio of DCs and T cells was co-cultured in 96-well round-bottom plates for 72 h and the quantity of IFN-γ in each supernatant was analysed through a mouse IFN-gamma Quantikine ELISA kit (R&D Systems) according to the manufacturer’s instructions. For in vivo cross-priming analysis, three days after the last injection of the above-described schedules, mice were further injected with 3 × 106 CFSE-labelled OT-I naïve CD8+ T cells. After three days, TDLNs were excised and the proliferation of CSFE-labelled OT-I T cells from TDLNs was analysed using an AccuriTM C6 Flow Cytometer (BD Biosciences).
Analysis of T cell infiltration into tumour tissues
Three days after the last injection, tumours were harvested. For flow cytometric analysis of T cell infiltration, excised tumour tissues were digested with a tumour dissociation kit (130-096-730, Miltenyi Biotec), dissociated to single cells, strained (40 μm), and subjected to RBC lysis. The obtained single cells were pre-incubated with 2 μg anti-mouse CD16/CD32 (BD Biosciences) for 5 min at 4°C and then stained with PerCP/Cy5.5-anti-CD45.2 (clone 104, 109827), APC-anti-CD3 (clone 17A2, 100235) and FITC-anti-CD8α (clone 53–6.7, 100705) antibodies. Data were obtained using an AccuriTM C6 Flow Cytometer and analysed with the FlowJo-V10 software (BD Biosciences). Immunofluorescence staining of tumour-infiltrated CD8+ T cells was performed as described below.
Immunofluorescence microscopy
Tumour tissues were embedded in OCT compound, frozen at −80°C, cryosectioned at 10 μm and mounted on slides. The sections were blocked with 3% BSA/PBS for 1 h at 25°C and incubated overnight at 4°C with the following primary antibodies: anti-HABP2 (Abcam, ab181837), anti-CD8 (BD Pharmingen, 550281), anti-PD-L1 (R&D Systems, AF1019) or anti-pan cytokeratin (Abcam, ab86734). The sections were washed with PBS and incubated for 1 h at 25°C with anti-rabbit Alexa Fluor 488-conjugated secondary antibody (711-545-152), anti-rat Alexa Fluor 488-conjugated secondary antibody (712-546-153), anti-goat Alexa Fluor 488-conjugated secondary antibody (705-545-003) or anti-mouse Alexa Fluor 647-conjugated secondary antibody (115-605-003, all from Jackson ImmunoResearch). DAPI-Fluoromount-G® (SouthernBiotech) was used to stain nuclei and mount the sections, and the results were analysed with a Leica fluorescence microscope.
Tumour-specific T cell activity
For detection of granzyme B expression of infiltrated CD8+ T cells, tumour tissues were extracted three days after the last injection and dissociated to single cells using a gentleMACSTM Octo Dissociator (Miltenyi Biotec). CD8+ T cells were selected using CD8a T cell isolation kit (Miltenyi Biotec). The isolated CD8+ T cells were pre-incubated with 2 μg anti-mouse CD16/CD32 (BD Biosciences) for 5 min at 4°C and then dyed with FITC-anti-CD8α (clone 53–6.7, 100705) and PerCP/Cy5.5-anti-Granzyme B (clone QA16A02, 372211) antibodies. Data were obtained through an AccuriTM C6 Flow Cytometer and analysed with the FlowJo-V10 software (BD Biosciences).
For detection of tumour-specific T cell activity, spleens were harvested three days after the last injection and dissociated to single cells using a gentleMACSTM Octo Dissociator (Miltenyi Biotec). The single cell suspensions were incubated with 10 μg/ml Ova peptide (SIINFEKL) for 48 h and the quantity of IFN-γ in each supernatant was analysed through a mouse IFN-gamma Quantikine ELISA kit (R&D Systems) according to the manufacturer’s instructions.
The analysis of immune cell population in TDLNs and tumour tissues
To analyse the change of immune cell population, TDLNs and tumour tissues were extracted three days after the last injection and dissociated to single cells. Among immune cells, macrophages were selected using anti-F4/80 bead (Miltenyi Biotec). The single cells and the isolated macrophages were pre-incubated with 2 μg anti-mouse CD16/CD32 (BD Biosciences) for 5 min at 4°C. Then each immune cells (MDSCs, Treg cells and macrophages) stained with fluorescence-conjugated antibodies. [MDSCs: PerCP/Cy5.5-anti-CD45.2 (clone 104, 109827), PE-anti-CD11b (clone M1/70, 101207), and FITC-anti-Gr1 (clone RB6-8C5, 108405). Treg: PerCP/Cy5.5-anti-CD45.2 (clone 104, 109827), APC-anti-CD3 (clone 17A2, 100,235), FITC-anti-CD4 (clone GK1.5, 100405), and PE-anti-CD25 (clone 3C7, 101903). Macrophages: APC-anti-F4/80 (clone BM8, 123116), FITC-anti-MHCII (clone M5/114.15.2, 107605) and PE-anti-CD206 (clone C068C2, 141705)] Data were obtained using an AccuriTM C6 Flow Cytometer and analysed with the FlowJo-V10 software (BD Biosciences).
Statistical analysis
To verify the significance of the differences (p value) between the groups, statistical analyses were performed through GraphPad Prism 5.1. Multiple comparisons were analysed using one-way ANOVA with Turkey’s post test, and the significance for Kaplan-Meier analysis was assessed using the log-rank test. The values are represented as mean ± SD for samples (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
HA fragments decomposed by Exo-PH20 induce DC maturation via TLR-4 pathway
We previously reported the development of a small EV with surface expression of PH20 (Exo-PH20) [Citation19]. Consistent with our previous report, our present analysis confirmed the expression of PH20 proteins and small EV markers (CD81, Tsg101 and Alix) on the small EV surface, showed that the small EVs are spherical in shape with an average diameter of ~100 nm (Figure S1a, b) and validated that Exo-PH20 can induce HA degradation in B16F10-Ova and 4T1 cancer cells (Figure S1c – f). We further used fluorophore-assisted carbohydrate electrophoresis (FACE) analysis [Citation24,Citation25] to measure the sizes of the degraded fragments and found that the HA oligosaccharides degraded from HMW-HA by Exo-PH20 were 4–8 mers, as assessed by comparison with a commercially available oligo-HA assortment (4–12 mer) ()).
