
ABSTRACT
Proliferation of vascular smooth muscle cells (VSMCs) plays crucial roles in vascular remodelling and stiffening in hypertension. Vascular adventitial fibroblasts are a key regulator of vascular wall function and structure. This study is designed to investigate the roles of adventitial fibroblasts-derived extracellular vesicles (EVs) in VSMC proliferation and vascular remodelling in normotensive Wistar-Kyoto rat (WKY) and spontaneously hypertensive rat (SHR), an animal model of human essential hypertension. EVs were isolated from aortic adventitial fibroblasts of WKY (WKY-EVs) and SHR (SHR-EVs). Compared with WKY-EVs, miR155-5p content was reduced, while angiotensin-converting enzyme (ACE) content was increased in SHR-EVs. WKY-EVs inhibited VSMC proliferation of SHR, which was prevented by miR155-5p inhibitor. SHR-EVs promoted VSMC proliferation of both strains, which was enhanced by miR155-5p inhibitor, but abolished by captopril or losartan. Dual luciferase reporter assay showed that ACE was a target gene of miR155-5p. MiR155-5p mimic or overexpression inhibited VSMC proliferation and ACE upregulation of SHR. WKY-EVs reduced ACE mRNA and protein expressions while SHR-EVs only increased ACE protein level in VSMCs of both strains. However, the SHR-EVs-derived from the ACE knockdown-treated adventitial fibroblasts lost the roles in promoting VSMC proliferation and ACE upregulation. Systemic miR155-5p overexpression reduced vascular ACE, angiotensin II and proliferating cell nuclear antigen levels, and attenuated hypertension and vascular remodelling in SHR. Repetitive intravenous injection of SHR-EVs increased blood pressure and vascular ACE contents, and promoted vascular remodelling in both strains, while WKY-EVs reduced vascular ACE contents and attenuated hypertension and vascular remodelling in SHR. We concluded that WKY-EVs-mediated miR155-5p transfer attenuates VSMC proliferation and vascular remodelling in SHR via suppressing ACE expression, while SHR-EVs-mediated ACE transfer promotes VSMC proliferation and vascular remodelling.
Introduction
Proliferation of vascular smooth muscle cells (VSMCs) is crucial in vascular remodelling and stiffening, and closely related to the progression of some cardiovascular diseases including hypertension, atherosclerosis, stroke, diabetic angiopathy [Citation1–Citation3]. Vascular adventitia acts as a biological processing centre for the retrieval, integration, storage and release of key regulators of vessel wall function [Citation4]. Excessive adventitial remodelling induces aortic maladaptation in angiotensin (Ang)-induced hypertension [Citation5]. The most abundant cell type in vascular adventitia is adventitial fibroblasts (AFs), which is a key regulator of vascular wall function and structure [Citation6]. AFs contribute to vascular remodelling [Citation7] and VSMC migration [Citation8] in hypertension.
Extracellular vesicles (EVs) are phospholipid membrane vesicles released by cells that play key roles in cell-to-cell communication in normal and diseased states [Citation9–Citation11]. EVs contain natural cargo molecules such as miRNA, mRNA and proteins, which are transferred to neighbouring or more distant cells to affect distinct signalling cascades [Citation12–Citation14]. These EVs encapsulate all vesicles released from cells, including microvesicles and exosomes. The microvesicles refer to plasma membrane shedding vesicles of 100–1000 nm, and the exosomes are defined as endosome-originated membrane vesicles with a diameter of 40–150 nm [Citation15,Citation16]. EVs are involved in modulating cardiovascular structure and function, and may be used as favourable strategies for diagnosis and treatment of cardiovascular diseases [Citation17].
MicroRNAs (miRNAs) are a class of naturally occurring, small non-coding RNA molecules, which bind to the 3′-untranslated regions (3′-UTRs) of a target mRNA sequence, and induce translation repression or mRNA degradation [Citation18]. Mature miRNA can be processed from either the 5′ or 3′ arm of miRNA precursors (pre-miRNA) to form miR-5p and miR-3p [Citation19]. It has been shown that miR155 promotes the proliferation and invasive abilities of colon cancer cells by targeting RNA-binding protein quaking [Citation20]. Conversely, silencing of miR155 increased proliferation and migration of human brain microvessel endothelial cells [Citation21]. Spontaneously hypertensive rat (SHR) is originally developed from Wistar-Kyoto rat (WKY), and is the best animal model of human essential hypertension [Citation22,Citation23]. Recently, we have found that angiotensin converting enzyme (ACE) content in EVs derived from aortic adventitial fibroblasts of SHR (SHR-EVs) is higher than that in the EVs derived from those of WKY (WKY-EVs). SHR-EVs promote migration of VSMCs via transferring ACE [Citation8]. The present study is designed to investigate the roles of EVs-mediated transfer of miR155-5p and ACE in VSMC proliferation and vascular remodelling in WKY and SHR.
Materials and methods
Experimental animals
Male WKY and SHR aged at 9 weeks were obtained from Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). The criterion for SHR used in the present study was that systolic blood pressure (SBP) was higher than 150 mm Hg. Experimental protocols were approved by the Experimental Animal Care and Use Committee of Nanjing Medical University, and were conformed to the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (NIH publication, 8th edition, 2011). Rats were housed in a 12 h light/12 h dark cycle with temperature-controlled room. The rats were fed with a standard chow and tap water ad libitum. At the end of the experiment, the rat was euthanized with an overdose of pentobarbital sodium (200 mg/kg, iv).
Culture of AFs and VSMCs
AFs and VSMCs were isolated and prepared from thoracic aorta of rats as we reported previously [Citation7,Citation24]. Briefly, thoracic aorta of WKY or SHR was isolated, and perivascular adipose tissues were removed. The aorta was cut open and stripped of intima, and adventitia was separated with media. Adventitia was treated with 0.2% collagenase in DMEM for 15 min, while the media was treated with 0.4% collagenase in DMEM for 30 min. After centrifugation at 200 × g for 10 min, the isolated cells were re-suspended in DMEM with 10% foetal bovine serum, 100 IU/mL penicillin and 10 mg/mL streptomycin in humidified atmosphere containing 5% CO2 at 37°C. AFs were confirmed by positive staining of vimentin (a marker of fibroblasts), and negative staining of PECAM-1 (a marker of endothelial cells) and α-SMA (a marker of VSMCs) [Citation7,Citation25].
Isolation of EVs from SHR-AF and WKY-AF cultures
EVs isolation and identification followed the MISEV 2018 guidelines [Citation26,Citation27]. The EVs were isolated from AFs medium with PureExo® Exosome Isolation Kit (101Bio, Palo Alto, CA, USA) according to the manual’s instruction [Citation28–Citation30]. After the density of AFs reached 80–90%, the AFs were incubated with serum free medium to avoid contamination of vesicles from serum for 48 h. The medium were collected and centrifuged at 3000 × g for 15 min at 4°C to remove cells and debris, and supernatant (2 mL) was transferred to a glass tube on ice. Solution A (0.125 mL), Solution B (0.125 mL) and Solution C (0.500 mL) from the Kit were mixed and vortexed for 10 s, and added into the supernatant. The tube was gently inverted at least 10 times to mix well, incubated at 4°C for 30 min. The upper colour layer was carefully pipet out without disturbing the lower cloudy layer (EVs in this layer). The left over in the tube was transferred to a 1.5 mL microcentrifuge tube and spun at 5000 × g for 3 min. The middle fluffy layer was transferred to a 0.5 mL microcentrifuge tube and spun at 5,000 × g for 3 min, and repeated this step one time. Then, the fluff pellet was left in the tube and its volume was about 25 μL. The tube cap was opened for air dry for 5 min, and 100 μL of 1× PBS was added to suspend the fluff pellet by pipetting up and down vigorously for 40 times. The tube was shaken on a horizontal shaker for 3 min with high speed, pipet up and down vigorously for 10 times, and then, this procedure was repeated for another 2 times. The tube was spun at 5000 × g for 5 min. The supernatant was carefully transferred into PureExo® Column supplied by the kit. The Column was spun at 1,000 × g for 5 min to collect all the “flow-through”. The “flow-through” is the isolated pure EVs (EVs suspended in PBS). EVs were utilized directly or stored at 4°C for up to 2 days, or at −80°C for up to 3 months [Citation31].