Figure 1. HA fragments decomposed by Exo-PH20 induced DC maturation. (a) Characterization of HA fragments generated by Exo-PH20. Top: Size determination of HA (> 10 kDa) by agarose gel electrophoresis. Bottom: FACE analysis of HA fragment from the centrifugation filtrate (< 10 kDa). Oligo-HA of defined sizes (4–12 disaccharides) were used as standards. (b) Flow cytometric analysis of the phenotypic maturation of DCs in vitro. BMDCs incubated with HMW-HA or Exo-PH20-decomposed HA fragments (< 10 kDa) for 24 h were stained with specific monoclonal antibodies against CD11c, CD40, CD86 and CCR7 and analysed by flow cytometry. The data represents the percentage (%) of CD40, CD86, CCR7 expression on BMDC. Each value represents the mean ± SD (*p < 0.05, ** p < 0.01, ***p < 0.001 versus the BMDC-only group as assessed by one-way ANOVA with Tukey’s post test). (c) Western blot analysis of the TLR-4 mediated-DC maturation pathway. BMDCs were incubated with HMW-HA or Exo-PH20-decomposed HA fragments (< 10 kDa) for 6 h. The contents of phospho-p38, phospho-p44/42, total p38, total p44/42 and GAPDH were detected in cell lysates and the NF-ĸB content was detected in nuclear extracts.
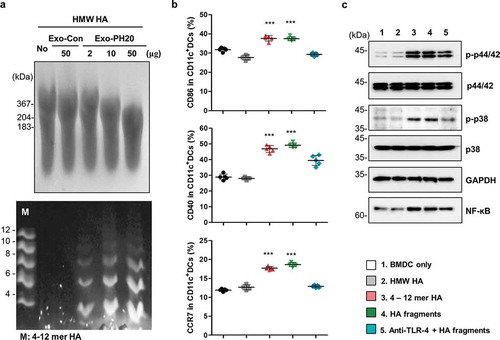
To test whether HA fragments digested from HMW-HA by Exo-PH20 can stimulate DC activation, oligo-HA were separated using Amicon® Ultra-0.5 column and incubated with immature bone marrow-derived DCs (BMDCs) for 24 h. Our results revealed that the collected oligo-HA, but not HMW-HA, induced BMDC maturation, as shown by the marked up-regulations of the co-stimulatory molecules, CD40, CD86 and CCR7 ( and Figure S2). Consistent with previous reports that the oligo-HA-triggered maturation of DCs is mediated by its binding to TLR-4 [Citation15], we found that blockade of TLR-4 function inhibited the expression levels of CD40, CD86 and CCR7 on HA-fragments-treated DCs ( and Figure S2). Next, we tested whether oligo-HA-induced DC maturation involves activation of the MAPK pathway and/or nuclear translocation of NF-KB, which are key mediators of TLR-4 pathway. As shown in , immature BDMCs incubated for 6 h with oligo-HA, but not HMW-HA, exhibited increased phosphorylation of p38 and p44/42 MAPK relative to the total content of these MAPKs in untreated control cells. The nuclear translocation of NF-KB was also increased in immature BMDCs exposed to Exo-PH20-degraded HA fragments (). Taken together, these results indicate that TLR-4 activation is involved in the ability of oligo-HA to trigger MAPK pathway activation, NF-KB nuclear translocation and DC maturation.
Exo-PH20-oligo-HA induced DC activation and migration potentiates tumour growth inhibition
The effect of Exo-PH20-degraded HA fragments was examined in the B16F10-Ova tumour mouse model. After the average tumour sizes had reached ~ 80 mm3, 50 μg of Exo-PH20 were intratumorally injected into mice a total of three times every three days. This dosing strategy was designed to counteract HA recombination and turnover. Intratumoral injection was chosen to ensure targeting of locoregional antigen-presenting cells (APCs), such as the DCs that circulate between the tumour and the draining lymph nodes (DLNs). As shown in , intratumoral administration of Exo-PH20 markedly decreased the tumour growth rate (~ 65% inhibition) of the B16F10-Ova tumours in comparison to that seen in the control groups. Weighing of tumours at the time of necropsy also revealed that Exo-PH20 treatment was more effective in halting cancer progression compared to all other groups. There was no significant body weight changes in any groups (Figure S3a, b).
Figure 2. Exo-PH20 induces anti-tumour immunity in the B16F10-Ova melanoma model. (a) Anti-tumour effect of Exo-PH20 in B16F10-Ova tumour-bearing C57BL/6 mice. Mice were injected (i.t.) with 50 μg Exo-Con or Exo-PH20 (black arrows). Each value represents the mean ± SD (n = 8 mice per group). (b) B16F10-Ova tumour-bearing mice were injected (i.t.) with 50 μg small EVs (black arrows) and further treated (i.p.) with 200 μg/kg of anti-TLR-4 on days 0, 8, 11 and 14. Each value represents the mean ± SD (n = 4 mice per group). (c – e) At day 18, TDLNs were extracted from mice of all groups and analysed by flow cytometry. (c) The percentage (%) of CD40, CD86, CD80 and CCR7 expression on CD11c+ DCs in TDLNs. Each value represents the mean ± SD (n = 5). (d) The number of CD103+ DCs in TDLNs, which was calculated as the % of CD103+CD11c+ DCs identified from among 106 TDLN cells. Each value represents the mean ± SD (n = 5). (e) The percentage (%) of H2Kb-Ova expression on CD11c+ DCs (Left) and CD103+CD11c+ DCs (Right) in TDLNs. The percentage (%) was determined with respect to the IgG. Each value represents the mean ± SD (n = 5). (f) B16F10-Ova tumour-bearing Batf3−/- mice were treated (i.t.) with PBS, 50 μg Exo-Con or Exo-PH20 (black arrows). Each value represents the mean ± SD (n = 3 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001, as assessed by one-way ANOVA with Tukey’s post test.
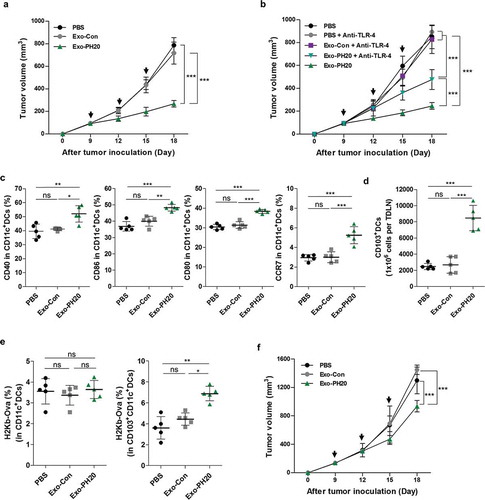
Consistent with previous studies [Citation19], we observed Exo-PH20-mediated HA degradation in tumour tissues (Figure S3c). However, we assumed that in vivo anti-tumour response noted above were significantly due to the DC activation by oligo-HA via TLR-4 signalling in addition to ECM remodelling. In support of this hypothesis, we pre-treated B16F10 tumour-bearing mice with neutralizing antibody against TLR-4 and performed small EV treatment. As shown in , our blockade of TLR-4 signalling significantly abrogated the Exo-PH20-induced anti-tumour response noted in mice that were not pretreated with TLR-4-neutralizing antibody ( and Figure S3d). In addition, there is no significantly difference in the DC number in tumour tissues by pre-treated TLR-4 antibody (Figure S3e). Of note, a previous study showed that the neutralizing-antibody-mediated blockade of TLR-4 did not affect tumour growth [Citation26].
Among the receptors of HA such as TLR-2, TLR-4 and CD44 [Citation27,Citation28], oligo-HA were reported to trigger T cell immunity by up-regulating and activating co-stimulatory molecules of DC depending on TLR-4 [Citation14,Citation15]. Studies have shown that lipopolysaccharide and its clinically relevant analogs (e.g. monophosphoryl lipid A) bind to TLR-4 complexes at the cell surface [Citation29,Citation30], triggering a signalling cascade that culminates in the induction of transcription factors (NF-κB and IRF3), MAPK kinases p38 and c-Jun NH2-terminal kinase [Citation31,Citation32]. During this process, DCs mature and exhibit partially up-regulation of surface proteins (CD40, CD80, and MHC molecules) and pro-inflammatory cytokines (IL-6, IL-12, and type I IFNs) [Citation33,Citation34]. To verify that Exo-PH20 treatment induces the maturation of DCs, we analysed the expression levels of CD40, CD86, and CD80 on DCs (CD11c+ cells) isolated from tumour draining lymph nodes (TDLNs). As expected, Exo-PH20 treatment up-regulated the surface expression levels of the co-stimulatory molecules, CD40, CD86 and CD80 in TDLN-derived DCs ( and Figure S4a-c).
Similar to results obtained for these surface markers, we found that Exo-PH20-decomposed HA fragments strongly up-regulated the expression of CCR7 in DCs of TDLNs ( and Figure S4c). CCR7 has been reported to be up-regulated on mature DCs and essentially required for their migration to DLNs; the main driving force of matured DC migration is elevating chemotactic concentrations of CCL19/21 (CCR7 ligands) [Citation35]. Expecially, migratory CD103+ DCs that are differentially distributed in the TME can migrate to LNs depending on CCR7 and efficiently cross-present tumour-specific antigens to CD8+ T cells [Citation36]. Accordingly, we observed that the number of CD103+ DCs was significantly increased in the TDLNs of mice treated with Exo-PH20 (). Collectively, during DC maturation, TLR-4-mediated markedly increased CCR7 expression resulted in large number of CD103+ DC migration to DLNs. In addition, these results indicate that DC migration towards lymphoid tissues mediated by the expression of chemokine receptors on DCs is responsible for tumour growth inhibition as much as DC maturation.
We also analysed the abundance of H-2Kb molecules associated with the Ova-derived peptide, SIINFEKL (residues 257–264), on DCs. As shown in , the density of H-2Kb/OVA complexes did not significantly different among the CD11c+ DCs of the various groups; however, the expression level of H-2Kb molecules on CD103+ DCs was elevated in the Exo-PH20 treatment group ( and Figure S5a, b). Intratumoral administration of Exo-PH20 was also tested in the 4T1 orthotopic mouse model, which yielded results comparable to those generated in the B16F10-Ova model (Figure S6). These results indicate that CD103+ DCs that are successfully matured by Exo-PH20 treatment transport tumour-specific antigens to the TLDNs to presumably for presentation to CD8+ T cells.
To directly confirm the effect of CD103+ DCs on tumour growth, we transplanted B16F10-Ova tumours into Batf3−/- mice. Mice deficient for basic leucine zipper transcriptional factor ATF-like 3 (BATF3) lack the two major DC subsets (lymphoid organ-resident CD8+ DCs and migratory CD103+ DCs) required for cross-presentation, and thus exhibit impaired CD8+ T cell responses [Citation37]. We found that the anti-tumour efficacy of Exo-PH20 was observed but not as potent as compared to the wild-type mice ( and Figure S3f). This suggests that the activation of Batf3-dependent CD103+ DCs is necessary for the ability of Exo-PH20-treated mice to mount a successful anti-tumour response. Collectively, these results indicate that Exo-PH20-mediated HA degradation stimulates DC maturation and accelerates the migration of CD103+ DCs, which are crucial for the initiation of cytotoxic T cell responses.