EVs were also isolated from the WKY-AF and SHR-AF medium with ultracentrifugation method [Citation32] to confirm the effects of EVs that isolated with the PureExo® Exosome Isolation Kit. Briefly, the collect medium was centrifuged at 300 × g for 5 min, 3000 × g for 30 min and then 10,000 × g for 60 min to remove dead cells, cell debris, apoptotic bodies and large vesicles (500–1000 nm). Then, the supernatants were ultracentrifuged at 120,000 × g for 70 min at 4°C to pellet the EVs. The pellets were washed three times with PBS, resuspended in defined amount of PBS for use.
Examination of EVs by transmission electron microscopy
Morphology of WKY-EVs and SHR-EVs were examined with transmission electron microscope as previously reported [Citation33]. Simply, 20 μL of EVs suspension was loaded onto a carbon-coated copper grid (Nisshin EM Corporation, Tokyo, Japan), and fixed at least 5 min. Afterwards, the EVs were stained with 2% uranyl acetate and dried for 10 min. Then, the grids were visualized with transmission electron microscope (Tecnai G2 Spirit TEM, Zeiss, Oberkochen, Germany) at 120 kV.
Identification of EVs by nanoparticle tracking analysis
Size distribution of EVs was analysed with Nanoparticle Tracking Analysis (NTA, NanoSight NS300, Malvern, Worcestershire, UK) [Citation34]. The particles are automatically tracked and sized based on Brownian motion and diffusion coefficient. EVs were diluted with deionized water to 1.0 mL. The measurement conditions were made at 23.75 ± 0.5°C, 25 frames per second (FPS) for 60 s with similar detection threshold for each sample. Three recordings were performed for each sample. Control medium and deionized water were used as controls.
Concentration of EVs
Protein content in EVs was measured with Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL, USA) following the manufacturer’s protocol, which was used as an index of the amount of EVs [Citation35–Citation37]. The final concentration of EVs in the VSMC medium was 30 μg protein/mL medium (about 0.56 × 106 particles of EVs/mL medium).
Evaluation of VSMC proliferation
VSMC proliferation was assessed by cell counting kit-8 (CCK-8) method, 5-ethynyl-2ʹ-deoxyuridine (EdU) incorporation assay and proliferating cell nuclear antigen (PCNA) expression as we previously reported [Citation38]. Firstly, the rate of VSMC proliferation was determined with CCK-8 kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. The absorbance was conducted at 450 nm using a microplate reader (ELX800, BioTek, Vermont, USA). Secondly, DNA synthesis was examined with EdU incorporation assay (Cell-Light™ EdU Apollo®567 In Vitro Imaging Kit, Guangzhou RiboBio, Guangzhou, China) to evaluate VSMC proliferation. The EdU positive cells were counted and normalized by the total number of Hoechst 33342 stained cells. Lastly, VSMC proliferation was evaluated by PCNA expression, which is a DNA clamp, acting on chromatin as a platform for various proteins involved in various aspects of DNA replication-linked processes.
Overexpression of miR155-5p in vitro and in vivo
Commercial AdmiRa-rno-miR-155-5p Virus and control adenovirus were obtained from Applied Biological Materials Inc. (Richmond, BC, Canada). For miR155-5p overexpression in VSMCs, the cells at 70% confluence in 6-well plates were transfected with control adenovirus or AdmiRa-rno-miR-155-5p Virus (40 MOI in 1 mL for each well) in an incubator. Measurements were performed 48 h after the transduction. For miR155-5p overexpression in rats, each rat received an intravenous injection of AdmiRa-rno-miR-155-5p Virus or control adenovirus (2 × 1011 plaque forming units/mL, 100 μL). Final experiment was performed 3 weeks after the transfection.
Transfection of miR-155 mimic and inhibitor
VSMCs in 6-well plates (about 5 × 105 cells/well) were cultured for 16 h. The cells were transfected with miR-155 mimic (50 nmol/L), or miR-155 inhibitor (100 nmol/L), or their corresponding negative controls. RNAifectin™ transfection reagent (6 μL) was simultaneously added into the medium for more efficient transfection. After 6 h, the culture medium was replaced to remove the transfection reagent. Detection was made 24 h after transfection. RNAifectin™ transfection reagent, miR-155 mimic, miR-155 inhibitor and their negative controls were purchased from Applied Biological Materials Inc. (Richmond, BC, Canada).
ACE knockdown in AFs
Lentiviral vectors targeting rat ACE (ACE-siRNA-lentivirus, 1 × 109 TU/mL) and scrambled siRNA-lentivirus (negative control) were constructed and verified by Shanghai Genechem (Shanghai, China). The nucleotide sequence in the ACE-siRNA-lentivirus was 5′-TGCCACGGAGGCCATGATAAA-3′. The effectiveness of the ACE-siRNA-lentivirus in down-regulation of ACE has been identified in our recent study [Citation8]. AFs were infected with ACE-siRNA-lentivirus (MOI = 80) containing polybrene for 24 h. Then, the medium was replaced with conventional culture medium for 72 h. AFs were trypsinized and washed with PBS, and seeded onto the cell culture bottle for 48 h. Then, the media was treated with serum-free medium for another 48 h. The culture medium was collected and EVs were isolated [Citation8].
Dual luciferase reporter assay
After VSMCs in white six-well plates were grown to 85–90% confluence, the cells were co-transfected with 2 µg of pLenti-UTR-GFP vector with rat ACE-3ʹUTR cloned behind the coding sequence and 2 µg of pre-miR155-5p or negative control in serum- and antibiotics-free DMEM with DNAfectin™ Plus Transfection Reagent (Applied Biological Materials Inc., Richmond, BC, Canada) for 6 h. Then, the medium was replaced with fresh culture medium, and the cells were incubated for 12 h. Firefly and Renilla luciferase were measured in cell lysates according to manufacturer’s protocol using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) on Luminometer 20/20n (Turmer Biosystems, Sunnyvale, CA, USA). Renilla luciferase activity was employed as an internal control for cellular density and transfection efficiency.
Measurement miR155-5p expression by qPCR
Measurement of miR155-5p was made in EVs, AFs, VSMCs, and transfected VSMCs, aorta and mesenteric artery of WKY and SHR. Total RNA was extracted using the miRcute miRNA isolation Kit (Tiangen Biotech, Beijing, China) and quantified using the NanoDrop 2000 Spectrophotometer (Thermo-Fisher Scientific, Wilmington, DE, USA). Identical starting concentrations of total RNA were used for all samples. Total RNA was reverse-transcribed to cDNA using a miRcute Plus miRNA First-Strand cDNA Kit (Tiangen Biotech) for miRNA. Changes in expression of various miRNA levels were determined quantitatively using Quantitative Reverse Transcriptase PCR (qRT-PCR). MiRcute Plus miRNA qPCR Kit (Tiangen Biotech) containing a QuantiTect SYBR Green PCR Master Mix and the miScript Universal Primer along with the miRNA-specific primer was used for the detection of mature miRNAs. Amplification and detection of the PCR products were performed on a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). U6 small RNA was used as an internal control to normalize the expression levels of the miR155-5p.
ACE and PCNA mRNA quantification by qPCR
ACE and PCNA mRNA were measured in transfected VSMCs, aorta and mesenteric artery. Total RNA was exacted with a Trizol reagent according to the manufacturer’s instructions (Life Technologies, Gaithersburg, MD, USA). RNA concentrations and purity were determined by the optical density at 260 and 280 nm. Reverse transcriptase reactions were made using the PrimeScript® RT reagent Kits (Takara, Otsu, Shiga, Japan) and StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). For quantitative PCR, cDNA fragments were subjected to SYBR Green RT-PCR (Takara Biotechnology Co., Ltd., Tokyo, Japan) using Stepone Plus system. The quantitative measures were obtained using the ΔΔCT method and normalized to the expression of GAPDH.
Western blot analysis
AFs, VSMCs, aorta and mesenteric artery were homogenized in lysis buffer. The supernatant was extracted for measurement of total protein using a BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL, USA). Equal amounts of total protein were separated by SDS-PAGE, and transferred to PVDF membrane in Trisglycine methanol buffer. Protein bands were visualized with Enhanced Chemiluminescence Detection Kit (Thermo Fisher Scientific, Rockford, IL, USA). Antibodies against calnexin, TSG101, APOA1, HSP70, and ACE were purchased from Abcam (Cambridge, MA, USA). Antibody against PCNA was obtained from Protein Tech Group Inc. (Chicago, IL, USA). Antibodies against CD9, CD63 and secondary antibodies were acquired from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Antibody against LC3B and GAPDH were bought from Cell Signalling Technology (Beverly, MA, USA). Considering that GAPDH is a more reliable housekeeping loading control [Citation39], GAPDH was used as an internal control of PCNA. Briefly, the primary antibody against PCNA was incubated with PVDF membrane at 4°C overnight and the target band was obtained. Then, the previous PCNA antibody was stripped from the membrane with the stripping buffer (Beyotime Biotechnology, Shanghai, China) for 5 min at 37°C under oscillating table. After being blocked, the membrane was incubated with the primary antibody against GAPDH and acquired band as an internal control of PCNA.