However, note that there would also be complex mechanisms of oligo HA-related signal specificity to other immune cells [Citation38,Citation39]. To verify this, we performed additional experiment for analysing the population of other immune cells: CD25+CD4+ T cells, M1-/M2- type macrophages, and myeloid suppressor cells (MDSCs). As shown in Fig. S3, while the changes in the number of MDSCs were hardly observed, CD25+CD4+ T cells (%) was shown to be reduced in the tumour and the TDLNs. We also found Exo-PH20-mediated an increase in the M1/M2 ratio of macrophages in the tumour tissues (Figure S3g-i). These results indicate not only DCs but also other immune cells that induce turning cold tumours to hot could contribute to the shrinking in tumour growth.
Exo-PH20 promotes DC-mediated T cell priming ability
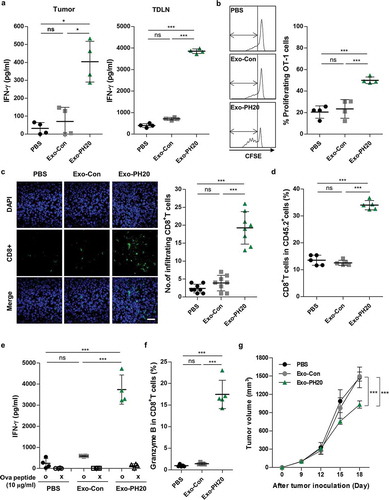
To evaluate the activity of DCs to prime anti-tumour T cell responses by Exo-PH20, we co-cultured DCs isolated from tumours or TLDNs of B16F10-Ova tumour-bearing mice with Ova-specific OT-I T cells treatment ex vivo. By measuring the IFN-γ production of these naïve T cells upon co-culture, we found that DCs from the tumours or TDLNs of Exo-PH20-treated mice yielded increased T cell activation (). To assess whether Exo-PH20-mediated HA degradation increases in vivo CD8+ T cell cross-priming, we performed adoptive transfer of CFSE-labelled Ova-specific naïve T (OT-I) cells into B16F10-Ova tumour-bearing mice via i.v. route, and measured the proliferation of OT-I T cells. As shown in , proliferating OT-I T cells (%) was remarkably increased in TDLNs of Exo-PH20-treated mice compared with other mouse groups (). These data suggest that ExoPH20-degraded oligo-HA can activate DCs to cross-prime T cells, thereby eliciting a strong tumour-specific immune response.
Figure 3. Exo-PH20 increases T cell cross-priming and CD8+ T cell infiltration. (a, c – f) Spleens, TDLNs and tumour tissues were harvested on day 18. (a) CD11c+ DCs were isolated from tumours or TDLNs and co-cultured ex vivo with OT-I naïve CD8+ T cells for 72 h. The amount of secreted IFN-γ in culture supernatants was analysed by ELISA. Each value represents the mean ± SD (n = 4). (b) At day 18, CFSE-labelled OT-I naïve CD8+ T cells were transferred (i.v.) into mice. After 72 h, TDLNs were harvested and the proliferation of OT-I CD8+ T cells was determined by flow cytometry. Left: Representative CFSE profiles of proliferating CD8+ T cells. Right: The percentages of gated proliferating CSFE+CD8+ T cells. Each value represents the mean ± SD (n = 4). (c) The accumulation of CD8+ T cells in B16F10-Ova tumour tissues. Left: Representative immunofluorescence images of the infiltration of CD8+ T cells into B16F10-Ova tumours. Tumour sections were stained with anti-mouse CD8 antibody (green) and nuclei were counterstained with DAPI (blue). Scale bar, 100 μm. Right: The number of tumour-infiltrating CD8+ T cells (per section at × 400 field of view) was counted in each group. Each value represents the mean ± SD (n = 10). (d) The percentages of CD8+ tumour-infiltrating lymphocytes in B16F10-Ova tumour tissues were analysed by flow cytometry. Each value represents the mean ± SD (n = 5). (e) The amount of IFN-γ in splenocyte culture supernatants was measured by ELISA. Each value represents the mean ± SD (n = 4). (f) The percentages of granzyme B in CD8+ T cells isolated from B16F10-Ova tumour tissues were analysed by flow cytometry. Each value represents the mean ± SD (n = 5). (g) Immune-deficient mice were transplanted with B16F10-Ova melanoma cells at day 0 and treated (i.t.) with PBS, 50 μg Exo-Con or Exo-PH20 (black arrows). Each value represents the mean ± SD (n = 4 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001, as assessed by one-way ANOVA with Tukey’s post test.
Exo-PH20-mediated tumour growth inhibition requires T cell immune responses
In agreement with previous reports [Citation19], we observed that enhanced T cell infiltration into tumour tissues in B16F10-Ova tumour-bearing mice () as well as 4T1 orthotopic mice (Figure S8) through immunofluorescence staining. Flow cytometric analysis of tumour-infiltrating leukocytes (TILs) also revealed that an escalation in the percentages of CD8+ cells among the TILs from B16F10-Ova tumour-bearing mice treated with Exo-PH20 ( and Figure S7a).
We also assessed the quality of the CD8+ T cell immune response against tumour-associated antigens, by stimulating splenocytes with Ova peptides. The IFN-γ production was markedly increased in the spleens (12-fold compared with PBS group) of mice treated with Exo-PH20 compared with the other groups (). Exo-PH20 also significantly enhanced the cytotoxicity of CD8+ T cells, as demonstrated by the increased expression of granzyme B () ans Figure S7b). Thess indicate that T cell immunity is important for the Exo-PH20-induced anti-tumour effects. Similarly, we found that Exo-PH20 treatment had a modest tumour growth inhibition effect in nude mice () and Figure S7c). Together, these results indicate that Exo-PH20 effectively triggers a local immune response in the tumour tissues, resulting in tumour regression through the activation of tumour-specific systemic immune responses.
Exo-PH20 in combination with anti-PD-L1 exhibits an improved anti-tumour immune response
When tumours encounter T cells, these tumour cells detect the high concentration of IFN-γ released by the T cells, and exhibit enhanced surface expression of PD-L1 in preparation for an immune-attack [Citation40,Citation41]. The local immune cells such as tumour-specific CTLs, become paralysed and cannot attack the tumours. As shown in ), we observed the expression of PD-L1 in both B16F10-Ova and 4T1 tumour tissues by immunofluorescence staining () and Figure S10a). These observations suggest that the IFN-γ-dependent PD-L1 expression can provide a strong immune-escape mechanism for cancer cells. Importantly, this contributes to our rationale for combining Exo-PH20 with PD-L1 blockades.
Figure 4. Anti-PD-L1 promotes anti-tumour immunity in the B16F10-Ova melanoma mouse model and the genetically engineered MMTV-PyMT mouse model. (a) Representative immunofluorescence images of PD-L1 expression in B16F10-Ova tumour sections. Each tumour section was stained with anti-mouse PD-L1 (green) and anti-mouse pan-cytokeratin (CK) (red). DAPI was used to counterstain nuclei (blue). Scale bar, 100 μm. (b) Mice were inoculated with 1 × 106 B16F10-Ova melanoma cells and injected with 50 μg Exo-PH20 (i.t.) and/or 2 mg/kg anti-PD-L1 (i.p.) as indicated. Each value represents the mean ± SD (n = 6 or 8 mice per group). (c) Mice exhibiting complete tumour regression were re-challenged with 2 × 106 B16F10-Ova cells on the opposite flank at day 28 after the initial tumour inoculation. Left: Each value represents the mean ± SD (n = 8 mice per group). Right: tumour volume growth curves of individual mice in each group. (d) CD11c+ DCs were isolated from tumours or TDLNs of B16F10-Ova tumour-bearing mice treated with Exo-PH20 and/or anti-PD-L1, and co-cultured ex vivo with OT-I naïve CD8+ T cells for 72 h. The amount of IFN-γ secreted from OT-I CD8+ T cells was analysed by ELISA. Each value represents the mean ± SD (n = 4). (e) The amount of IFN-γ in splenocyte culture supernatants was analysed by ELISA. Each value represents the mean ± SD (n = 4). (f – g) MMTV-PyMT mice were treated with 200 μg Exo-PH20 (i.v.) and/or 2 mg/kg anti-PD-L1 (i.p.) as indicated. (f) The change in tumour volume relative to that at the initiation of treatment in MMTV-PyMT mice. Each value represents the mean ± SD (n = 6 mice per group). (g) Kaplan-Meier survival curves of MMTV-PyMT mice subjected to co-treatment with Exo-PH20 and anti-PD-L1 (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001, as assessed by one-way ANOVA with Tukey’s post test (a – f) and the log-rank test (g).
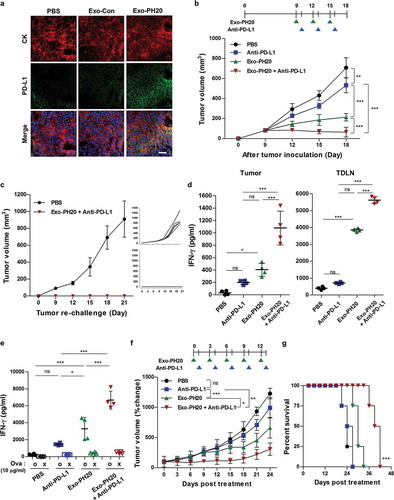
To evaluate the therapeutic efficacy of Exo-PH20 in combination with anti-PD-L1 in vivo, we treated B16F10-Ova tumour-bearing mice intraperitoneally with anti-PD-L1 followed by intratumoral injection of Exo-PH20, every three days for three cycles. As shown in ), the combined treatment exerted a more dramatic effect on tumour suppression than either monotherapy, leading to complete tumour regression in 50% (4 of 8) of the mice in the co-treated group () and Figure S9). To determine whether combined treatment provided protective immune memory, a greater number of tumour cells was used to re-challenge the contralateral flank of B16F10-Ova mice 10 days after primary tumour rejection. Notably, no tumour recurrence was observed in either flanks upon termination of the study (day 21), suggesting that Exo-PH20-generated HA fragment-mediated DC activation contributes to inducing a durable systemic immune memory and preventing tumour relapse ()).
We further assessed whether the combined therapy could potentiate the cross-prime ability of DCs to CD8+ T cells ex vivo. To verify that, we co-culcutred DCs from tumours or TDLNs with OVA-specific OT-I T cells and analysed T cell function by measuring IFN-γ production. As expected, the combined therapy resulted in more IFN-γ secretion than either monotherapy (). This result was paralleled by the increased IFN-γ production seen in splenocytes stimulated with Ova peptides (). Together, our findings suggest that Exo-PH20 plus anti-PD-L1 can effectively activate CD8+ T cell immunity.
The therapeutic effects of the combination therapy were further validated in the 4T1 breast cancer mouse model (Figure S10). Similar to the above results, we found that combined treatment with Exo-PH20 plus anti-PD-L1 was more effective than either monotherapy in suppressing tumours and activating IFN-γ-producing CD8+ T cells in the 4T1 orthotopic mouse model (Figure S10).