Immunohistochemistry for ACE
Aorta and mesenteric artery were isolated and fixed in 4% paraformaldehyde, embedded in paraffin and transversely cut into 5 μm sections with a cryostat (Leica, Solms, Germany). The sections were washed three times with 0.1 M PBS after de-paraffinization, blocked with blocking buffer for 5 min, and then incubated with primary anti-ACE antibody (1:100; Abcam, Cambridge, MA, USA) for 24 h at 4°C, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit antibody for 30 min in room temperature. 3,3-Diaminobenzidine was utilized to develop the positive cells. Sections were counterstained with haematoxylin.
Immunofluorescent analysis for ACE
Immunofluorescence analysis was performed as we previously described [Citation40]. Briefly, VSMCs were plated on glass coverslips in 6-well plates and treated with 4% paraformaldehyde for 15 min. The cells were washed with 1× PBS and permeabilized with 0.2% Triton X-100 for 5 min, and then, blocked with 1% BSA at room temperature for 1 h, incubated with the primary anti-body against ACE (1:100; Abcam, Cambridge, MA, USA) overnight. The cells were washed three times with 1× PBS and incubated with the secondary fluorescein anti-rabbit IgG (1:1000) for 1 h. The nucleus was stained with DAPI (Southern Biotech, Birmingham, AL, USA). The immunofluorescence images were acquired with an Olympus BX51 microscope (Olympus, Tokyo, Japan) coupled with an Olympus DP70 digital camera.
ELISA
Ang II and ACE contents were determined using commercial ELISA kits (Ang II: LifeSpan BioSciences, Seattle, WA, USA; ACE: Uscn Life Sciences Inc., Wuhan, China) according to the manufacturer’s protocols. Optical density was read at 450 nm using Microplate Reader (STNERGY/H4, BioTek, Vermont, USA).
Masson’s staining
Aorta and mesenteric artery were prefixed, and the paraffin-embedded sections were stained with Masson’s trichrome staining [Citation41]. The images were collected with a light microscope (BX-51, Olympus, Tokyo, Japan). Media thickness, lumen diameter and their ratio were used as indexes of vascular remodelling.
Blood pressure measurement
Blood pressure of tail artery was measured in conscious state with a non-invasive computerized tail-cuff system (NIBP, ADInstruments, Sydney, New South Wales, Australia). The rats were warmed for 10–20 min at 28ºC before the measurements in order to allow detection of tail artery pulsations at the steady pulse level. SBP was obtained by averaging 10 measurements [Citation42].
Flow cytometric analysis of cell apoptosis
Apoptosis rate was examined using Annexin V-FITC apoptosis detection kit (Beyotime Biotechnology, China) following the manufacturer’s instructions. Simply, the AFs were washed twice with PBS, and harvested by Trypsin without EDTA. Afterwards, the cells were resuspended in 195 μL binding buffer, 5 μL Annexin V-FITC regent and 10 μL propidium iodide (PI) regent, and incubated for 15 min at room temperature in the dark. The cell apoptosis was detected by a flow cytometry (BD FACSCalibur cytometer, Becton Dickinson, San Jose, CA, USA).
Statistical analysis
Trials were conducted in a double-blinded and randomized fashion. Data are given in mean±SE. Student’s unpaired t-Test was used for comparisons between two groups. One-way or two-way ANOVA followed by post hoc Bonferroni test was used for multiple comparisons. A value of P < 0.05 was considered statistically significant.
Results
Identification and comparison of WKY-EVs and SHR-EVs
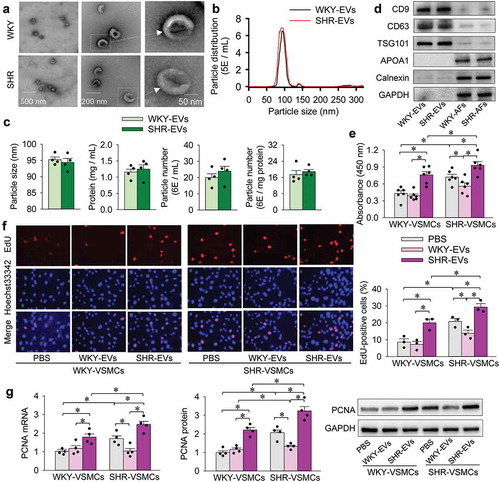
Transmission electron microscopic images showed that both WKY-EVs and SHR-EVs had a similar cup-shaped or spherical morphology (). According to nanoparticle tracking analysis, the distribution curve of the particle size of WKY-EVs and SHR-EVs were similar (). There were no significant differences in mean particle size of EVs, total protein content in EVs, particle number per mL and particle number per mg protein (). Protein content in the solution of EVs was 1.17 ± 0.16 mg protein/mL. The concentration of EVs was (21.94 ± 3.01)×106 particles/mL, which was equivalent to 18.7 × 106 particles/mg protein. In the present study, the final concentration of EVs used in the VSMC medium was 30 μg protein/mL medium (about 0.56 × 106 particles of EVs/mL medium). EVs-associated protein markers, two transmembrane/lipid-bound proteins (CD9 and CD63) and a cytosolic protein (TSG101) were found in both WKY-EVs and SHR-EVs. Two negative protein markers, apolipoprotein A1 (APOA1) and calnexin (an endoplasmic reticulum marker) were not found in the both WKY-EVs and SHR-EVs (). The isolated EVs are very likely to be exosomes in the present study according to the characteristic of the EVs.
Figure 1. Characterization of EVs and effects of EVs on VSMC proliferation. (a–b), the isolated EVs were immediately used for the characterization. (a), transmission electron microscopic images of EVs. (b), nanoparticle tracking analysis showing the particle distribution of different size of EVs. The value of the ordinate represents the mean particle number within every nanometre diameter range. (c), mean particle size of EVs, protein content in EVs, particle number per mL and particle number per mg protein. (d), Western blot analysis showing the biomarkers of EVs including CD9, CD63 and TSG101. APOA1 and calnexin were used as negative controls. (e–g), effects of WKY-EVs and SHR-EVs on VSMC proliferation. The VSMC proliferation was evaluated with CCK-8 kits (e), EdU-positive cells (f), PCNA mRNA and protein expression (g). The isolated EVs were immediately used for the evaluation of VSMC proliferation. The measurements were made 24 h after transferring WKY-EVs (30 μg/mL) or SHR-EVs (30 μg/mL) to the media of WKY-VSMCs and SHR-VSMCs. Values are mean±SE. n = 6 per group in (e); n = 3 per group in F; n = 4 per group in other groups. The cells were obtained from 6 WKY and 6 SHR.
Effects of EVs on VSMC proliferation
Effects of EVs on VSMC proliferation were evaluated with CCK-8 kits, EdU staining, PCNA mRNA and protein expression. The measurements were carried out 24 h after transferring PBS, WKY-EVs (30 μg/mL) or SHR-EVs (30 μg/mL) to the media of WKY-VSMCs and SHR-VSMCs. WKY-EVs had no significant effect on WKY-VSMC proliferation, but attenuated SHR-VSMC proliferation. SHR-EVs promoted the proliferation of both WKY-VSMCs and SHR-VSMCs, although the SHR-VSMC proliferation was enhanced compared with that of WKY-VSMCs (). It is probable that proliferation-inhibiting factor is dominant in WKY-EVs, and proliferation-promoting factor is dominant in SHR-EVs. We speculated that the effects of EVs on VSMC proliferation may not be caused by some possible signal molecules outside the EVs, because the EVs were washed and diluted with PBS several times. To support our speculation, the supernatant of EVs was obtained by ultracentrifugation to remove EVs. The supernatant of EVs had no significant effect on VSMC proliferation (Supplementary Figure 1), and no detectable miR155-5p can be found in the supernatant of EVs (Supplementary Figure 2), which confirm our speculation. On the other hand, the effects of EVs from the AFs cultured in EV-depleted serum on the VSMC proliferation were similar to these cultured in the serum without EV depletion, suggesting that the observed effects of EVs are caused by the AFs-derived EVs rather than the serum-derived EVs (Supplementary Figure 3). We noted that AFs can grow well in serum free medium for 48 h. AFs cultured in serum free medium for 48 h had no significant effects on the heat shock protein 70 (HSP70) expression and cell apoptosis compared with the AFs cultured in the medium with serum, suggesting that no obvious stress and cell death were observed in the AFs cultured in serum free medium for 48 h (Supplementary Figure 4). The starvation due to the serum-free medium for 48 h increased the ratio of LC3B II to LC3B I, an index of autophagy, but had no significant effects on the proliferation and lactate dehydrogenase (LDH) activity in AFs (Supplementary Figure 5A–C). The starvation-induced autophagy may degrade large intracellular aggregates and dysfunctional organelles, promote nutrient recycling and play an important protective role in the cell survival.
MiR155-5p levels and effects of miR155-5p on VSMC proliferation
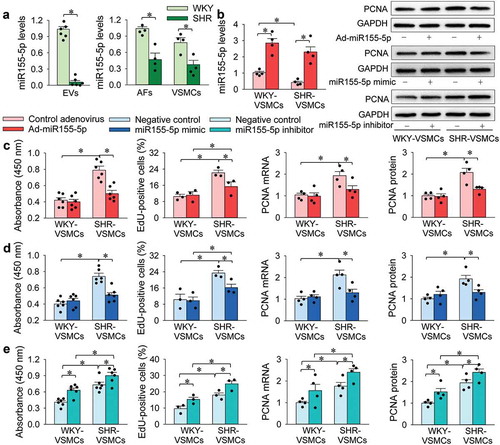
It has been found that miR155 levels are negatively correlated with blood pressure in SHR [Citation43], and miR155 in AFs regulates the differentiation of AFs [Citation44]. In our preliminary study, we found that miR155-5p was rich in WKY-EVs, which caused our great interest that EVs-mediated miR155-5p transfer might be a critical factor in inhibiting VSMC proliferation. Thus, a series of experiments were designed to test the hypotheses. We found that miR155-5p levels in EVs, AFs and VSMCs of SHR were lower than those of WKY. It is worth noting that miR155-5p in the SHR-EVs reduced to a very low level compared with that of WKY-EVs (). We further examined the effects of miR155-5p overexpression on VSMC proliferation. The efficiency of miR155-5p overexpression was confirmed by the miR155-5p upregulation in WKY-VSMCs and SHR-VSMCs (). The miR155-5p overexpression in VSMCs had no significant effect on WKY-VSMC proliferation, but attenuated SHR-VSMC proliferation (). Similarly, miR155-5p mimic attenuated SHR-VSMC proliferation, but was unable to attenuate WKY-VSMC proliferation (). Furthermore, miR155-5p inhibitor enhanced the proliferation of WKY-VSMCs and SHR-VSMCs (). These results indicate that miR155-5p inhibits VSMC proliferation of SHR, and endogenous miR155-5p plays a role in attenuating VSMC proliferation of WKY and SHR.
Figure 2. MiR155-5p levels and effects of miR155-5p on WKY-VSMC and SHR-VSMC proliferation. The proliferation was respectively evaluated with CCK-8 kits, EdU incorporation assay, PCNA mRNA and protein expression. (a) miR155-5p levels in EVs, AFs and VSMCs. (b) effects of miR155-5p overexpression on miR155-5p levels of VSMCs. (c) effects of miR155-5p overexpression on VSMC proliferation. The measurements were carried out 48 h after treatment with control adenovirus or miR155-5p adenovirus (40 MOI). (d) effects of miR155-5p mimic (50 nmol/L) or its negative control (50 nmol/L) on VSMC proliferation. The measurements were carried out 24 h after negative control or miR155-5p mimic treatment. (e) effects of miR155-5p inhibitor on VSMC proliferation. Measurements were performed 24 h after the VSMC treatment with miR155-5p inhibitor (100 nmol/L) or its negative control (100 nmol/L). Values are mean±SE. n = 6 per group for measuring miR155-5p levels in EVs and absorbance; n = 3 per group for measuring EdU-positive cells. n = 4 per group in other groups. The cells were obtained from 6 WKY and 6 SHR.
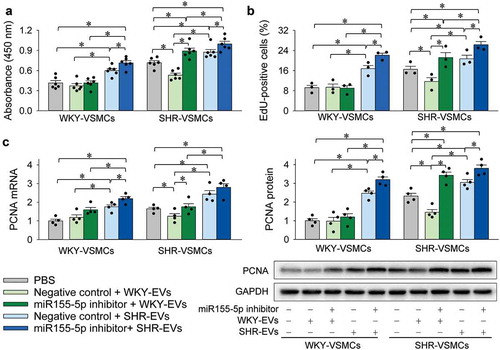
Interaction of miR155-5p inhibitor and EVs in VSMC proliferation
Inhibitor of miR155-5p was used to determine whether miR155-5p is involved in the effects of EVs on VSMC proliferation. The inhibitory effect of WKY-EVs on the proliferation of SHR-VSMCs was reversed by the miR155-5p inhibitor. The promoting effects of SHR-EVs on the proliferation of both WKY-VSMCs and SHR-VSMCs were enhanced by the miR155-5p inhibitor (). These results suggest that miR155-5p in EVs inhibits VSMC proliferation.
Figure 3. Interaction of miR155-5p inhibitor and EVs on VSMC proliferation. The VSMC proliferation was evaluated with CCK-8 kits (a), EdU-positive cells (b), PCNA mRNA and protein expressions (c). Measurements were performed 24 h after the VSMC treatment with PBS, combined negative control (100 nmol/L) and EVs (30 μg/mL), or combined miR155-5p inhibitor (100 nmol/L) and EVs (30 μg/mL). Values are mean±SE. n = 6 per group in A; n = 3 per group in (b); n = 4 per group in (c). The cells were obtained from 6 WKY and 6 SHR.
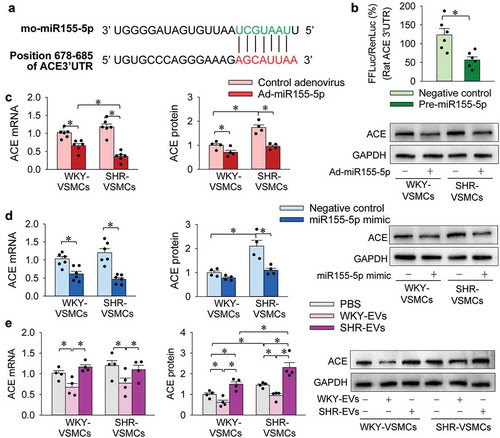
Effects of miR155-5p and EVs on ACE expression
A recent study in our lab has shown that ACE contents were increased in SHR-EVs compared with that in WKY-EVs, and the ACE in SHR-EVs promotes the migration of both WKY-VSMCs and SHR-VSMCs [Citation8]. We suspected that the increased ACE in SHR-EVs might be an important proliferation-promoting factor in the EVs. According to the TargetScanHuman, an online prediction of microRNA targets (http://www.targetscan.org/), ACE might be a target of miR155-5p. The predicted location of the combination is in position 678–685 of ACE 3ʹ-UTR (). To test this hypothesis, dual luciferase reporter assay was performed in VSMCs from WKY. Potential binding sites of miR155-5p in the 3′UTR of the rat ACE mRNA were determined by TargetScan. Firefly luciferase activity was significantly inhibited when co-transfected with pre-miR155-5p, indicating that rat ACE is a target of miR155-5p (). More importantly, miR155-5p overexpression caused by Ad-miR155-5p transfection reduced ACE mRNA and protein expressions in VSMCs of WKY and SHR (). Similar inhibitory effects of miR155-5p mimic on ACE expression were observed in VSMCs of WKY and SHR (). Thus, we further examined the effects of EVs on ACE mRNA and protein expressions in VSMCs. WKY-EVs inhibited ACE mRNA and protein expressions in both WKY-VSMCs and SHR-VSMCs. SHR-EVs had no significant effects on ACE mRNA expression, but up-regulated the ACE protein in both WKY-VSMCs and SHR-VSMCs (). These results provide evidence that miR155-5p inhibits ACE expression in VSMCs. WKY-EVs inhibit ACE expression by transferring miR155-5p, while SHR-EVs promote ACE expression by directly transferring ACE.