Genetically engineered, autochthonous tumour models that produce immunosuppressive tumour microenvironments are typically resistant to cancer immunotherapy [Citation42,Citation43]. To verify the potency of our combined therapy in an autochthonous tumour model, we used the MMTV-PyMT mouse is the model of breast cancer metastasis. In this model, MMTV-LTR is used to drive the expression of mammary gland-specific polyomavirus middle T-antigen (PyMT), leading to the rapid growth of highly metastatic tumours [Citation44]. When the first palpable tumour (~ 50–110 mm3) was found, mice were administrated with Exo-PH20 (i.v.) or anti-PD-L1 (i.p.) alone or in combination with the anti-PD-L1 (). In control mice, progressive tumour growth was observed and tumour size reached greater than ~1500 mm3 at 24 days from the start of treatment. Treatment with Exo-PH20 had a modest effect in regressing the tumour burden, while the anti-PD-L1 had only a slight tumour-regressing effect (). In contrast, combined therapy with Exo-PH20 plus anti-PD-L1 led to the efficient regression of induced tumours and was associated with a nearly 1.6-fold increase in overall survival (). Therefore, these findings clearly demonstrate that the combination of Exo-PH20 and anti-PD-L1 provides therapeutic benefits in terms of enhanced tumour suppression, even in a genetically engineered MMTV-PyMT mouse model.
Discussion
In this paper, we describe a new mechanism through which PH20 hyaluronidase can act against tumours. Our strategy of targeting HA seeks to ‘kill two birds with one stone’, as Exo-PH20 primarily degrades highly accumulated extracellular HA while the resulting oligo-HA molecules act as TLR-4 agonists to stimulate DCs, which subsequently prime CD8+ T cells. As expected, DC activation was found to be crucial for the induction of tumour-specific immune responses. Moreover, we found that combining ICB with Exo-PH20 yielded a dramatically increased anti-tumour efficacy.
PEGPH20 could be expected to have clinical activity in patients with high levels of HA in their tumours (immune-excluded tumours) [Citation23]. They are also currently being examined in clinical trials (phase I/II) in combination with anti-PD-1/PD-L1 antibodies (NCT03634332, NCT02563548, NCT03481920, NCT03193190, and NCT03281369), showing significantly suppressed tumour growth in high-HA-expressing pancreatic tumour mice models [Citation45,Citation46]. However, degradation of the ECM may be not sufficient to induce tumour-specific T cell immune responses in non-T cell inflamed tumours (immunologically cold; exhibit poor immune cell infiltration due to their complex stromal environments, low mutational load and the absence of the danger signals required for T cell priming). Fortunately, our results show that Exo-PH20 could target non-inflamed tumour (not only immune-excluded but also immune-desert tumour) by: 1) enhancing the activities of APCs to awaken intrinsic immunity against tumour; and 2) degrading the physiological and immunosuppressive barriers of HMW-HA. Our finding provides a new insight for HA degradation by Exo-PH20 that functions as pro-inflammatory DAMPs to activate DCs, thereby overcoming immunosuppressive TME.
As emerging cancer immunotherapies using ICBs work predominantly in inflamed (hot) tumours that are infiltrated by T cells [Citation47,Citation48], patients with a “cold” tumour rarely respond to single-agent ICB using anti-PD-1/PD-L1 therapeutic antibodies [Citation49]. Therefore, effective cancer immunotherapies will require that T cells successfully passage through the early stages of T cell priming, activation and tumour infiltration. Here we provide an additional benefit using a hyaluronidase for cancer immunotherapy; targeting HA that promotes HA degradation may induce turning a cold tumour into a hot tumour, increasing sensitivity to immune checkpoint blockade therapies.
Based on our present results, we suggest a small EV-based strategy that can overcome the activation energy threshold of the immunosuppressive TME. The complex composition of an EV means that they can show both immunostimulatory and immune-suppressive effects, which must be considered when EVs are employed for cancer immunotherapy [Citation50]. Although small EVs are considered to be safe, biocompatible and non-immunogenic, owing to their endogenous origin, the transfer of an unknown mix of bioactive molecules can add risks to immunotherapy [Citation51]. Further studies will be needed to improve our understanding of the content, and potential toxicity of the any EV intended for clinical use [Citation52]. Despite these obstacles, research on EV-based immunotherapy is bound to increase given the recent reports of promising results [Citation53]. In addition, considering that small EVs may be engineered on the nanoscale, their combination with other drugs or nanomaterials is expected to be a winning strategy for cancer immunotherapy [Citation54]. Therefore, our approach will also be expected to result in a rising trend of EV-based cancer immunotherapy.
In summary, our results provide mechanistic insights into the anti-tumour effects of oligo-HA generated from the degradation of HMW-HA, and demonstrate that targeting HA has therapeutic potential for the treatment of cold tumours.
Author contributions
Y.Y. and I.-S.K. designed the experiments; Y.H., Y.K.K., G.B.K., G.-H.N., and S.A.K. performed the experiments; Y.H., Y.K.K., and Y.P. analysed the data and Y.H., Y.Y., and I.-S.K. wrote the manuscript.
Supplemental Material
Download MS Word (7.3 MB)Acknowledgments
This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (2019R1A2B5B03004360 and 2017R1A3B1023418), the KU-KIST Graduate School of Converging Science and Technology Program, and the KIST Institutional Program.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Paulos CM, Kaiser A, Wrzesinski C, et al. Toll-like receptors in tumor immunotherapy. Clin Cancer Res. 2007 Sep 15;13(18 Pt 1):5280–15. PubMed PMID: 17875756; PubMed Central PMCID: PMCPMC2131730.
- Cen X, Liu S, Cheng K. The role of toll-like receptor in inflammation and tumor immunity. Front Pharmacol. 2018;9:878. PubMed PMID: 30127747; PubMed Central PMCID: PMCPMC6088210.
- Krysko O, Love Aaes T, Bachert C, et al. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013 May 16;4:e631. PubMed PMID: 23681226; PubMed Central PMCID: PMCPMC3674363.
- Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010. PubMed PMID: 20706656; PubMed Central PMCID: PMCPMC2913853. DOI:10.1155/2010/672395
- Kelsh RM, McKeown-Longo PJ. Topographical changes in extracellular matrix: activation of TLR4 signaling and solid tumor progression. Trends Cancer Res. 2013 Jan 1;9:1–13. PubMed PMID: 24634571; PubMed Central PMCID: PMCPMC3952558.
- Fuchs K, Hippe A, Schmaus A, et al. Opposing effects of high- and low-molecular weight hyaluronan on CXCL12-induced CXCR4 signaling depend on CD44. Cell Death Dis. 2013 Oct 03;4:e819. PubMed PMID: 24091662; PubMed Central PMCID: PMCPMC3824673.
- Cyphert JM, Trempus CS, Garantziotis S. Size matters: molecular weight specificity of hyaluronan effects in cell biology. Int J Cell Biol. 2015;2015:563818. PubMed PMID: 26448754; PubMed Central PMCID: PMC4581549.
- Murai T. Lipid raft-mediated regulation of hyaluronan-CD44 interactions in inflammation and cancer. Front Immunol. 2015;6:420. PubMed PMID: 26347743; PubMed Central PMCID: PMCPMC4542320.
- Alaniz L, Garcia M, Rizzo M, et al. Altered hyaluronan biosynthesis and cancer progression: an immunological perspective. Mini Rev Med Chem. 2009 Nov 1;9(13):1538–1546. PubMed PMID: 20205635.
- Kuang DM, Wu Y, Chen N, et al. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood. 2007 Jul 15;110(2):587–595. PubMed PMID: 17395778.
- Itano N, Zhuo L, Kimata K. Impact of the hyaluronan-rich tumor microenvironment on cancer initiation and progression. Cancer Sci. 2008 Sep;99(9):1720–1725. PubMed PMID: 18564137.
- Wu RL, Huang L, Zhao HC, et al. Hyaluronic acid in digestive cancers. J Cancer Res Clin Oncol. 2017 Jan;143(1):1–16. 10.1007/s00432-016-2213-5. PubMed PMID: 27535565.
- Alaniz L, Rizzo M, Garcia MG, et al. Low molecular weight hyaluronan preconditioning of tumor-pulsed dendritic cells increases their migratory ability and induces immunity against murine colorectal carcinoma. Cancer Immunol Immunother. 2011 Oct;60(10):1383–1395. PubMed PMID: 21638126.
- Muto J, Morioka Y, Yamasaki K, et al. Hyaluronan digestion controls DC migration from the skin. J Clin Invest. 2014 Mar;124(3):1309–1319. PubMed PMID: 24487587; PubMed Central PMCID: PMC3934161.
- Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002 Jan 7;195(1):99–111. PubMed PMID: 11781369; PubMed Central PMCID: PMC2196009.
- Olsson M, Bremer L, Aulin C, et al. Fragmented hyaluronan has no alarmin function assessed in arthritis synovial fibroblast and chondrocyte cultures. Innate Immun. 2018 Feb;24(2):131–141. PubMed PMID: 29495940.
- Dong Y, Arif A, Olsson M, et al. Endotoxin free hyaluronan and hyaluronan fragments do not stimulate TNF-alpha, interleukin-12 or upregulate co-stimulatory molecules in dendritic cells or macrophages. Sci Rep. 2016 Nov 21;6:36928. PubMed PMID: 27869206; PubMed Central PMCID: PMCPMC5116629.
- Ebid R, Lichtnekert J, Anders H-J. Hyaluronan is not a ligand but a regulator of toll-like receptor signaling in mesangial cells: role of extracellular matrix in innate immunity. ISRN Nephrol. 2014;2014:11.
- Hong Y, Nam G-H, Koh E, et al. Exosome as a vehicle for delivery of membrane protein therapeutics, PH20, for enhanced tumor penetration and antitumor efficacy. Adv Funct Mater. 2018;28(5):1703074.
- Yang Y, Hong Y, Cho E, et al. Extracellular vesicles as a platform for membrane-associated therapeutic protein delivery. PubMed PMID: 29535849; PubMed Central PMCID: PMC5844050 J Extracell Vesicles. 2018;7(1):1440131.
- Kultti A, Li X, Jiang P, et al. Therapeutic targeting of hyaluronan in the tumor stroma. Cancers (Basel). 2012 Sep 6;4(3):873–903. PubMed PMID: 24213471; PubMed Central PMCID: PMCPMC3712709.
- Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012 Mar 20;21(3):418–429. PubMed PMID: 22439937; PubMed Central PMCID: PMC3371414.
- Thompson CB, Shepard HM, O’Connor PM, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther. 2010 Nov;9(11):3052–3064. PubMed PMID: 20978165.
- Midura RJ, Cali V, Lauer ME, et al. Quantification of hyaluronan (HA) using a simplified fluorophore-assisted carbohydrate electrophoresis (FACE) procedure. Methods Cell Biol. 2018;143:297–316. PubMed PMID: 29310784.
- Gao N. Fluorophore-assisted carbohydrate electrophoresis: a sensitive and accurate method for the direct analysis of dolichol pyrophosphate-linked oligosaccharides in cell cultures and tissues. Methods. 2005 Apr;35(4):323–327. PubMed PMID: 15804603.
- Yang HZ, Cui B, Liu HZ, et al. Blocking TLR2 activity attenuates pulmonary metastases of tumor. PloS One. 2009 Aug 5;4(8):e6520. PubMed PMID: 19654875; PubMed Central PMCID: PMC2716531.