Figure 4. Effects of miR155-5p on ACE expressions in VSMCs. (a) predicted location of the miR155-5p combination according to TargetScanHuman. (b) dual luciferase reporter assay shows that ACE is a target of miR155-5p in VSMCs. FFLuc, Firefly luciferase; RenLuc, Renilla luciferase. (c) effects of miR155-5p overexpression on ACE mRNA and protein expressions in VSMCs. The measurements were carried out 48 h after treatment with control adenovirus or Ad-miR155-5p (40 MOI). (d) effects of miR155-5p mimic on ACE mRNA and protein expressions in VSMCs. The measurements were carried out 24 h after treatment with negative control or miR155-5p mimic (50 nmol/L). E, effects of WKY-EVs and SHR-EVs on ACE mRNA and protein expressions in VSMCs. The measurements were made 24 h after transferring WKY-EVs (30 μg/mL) or SHR-EVs (30 μg/mL) to the media of WKY-VSMCs and SHR-VSMCs. Values are mean±SE. n = 6 per group in (b); n = 4 or 6 per group in (c–e). The cells were obtained from 6 WKY and 6 SHR.
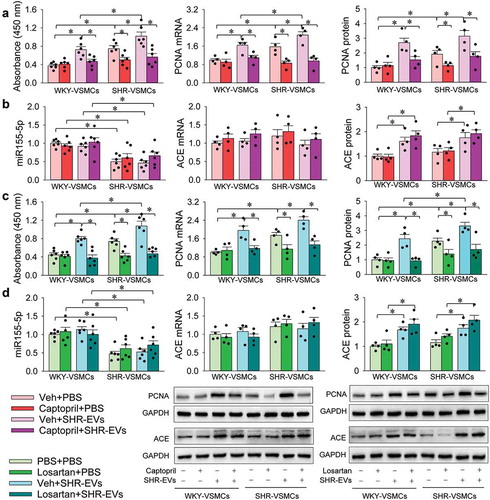
Captopril or losartan inhibits SHR-EVs-induced VSMC proliferation
As we recently reported, ACE content and activity were increased in SHR-EVs compared with those of WKY-EVs [Citation8]. The increased ACE content in SHR-EVs was further confirmed with Western blot () and ELISA (Supplementary Figure 6) in the present study. To determine the roles of SHR-EVs in promoting VSMC proliferation were mediated by the ACE in the EVs, the effects of an ACE inhibitor captopril and an AT1 receptor antagonist losartan on the SHR-EVs-induced VSMC proliferation were examined. Captopril inhibited the SHR-EVs-induced proliferation of WKY-EVs and SHR-EVs (), but had no significant effects on miR155-5p, ACE mRNA and protein expressions (). Similarly, losartan abolished the SHR-EVs-induced proliferation of WKY-EVs and SHR-EVs (), but had no significant effects on miR155-5p, ACE mRNA and protein expressions (). These results indicate that EVs-mediated ACE transfer contributes to the proliferation-promoting effect of SHR-EVs.
Figure 5. Effects of captopril and losartan on the VSMC responses to SHR-EVs. Measurements were carried out 24 h after treatment with SHR-EVs (30 µg/mL) plus captopril (200 µmol/L) or losartan (10 µmol/L). Captopril was dissolved in PBS containing 1% DMSO, and the PBS containing 1% DMSO was used as control (Veh). The VSMC proliferation was evaluated with CCK-8 kits, PCNA mRNA and protein expressions. (a) effects of captopril on the SHR-EVs-induced VSMC proliferation. (b) effects of captopril on the SHR-EVs-induced changes of miR155-5p, ACE mRNA and protein expressions. (c) effects of losartan on the SHR-EVs-induced VSMC proliferation. (d) effects of losartan on the SHR-EVs-induced changes of miR155-5p, ACE mRNA and protein expressions. Values are mean±SE. n = 6 per group for measuring absorbance and miR155-5p levels; n = 4 per group for other groups. The cells were obtained from 6 WKY and 6 SHR.
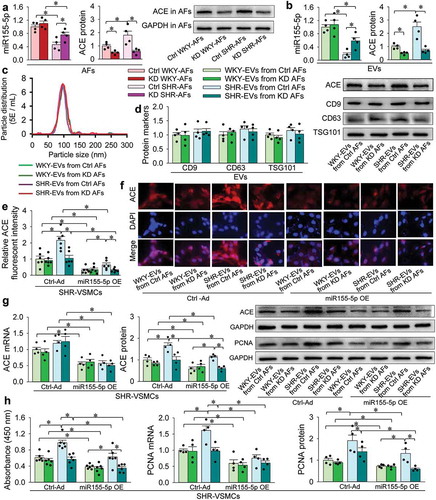
ACE knockdown in SHR-AFs abolishes the roles of the SHR-EVs in promoting VSMC proliferation and ACE protein upregulation
WKY-AFs and SHR-AFs were infected with ACE-siRNA lentiviral vectors for ACE knockdown (KD) or negative control lentivirus (Ctrl) for 5 days. WKY-EVs and SHR-EVs were respectively isolated from the Ctrl-treated or KD-treated AFs. SHR-VSMCs were infected with adenovirus-mediated miR155-5p overexpression (miR155-5p OE) or negative control adenovirus (Ctrl-Ad) for 2 days. And then, the isolated WKY-EVs and SHR-EVs were, respectively, added into the Ctrl-Ad-treated or Ad-miR155-5p-treated SHR-VSMCs for 1 day. The protocols are shown in a schematic diagram (Supplementary Figure 7). ACE knockdown in AFs had no significant effects on miR155-5p levels in WKY-AFs and WKY-EVs, but attenuated the miR155-5p down-regulation in SHR-AFs and SHR-EVs. As expected, ACE knockdown in AFs reduced ACE contents not only in SHR-AFs and SHR-EVs, but also WKY-AFs and WKY-EVs, indicating the effectiveness of ACE knockdown (). ACE knockdown had no significant effects on the EV size distribution (), as well as EV marker protein expressions including CD9, CD63 and TSG101 (). Moreover, the ACE knockdown did not affect the proliferation of WKY-AFs and SHR-AFs (Supplementary Figure 8). These results suggest that the ACE knockdown may not affect the production of EVs. WKY-EVs and SHR-EVs from ACE knockdown AFs had no significant effects on ACE mRNA expressions in SHR-VSMCs (). However, SHR-EVs from ACE knockdown AFs lost the roles in increasing the ACE protein contents in SHR-VSMCs, and miR155-5p overexpression reduced ACE expression in all groups evidenced by the ACE protein expression () and the ACE immunofluorescent intensity (). As we expected, overexpression of miR155-5p inhibited VSMC proliferation in all groups (). These results indicate that SHR-EVs-mediated ACE transfer contributes to the ACE upregulation and proliferation in VSMCs. The findings show the importance of miR155-5p in inhibiting ACE expressions and proliferation of the SHR-VSMCs. The reduced miR155-5p and increased ACE in SHR-EVs are responsible for the ACE upregulation and proliferation of SHR-VSMCs.
Figure 6. Effects of ACE knockdown in AFs and its interaction with miR155-5p overexpression in VSMCs. AFs were treated with scrambled siRNA-lentivirus (Ctrl) or ACE-siRNA-lentivirus (80 MOI) for ACE knockdown (KD). After 5 days, EVs were isolated from AFs. (a) effects of the KD on the miR155-5p and ACE protein contents in AFs. (b) effects of the KD on the miR155-5p and ACE protein contents in EVs. (c) effects of the KD on the particle distribution of different size of EVs determined by nanoparticle tracking analysis. The value of the ordinate represents the mean particle number within every nanometre diameter range. (d) effects of the KD on the marker protein expressions of EVs. E-H, comparison of the roles of the EVs from Ctrl AFs and the EVs from KD AFs and its interaction with miR155-5p overexpression (OE) in SHR-VSMCs. SHR-VSMCs were treated with Ad-scrambled miR155-5p (Ctrl) and Ad-miR155-5p (miR155-5p OE, 40 MOI) for 48 h followed by EVs treatment for 24 h. (e) bar graph showing relative ACE fluorescent intensity in SHR-VSMCs. (f) representative images showing the ACE immunofluorescence (red) and DAPI immunofluorescence (blue) in SHR-VSMCs. (g) ACE mRNA and protein expressions in SHR-VSMCs. H, VSMC proliferation evaluated with CCK-8 kits, PCNA mRNA and protein expressions. Values are mean±SE. n = 6 per group in measuring absorbance and ACE immunofluorescence; n = 4 per group for other measurements. The cells were obtained from 6 WKY and 6 SHR.
Effects of EVs isolated with ultracentrifugation method on VSMC proliferation
To confirm the effect of EVs on VSMC proliferation, WKY-EVs and SHR-EVs were isolated with ultracentrifugation. Transmission electron microscopic images showed that both WKY-EVs and SHR-EVs had a similar spherical morphology (Supplementary Figure 9A). There were no significant differences in the particle size distribution (Supplementary Figure 9B), mean particle size of EVs, total protein content in EVs, particle number per mL and particle number per mg protein (Supplementary Figure 9C) between WKY-EVs and SHR-EVs. The EVs isolated with ultracentrifugation method were further confirmed by the EVs-associated protein markers, CD9, CD63 and TSG101 as well as the negative protein markers, APOA1 and calnexin (Supplementary Figure 9D). The miR155-5p levels were reduced, while ACE protein contents were increased in the SHR-EVs (Supplementary Figure 10). The WKY-EVs attenuated SHR-VSMC proliferation, but SHR-EVs enhanced the proliferation of both WKY-VSMCs and SHR-VSMCs (Supplementary Figure 11). The results further support our findings in the EVs isolated with PureExo® Exosome Isolation Kit.
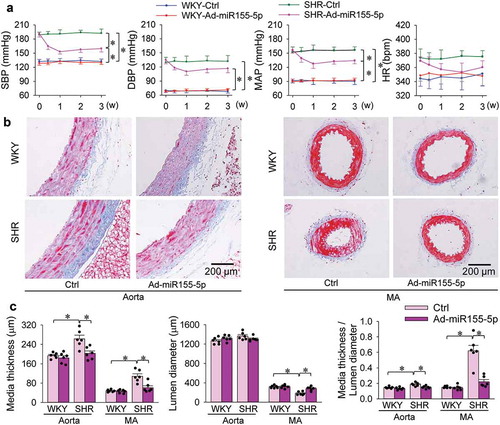
Effects of miR155-5p overexpression in SHR
Therapeutic effects of miR155-5p overexpression were examined in SHR. Either WKY and SHR were, respectively, subjected to intravenous injection of Ad-scrambled miR155-5p (Ctrl) or Ad-miR155-5p. Blood pressure and heart rate were measured every week. All other measurements were performed at the end of 3 weeks. The effectiveness of miR155-5p overexpression was confirmed by the increased miR155-5p levels in the aorta and mesenteric artery of WKY and SHR (). The miR155-5p overexpression inhibited ACE mRNA expressions in both WKY and SHR. However, it only decreased ACE protein expressions in SHR but not in WKY (). The findings were further confirmed by ACE immunohistochemistry analysis that the upregulation of ACE in the aorta of SHR were attenuated by miR155-5p overexpression (). Similarly, miR155-5p overexpression reduced Ang II contents in aorta and mesenteric artery of SHR, but not in these of WKY (). PCNA mRNA and protein expressions were upregulated in the aorta and mesenteric artery of SHR, which were attenuated by miR155-5p overexpression (). Overexpression of miR155-5p had no significant effect on blood pressure in WKY, but had significantly anti-hypertensive effect in SHR, reaching its maximal effect at the end of the first week, and lasting at least 3 weeks (). Masson’s staining of the aorta and mesenteric artery showed that miR155-5p overexpression attenuated vascular remodelling (), and reduced media thickness and ratio of media thickness to lumen diameter in aorta and mesenteric artery of SHR (). These results indicate that increased miR155-5p expression in SHR attenuates hypertension, vascular remodelling and proliferation, and reduced vascular ACE and Ang II levels.
Figure 7. Effects of miR155-5p overexpression on the levels of miR155-5p, ACE, Ang II and PCNA expressions in aorta and mesenteric artery (MA) of WKY and SHR. Measurements were carried out 3 weeks after intravenous administration of Ad-scrambled miR155-5p (Ctrl) or Ad-miR155-5p (2 × 1011 plaque forming units/mL, 100 µL). (a) miR155-5p levels. (b) ACE mRNA and protein expressions. (c) representative images of immunohistochemistry for ACE (brown colour) in aorta. (d) bar graph showing the relative density of ACE staining in aorta. (e) Ang II contents. (f) PCNA mRNA and protein levels. Values are mean±SE. The data were obtained from 12 WKY and 12 SHR. n = 6 per group in (d) and (e); n = 4 per group in (a), (b) and (f).
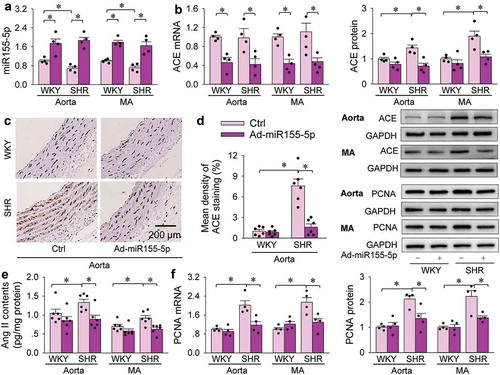
Figure 8. Effects of Ad-miR155-5p overexpression on blood pressure and vascular remodelling in aorta and mesenteric artery (MA) of WKY and SHR. Measurement was carried out 3 weeks after intravenous administration of Ad-scrambled miR155-5p (Ctrl) or Ad-miR155-5p (2 × 1011 plaque forming units/mL, 100 µL). (a) systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial pressure (MAP) and heart rate (HR) measured once a week in awake state. (b) representative images of Masson’s staining of aorta and MA. (c) bar graph showing the Masson’s staining analysis for media thickness, lumen diameter and their ratio in aorta and MA. Values are mean±SE. The data were obtained from 12 WKY and 12 SHR. n = 6 per group.
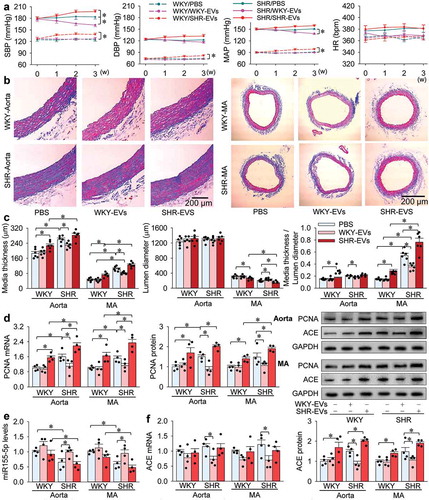
Effects of intravenous injection of WKY-EVs and SHR-EVs in WKY and SHR
We further examined the roles of WKY-EVs and SHR-EVs in vivo. Either WKY or SHR were randomly divided into three groups, which were subjected to intravenous injection of PBS, WKY-EVs or SHR-EVs every 2 days for 10 times. Blood pressure and heart rate were measured every week. All other measurements were performed at the end of 3 weeks. WKY-EVs had a mild anti-hypertensive effect in SHR, but had no significant effect on blood pressure in WKY. SHR-EVs increased blood pressure in WKY and aggravated hypertension in SHR (). Masson’s staining of the aorta and mesenteric artery showed that WKY-EVs attenuated vascular remodelling while SHR-EVs deteriorated vascular remodelling (). Consistently, WKY-EVs reduced vascular PCNA expressions in SHR, and SHR-EVs increased vascular PCNA expressions in both WKY and SHR (). On the other hand, WKY-EVs increased vascular miR155-5p and reduced ACE mRNA and protein expressions in SHR. SHR-EVs had no significant effects on vascular miR155-5p and ACE mRNA levels, but increased vascular ACE protein contents in WKY and SHR ().
Figure 9. Effects of intravenous injection of WKY-EVs and SHR-EVs in WKY and SHR. Either WKY or SHR were randomly divided into 3 groups, which were subjected to intravenous injection of 0.5 mL of PBS, WKY-EVs (100 μg) or SHR-EVs (100 μg) every 2 days for 10 times. (a) systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial pressure (MAP) and heart rate (HR) measured once a week in awake state. (b) representative images of Masson’s staining of aorta and MA. (c) bar graph showing the Masson’s staining analysis for media thickness, lumen diameter and their ratio in aorta and MA. (d) PCNA mRNA and protein in the media of aorta and MA. (e) miR155-5p levels in the media of aorta and MA. (f) ACE mRNA and protein expressions in the media of aorta and MA. Values are mean±SE. The data were obtained from 18 WKY and 18 SHR. n = 6 per group in A-C; n = 4 per group in (d–f).