- Toole BP. Hyaluronan-CD44 interactions in cancer: paradoxes and possibilities. Clin Cancer Res. 2009 Dec 15;15;(24):7462–7468. PubMed PMID: 20008845; PubMed Central PMCID: PMC2796593. Doi:10.1158/1078-0432.CCR-09-0479
- Misra S, Hascall VC, Markwald RR, et al. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol. 2015;6:201. PubMed PMID: 25999946; PubMed Central PMCID: PMC4422082.
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008 May;42(2):145–151. PubMed PMID: 18304834.
- Kuzmich NN, Sivak KV, Chubarev VN, et al. TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines (Basel). 2017 Oct 4;5(4). PubMed PMID: 28976923; PubMed Central PMCID: PMC5748601.
- An H, Yu Y, Zhang M, et al. Involvement of ERK, p38 and NF-kappaB signal transduction in regulation of TLR2, TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology. 2002 May;106(1):38–45. PubMed PMID: 11972630; PubMed Central PMCID: PMC1782697.
- Fitzgerald KA, Rowe DC, Barnes BJ, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. 2003 Oct 6;198(7):1043–1055. PubMed PMID: 14517278; PubMed Central PMCID: PMC2194210.
- Hubo M, Trinschek B, Kryczanowsky F, et al. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front Immunol. 2013;4:82. PubMed PMID: 23565116; PubMed Central PMCID: PMC3615188.
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004 Oct;5(10):987–995. PubMed PMID: 15454922.
- Jang MH, Sougawa N, Tanaka T, et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J Immunol. 2006 Jan 15;176(2):803–810. PubMed PMID: 16393963.
- Roberts EW, Broz ML, Binnewies M, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in Melanoma. Cancer Cell. 2016 Aug 8;30(2):324–336. PubMed PMID: 27424807; PubMed Central PMCID: PMC5374862.
- Hildner K, Edelson BT, Purtha WE, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008 Nov 14;322(5904):1097–1100. PubMed PMID: 19008445; PubMed Central PMCID: PMC2756611.
- Bollyky PL, Falk BA, Long SA, et al. CD44 costimulation promotes FoxP3+ regulatory T cell persistence and function via production of IL-2, IL-10, and TGF-beta. J Immunol. 2009 Aug 15;183(4):2232–2241. PubMed PMID: 19635906; PubMed Central PMCID: PMC3057032.
- Rayahin JE, Buhrman JS, Zhang Y, et al. High and low molecular weight hyaluronic acid differentially influence macrophage activation. ACS Biomater Sci Eng. 2015 Jul 13;1(7):481–493. PubMed PMID: 26280020; PubMed Central PMCID: PMC4533115.
- Mandai M, Hamanishi J, Abiko K, et al. Dual faces of IFNgamma in cancer progression: a role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin Cancer Res. 2016 May 15;22(10):2329–2334. 10.1158/1078-0432.CCR-16-0224. PubMed PMID: 27016309.
- Garcia-Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017 May 9;19(6):1189–1201. PubMed PMID: 28494868.
- Moynihan KD, Opel CF, Szeto GL, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 2016 Dec;22(12):1402–1410. PubMed PMID: 27775706; PubMed Central PMCID: PMCPMC5209798.
- Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell. 2017 Mar 13;31(3):311–325. PubMed PMID: 28292435; PubMed Central PMCID: PMCPMC5423788.
- Jonkers J, Derksen PW. Modeling metastatic breast cancer in mice. J Mammary Gland Biol Neoplasia. 2007 Sep;12(2–3):191–203. PubMed PMID: 17587153.
- Rodriguez LM, Guillén-Ponce C, Feliu J, et al. P-183 A pilot trial of PEGPH20 (pegvorhyaluronidase alfa) in combination with avelumab (anti-PD-L1 MSB0010718C) in chemotherapy resistant pancreatic cancer (PDAC)-trial in progress. Ann Oncol. 2018;29(suppl_5):mdy151.182.
- Thompson B, Kimbler T, Blouw B, et al. Abstract B38: increasing tumor-infiltrating CD8+ T cell response and checkpoint inhibitor efficacy by enzymatic reduction of tumor hyaluronan in a murine syngeneic pancreatic cancer model. Cancer Immunol Res. 2018;6(9 Supplement):B38–B38.
- Adachi K, Kano Y, Nagai T, et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018 Apr;36(4):346–351. PubMed PMID: 29505028.
- Tang H, Wang Y, Chlewicki LK, et al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell. 2016 Mar 14;29(3):285–296. PubMed PMID: 26977880; PubMed Central PMCID: PMCPMC4794755.
- Bu X, Mahoney KM, Freeman GJ. Learning from PD-1 resistance: new combination strategies. Trends Mol Med. 2016 Jun;22(6):448–451. PubMed PMID: 27174038.
- Tran TH, Mattheolabakis G, Aldawsari H, et al. Exosomes as nanocarriers for immunotherapy of cancer and inflammatory diseases. Clin Immunol. 2015 Sep;160(1):46–58. PubMed PMID: 25842185.
- Zhang B, Yin Y, Lai RC, et al. Immunotherapeutic potential of extracellular vesicles. Front Immunol. 2014;5:518. PubMed PMID: 25374570; PubMed Central PMCID: PMC4205852.
- Ingato D, Lee JU, Sim SJ, et al. Good things come in small packages: overcoming challenges to harness extracellular vesicles for therapeutic delivery. J Control Release. 2016 Nov 10;241:174–185. PubMed PMID: 27667180.
- Syn NL, Wang L, Chow EK, et al. Exosomes in cancer nanomedicine and immunotherapy: prospects and challenges. Trends Biotechnol. 2017 Jul;35(7):665–676. PubMed PMID: 28365132.
- Gilligan KE, Dwyer RM. Engineering exosomes for cancer therapy. Int J Mol Sci. 2017 May 24;18(6). PubMed PMID: 28538671; PubMed Central PMCID: PMCPMC5485946. DOI: 10.3390/ijms18061122