Discussion
EVs have been recognized to be important in cell-to-cell communication due to their ability to transfer important cellular cargoes such as mRNA, miRNA and proteins to neighbouring cells or more distant cells through circulation [Citation9]. The present findings showed a novel mechanism that AFs modulate VSMC proliferation via EVs-mediated transfer of miR155-5p and ACE, a proliferation-inhibiting factor and a proliferation-promoting factor, respectively. WKY-EVs or miR155-5p attenuates but SHR-EVs promote VSMC proliferation, vascular remodelling and hypertension in SHR.
MiR155 is negatively related to the blood pressure in SHR [Citation43], and is involved in the differentiation of AFs [Citation44]. In this study, we showed that miR155-5p played beneficial roles in attenuating VSMC proliferation, vascular remodelling and hypertension in hypertensive rats, suggesting that miR155-5p might be taken as a potential therapeutic strategy in attenuating VSMC proliferation and vascular remodelling in some vascular diseases. It is interesting that the inhibitory effect of WKY-EVs on SHR-VSMC proliferation was prevented by miR155-5p inhibitor. Repeated intravenous injection of WKY-EVs attenuated vascular remodelling and hypertension in SHR. The results reveal that AFs inhibit VSMC proliferation via EVs-mediated miR155-5p transfer, which may be favourable to the attenuation of vascular remodelling at a very early stage of hypertension. ACE is well known to convert Ang I to Ang II, which is crucial in cardiovascular remodelling and hypertension [Citation45,Citation46]. Blockage of angiotensin receptors reduces cardiovascular events [Citation47]. Inhibition of ACE reduces VSMC proliferation in rats [Citation48], and lowers morbidity/mortality in hypertensive patients [Citation49]. Bioinformatics analysis and dual luciferase reporter gene assay showed that ACE was one of targets of miR155-5p, which was confirmed by the findings that miR155-5p inhibited ACE mRNA and protein expressions in SHR-VSMCs and in arteries of SHR. On the other hand, WKY-EVs inhibited ACE mRNA and protein expressions and increased miR155-5p levels in SHR-VSMCs and in arteries of SHR. These findings indicate that miR155-5p inhibits SHR-VSMC proliferation via downregulation of ACE expression, and the anti-proliferation role of WKY-EVs was mediated by the inhibitory effect of miR155-5p on ACE expressions in SHR-VSMCs.
SHR-EVs stimulated VSMC proliferation, and promoted vascular remodelling of both WKY and SHR. ACE content increased while miR155-5p content reduced in the SHR-EVs. SHR-EVs had no significant effects on miR155-5p and ACE mRNA levels but increased ACE protein levels in VSMCs and arteries of both WKY and SHR. The SHR-EVs-induced VSMC proliferation was abolished by either ACE inhibitor captopril or AT1 receptor antagonist losartan. More importantly, the SHR-EVs isolated from the AFs with ACE knockdown pretreatment lost their effect on VSMC proliferation accompanied by reduction of ACE content in the SHR-EVs. These findings provide solid evidence that increased ACE content is mainly responsible for the proliferation and vascular remodelling effects of the SHR-EVs. The reduced miR155-5p in SHR-EVs may only partially contribute to the effects of the SHR-EVs. It is noted that captopril or losartan treatment inhibited SHR-EVs-induced PCNA mRNA and protein expressions, rather than ACE mRNA and protein expressions. The discrepancy attributed to the SHR-EVs-mediated transfer ACE protein to the VSMCs. The increased ACE protein in VSMCs promoted Ang II production, which activated AT1 receptors, and then promoted both mRNA and protein expressions of PCNA in the VSMCs. Therefore, inhibition of ACE with captopril or blockage of AT1 receptors with losartan had no significant effects on ACE expressions, but abolished the effects of SHR-EVs on the PCNA mRNA and protein expressions.
Remodelling of large and small arteries contributes to the development and complications of hypertension. In the present study, miR155-5p overexpression attenuated hypertension and vascular remodelling accompanied with reduced vascular ACE and Ang II contents in SHR. The anti-hypertension effect of miR155-5p may be attributed to the reduced ACE and Ang II levels, as well as attenuated vascular remodelling in SHR. These findings shed a light on future studies that upregulation of miR155-5p might be taken as a new strategy for attenuating hypertension and vascular remodelling in hypertension or some other vascular diseases.
ACE inhibitors are effective drugs used in the treatment of hypertension. Millions of people take ACE inhibitors to lower their blood pressure. These drugs are relatively safe in short-term treatment, but concerns have been raised about a possible association of their long-term use with an increased risk of cancer. In a recent large, population based cohort study, the use of ACE inhibitors was associated with an increased risk of lung cancer. Among people using ACE inhibitors for more than five years, the increased risk may be as high as 14% [Citation50]. Our study shows that miR155-5p might be used to inhibit ACE expression. It is very interesting that repetitive intravenous injection of WKY-EVs attenuated hypertension and vascular remodelling accompanied with increased vascular miR155-5p and reduced ACE in the SHR. The anti-hypertension effect of WKY-EVs seems a little slow and mild compared with that of miR155-5p overexpression, which may be attributed to the rapidly and greatly increased miR155-5p level in the rats with miR155-5p overexpression. We speculate that the EVs containing more miR155-5p might cause greater effects than the isolated EVs from AFs. There might be a potential prospect that a special kind of exosomes for miR155-5p delivery was designed and used to reducing ACE expression, attenuating vascular remodelling in hypertension, which need further investigation.
Although we found that miR155-5p inhibited proliferation of SHR-VSMCs via targeting ACE, we cannot rule out a possibility that other targets of miR155-5p might be partially involved in the inhibitory effect of miR155-5p on VSMC proliferation. Moreover, the possibility that some factors in or associated with EVs might be contributing to the effect on VSMC proliferation cannot be completely ruled out. Some proteins including lipoproteins are major constituents of non-EV structures often co-isolated with EVs [Citation26]. APOA1 was not detected in the EVs, suggesting a high purity of the EV preparation was obtained in the present study and excluding the possibility that non-vesicular material such as lipoproteins in the EV preparation might carry miR155-5p. On the other hand, the size of most EVs from AFs was in the range of 80–110 nm, suggesting that the EVs were very likely to be exosomes.
In summary, AFs-derived EVs play an important role in modulating VSMC proliferation. The miR155-5p in WKY-EVs inhibits proliferation of SHR-VSMCs via suppressing ACE expression. The ACE in SHR-EVs promotes proliferation of WKY-VSMCs and SHR-VSMCs. Both increased ACE and reduced miR155-5p in the SHR-EVs contribute to the proliferation of SHR-VSMCs. Overexpression of miR155-5p reduces vascular ACE contents and attenuates vascular remodelling and hypertension in SHR. Repetitive intravenous injection of WKY-EVs reduced vascular ACE and attenuated hypertension and vascular remodelling in SHR, while SHR-EVs increased blood pressure and vascular ACE contents, and promoted vascular remodelling in both WKY and SHR. These findings show the importance of EVs-mediated miR155-5p and ACE acting as crucial signalling mediators in VSMC proliferation. MiR155-5p or exosomes-mediated miR155-5p transfer may be used as a novel strategy for intervention of VSMC proliferation, vascular remodelling and hypertension.
Supplemental Material
Download MS Word (1.3 MB)Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Rizzoni D, Agabiti RE. Small artery remodeling in hypertension and diabetes. Curr Hypertens Rep. 2006;8(1):90–19.
- Ponticos M, Smith BD. Extracellular matrix synthesis in vascular disease: hypertension, and atherosclerosis. J Biomed Res. 2014;28(1):25–39.
- Xu J, Shi GP. Vascular wall extracellular matrix proteins and vascular diseases. Biochim Biophys Acta. 2014;1842(11):2106–2119.
- Stenmark KR, Yeager ME, El Kasmi KC, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75:23–47.
- Bersi MR, Bellini C, Wu J, et al. Excessive adventitial remodeling leads to early aortic maladaptation in angiotensin-induced hypertension. Hypertension. 2016;67(5):890–896.
- Stenmark KR, Nozik-Grayck E, Gerasimovskaya E, et al. The adventitia: essential role in pulmonary vascular remodeling. Compr Physiol. 2011;1(1):141–161.
- Ling L, Chen D, Tong Y, et al. Fibronectin type III domain containing 5 attenuates inflammasome activation and phenotypic transformation of adventitial fibroblasts in spontaneously hypertensive rats. J Hypertens. 2018;36(5):1104–1114.
- Tong Y, Ye C, Ren XS, et al. Exosome-mediated transfer of ACE (angiotensin-converting enzyme) from adventitial fibroblasts of spontaneously hypertensive rats promotes vascular smooth muscle cell migration. Hypertension. 2018;72(4):881–888.
- Javeed N, Mukhopadhyay D. Exosomes and their role in the micro-/macro-environment: a comprehensive review. J Biomed Res. 2017;31(5):386–394.
- Nieuwland R, Falcon-Perez JM, Soekmadji C, et al. Essentials of extracellular vesicles: posters on basic and clinical aspects of extracellular vesicles. J Extracell Vesicles. 2018;7(1):1548234.
- Roy S, Hochberg FH, Jones PS. Extracellular vesicles: the growth as diagnostics and therapeutics; a survey. J Extracell Vesicles. 2018;7(1):1438720.
- Nguyen MA, Karunakaran D, Geoffrion M, et al. Extracellular vesicles secreted by atherogenic macrophages transfer microRNA to inhibit cell migration. Arterioscler Thromb Vasc Biol. 2018;38(1):49–63.
- Buschmann D, Kirchner B, Hermann S, et al. Evaluation of serum extracellular vesicle isolation methods for profiling miRNAs by next-generation sequencing. J Extracell Vesicles. 2018;7(1):1481321.
- Wen C, Seeger RC, Fabbri M, et al. Biological roles and potential applications of immune cell-derived extracellular vesicles. J Extracell Vesicles. 2017;6(1):1400370.
- Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232.
- Torrano V, Royo F, Peinado H, et al. Vesicle-MaNiA: extracellular vesicles in liquid biopsy and cancer. Curr Opin Pharmacol. 2016;29:47–53.
- Sun HJ, Zhu XX, Cai WW, et al. Functional roles of exosomes in cardiovascular disorders: a systematic review. Eur Rev Med Pharmacol Sci. 2017;21(22):5197–5206.
- Hartmann D, Fiedler J, Sonnenschein K, et al. MicroRNA-based therapy of GATA2-deficient vascular disease. Circulation. 2016;134(24):1973–1990.
- Griffiths-Jones S. The microRNA registry. Nucleic Acids Res. 2004;32(Database issue):D109–D111.
- He B, Gao SQ, Huang LD, et al. MicroRNA-155 promotes the proliferation and invasion abilities of colon cancer cells by targeting quaking. Mol Med Rep. 2015;11(3):2355–2359.
- Liu Y, Pan Q, Zhao Y, et al. MicroRNA-155 regulates ROS production, NO generation, apoptosis and multiple functions of human brain microvessel endothelial cells under physiological and pathological conditions. J Cell Biochem. 2015;116(12):2870–2881.
- Bell D, Kelso EJ, Argent CC, et al. Temporal characteristics of cardiomyocyte hypertrophy in the spontaneously hypertensive rat. Cardiovasc Pathol. 2004;13(2):71–78.
- Graham D, McBride MW, Brain NJ, et al. Congenic/consomic models of hypertension. Methods Mol Med. 2005;108:3–15.
- Sun HJ, Zhao MX, Ren XS, et al. Salusin-beta promotes vascular smooth muscle cell migration and intimal hyperplasia after vascular injury via ROS/NFkappaB/MMP-9 pathway. Antioxid Redox Signal. 2016;24(18):1045–1057.
- Yuen CY, Wong SL, Lau CW, et al. From skeleton to cytoskeleton: osteocalcin transforms vascular fibroblasts to myofibroblasts via angiotensin II and Toll-like receptor 4. Circ Res. 2012;111(3):e55–e66.
- Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750.
- Van DJ, Hendrix A. Is your article EV-TRACKed? J Extracell Vesicles. 2017;6(1):1379835.
- Busato A, Bonafede R, Bontempi P, et al. Magnetic resonance imaging of ultrasmall superparamagnetic iron oxide-labeled exosomes from stem cells: a new method to obtain labeled exosomes. Int J Nanomedicine. 2016;11:2481–2490.
- Takahara K, Ii M, Inamoto T, et al. microRNA-145 mediates the inhibitory effect of adipose tissue-derived stromal cells on prostate cancer. Stem Cells Dev. 2016;25(17):1290–1298.
- Bonafede R, Scambi I, Peroni D, et al. Exosome derived from murine adipose-derived stromal cells: neuroprotective effect on in vitro model of amyotrophic lateral sclerosis. Exp Cell Res. 2016;340(1):150–158.
- Park SJ, Jeon H, Yoo SM, et al. The effect of storage temperature on the biological activity of extracellular vesicles for the complement system. In Vitro Cell Dev Biol Anim. 2018;54(6):423–429.
- Gao F, Jiao F, Xia C, et al. A novel strategy for facile serum exosome isolation based on specific interactions between phospholipid bilayers and TiO2. Chem Sci. 2019;10(6):1579–1588.
- Sobue A, Ito N, Nagai T, et al. Astroglial major histocompatibility complex class I following immune activation leads to behavioral and neuropathological changes. Glia. 2018;66(5):1034–1052.
- Xiao J, Pan Y, Li XH, et al. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis. 2016;7(6):e2277.
- Gallet R, Dawkins J, Valle J, et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J. 2017;38(3):201–211.
- Li W, Zhang X, Wang J, et al. TGFbeta1 in fibroblasts-derived exosomes promotes epithelial-mesenchymal transition of ovarian cancer cells. Oncotarget. 2017;8(56):96035–96047.
- Dioufa N, Clark AM, Ma B, et al. Bi-directional exosome-driven intercommunication between the hepatic niche and cancer cells. Mol Cancer. 2017;16(1):172.
- Sun HJ, Ren XS, Xiong XQ, et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017;8(10):e3074.
- Yu HR, Kuo HC, Huang HC, et al. Glyceraldehyde-3-phosphate dehydrogenase is a reliable internal control in Western blot analysis of leukocyte subpopulations from children. Anal Biochem 2011;413(1):24–29.
- Zang YH, Chen D, Zhou B, et al. FNDC5 inhibits foam cell formation and monocyte adhesion in vascular smooth muscle cells via suppressing NFkappaB-mediated NLRP3 upregulation. Vascul Pharmacol. 2019;121:106579.
- Fan ZD, Zhang L, Shi Z, et al. Artificial microRNA interference targeting AT1a receptors in paraventricular nucleus attenuates hypertension in rats. Gene Ther. 2012;19(8):810–817.
- LL Z, Ding L, Zhang F, et al. Salusin-beta in rostral ventrolateral medulla increases sympathetic outflow and blood pressure via superoxide anions in hypertensive rats. J Hypertens. 2014;32(5):1059–1067.
- Xu CC, Han WQ, Xiao B, et al. Differential expression of microRNAs in the aorta of spontaneously hypertensive rats. Sheng Li Xue Bao. 2008;60(4):553–560.
- Zheng L, Xu CC, Chen WD, et al. MicroRNA-155 regulates angiotensin II type 1 receptor expression and phenotypic differentiation in vascular adventitial fibroblasts. Biochem Biophys Res Commun. 2010;400(4):483–488.
- Bihl JC, Zhang C, Zhao Y, et al. Angiotensin-(1-7) counteracts the effects of Ang II on vascular smooth muscle cells, vascular remodeling and hemorrhagic stroke: role of the NFsmall ka, CyrillicB inflammatory pathway. Vascul Pharmacol. 2015;73:115–123.
- Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342(3):145–153.
- Messerli FH, Bangalore S. Angiotensin receptor blockers reduce cardiovascular events, including the risk of myocardial infarction. Circulation. 2017;135(22):2085–2087.
- Uehara Y, Numabe A, Kawabata Y, et al. Inhibition of protein synthesis and antiproliferative effect of the angiotensin converting enzyme inhibitor trandolaprilat in rat vascular smooth muscle cells. J Hypertens. 1993;11(10):1073–1081.
- van Vark LC, Bertrand M, Akkerhuis KM, et al. Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin-angiotensin-aldosterone system inhibitors involving 158,998 patients. Eur Heart J. 2012;33(16):2088–2097.
- Hicks BM, Filion KB, Yin H, et al. Angiotensin converting enzyme inhibitors and risk of lung cancer: population based cohort study. BMJ. 2018;363:k4209.