
ABSTRACT
Exosomes play a critical role in intercellular communication since they contain signalling molecules and genetic materials. During tumorigenesis, tumour-derived exosomes have been demonstrated to promote tumour angiogenesis and metastasis. However, how the exosomes facilitate tumour metastasis is not clear. Here we explored the effect of HeLa cell-derived exosomes (ExoHeLa) on endothelial tight junctions (TJ) and the related mechanisms. After human umbilical vein endothelial cells (HUVEC) were treated with ExoHeLa, TJ proteins zonula occludens-1 (ZO-1) and Claudin-5 in HUVEC were significantly reduced as compared with that treated with exosomes from human cervical epithelial cells, while mRNA levels of ZO-1 and Claudin-5 remained unchanged. Consequently, permeability of endothelial monolayer was increased after the treatment with ExoHeLa. Injection of ExoHeLa into mice also increased vascular permeability and tumour metastasis in vivo. Neither knocking down of Dicer nor use of inhibitors of microRNAs targeting at mRNAs of ZO-1 and Claudin-5 could block the inhibitory effect of ExoHeLa on ZO-1 and Claudin-5. The expression of genes involved in endoplasmic reticulum (ER) stress was significantly increased in HUVECs after treated with ExoHeLa. Inhibition of ER stress by knocking down protein kinase RNA-like endoplasmic reticulum kinase prevented the down-regulation of ZO-1 and Claudin-5 by ExoHeLa. Our study found that HeLa cell-derived exosomes promote metastasis by triggering ER stress in endothelial cells and break down endothelial integrity. Such effect of exosomes is microRNA-independent.
Introduction
Metastasis is the leading cause of mortality in cancer patients. According to the “seed and soil” hypothesis, migratory tumour cells leave the primary tumour by intravasation, disseminating throughout the body via the circulation, and finally engraft in a distant organ which provides an appropriate microenvironment. The last step requires extravasation: tumour cells move out of the circulatory system by penetrating through the endothelial layer. It is still not clear how tumour cells break down vascular barrier although the evidences show that tumour cells can interact with local microenvironment by cell–cell interactions and paracrine effects through the release of a variety of growth factors, chemokines and matrix-degrading enzymes that can enhance the invasion of tumour [Citation1]. Recently, an emerging mechanism by which tumour cells interact with the local microenvironment by releasing extracellular vesicles (EVs) such as exosomes has been proposed [Citation2–Citation4].
Exosomes are 40–150 nm membrane-encapsulated vesicles that are released into the extracellular environment by many cell types, including tumour cells [Citation5–Citation7]. It contains DNA, non-coding RNAs, mRNAs, as well as proteins [Citation8–Citation10]. Several roles of tumour cell-derived exosomes in modulating tumour microenvironment have been described, including biological behaviour “education” [Citation11], immune modulation [Citation12,Citation13], and proangiogenic effect [Citation14–Citation16].
So far, most of the research about the effect of tumour-derived exosomes on endothelial cells (ECs) is focused on the proangiogenic effect. For example, exosomes from glioblastoma cells stimulate EC proliferation [Citation5]; exosomes released by a pancreatic cell line transfected with D6.1A tetraspanin stimulate endothelial tubulogenesis [Citation17]. However, growing evidence shows that tumour cells also break down the vascular integrity by direct interaction with ECs [Citation18] or by means of exosomes [Citation14,Citation19,Citation20]. Disruption of vascular integrity could then facilitate cancer cell dissemination and growth at distant sites [Citation19].
Tight junctions (TJs) play important roles in vascular integrity and control the permeability of endothelial monolayer. Down-regulation or loss of TJs contributes to cancer progression by altering cell migration, proliferation, polarity and differentiation [Citation21,Citation22]. For example, reduction of TJ-associated zonula occludens 1 (ZO-1) in ECs by primary breast tumours is associated with metastasis in breast cancer patients [Citation23]. TJ protein Claudin-5 (CLDN5) not only regulates paracellular ionic selectivity but also plays a role in the regulation of tumour cell motility, suggesting that CLDN5 contributes to the control of brain metastasis [Citation24]. MicroRNAs have also been identified as one mechanism of TJ disruption [Citation14,Citation19]. However, the effects of cancer cell-derived exosomes on TJs are currently yet to be unveiled.
In the present study, we investigated the effect of exosomes derived from HeLa cell (ExoHeLa) on the expression of TJ-associated proteins in ECs and explored the possible mechanism. Our research reveals that ExoHeLa disrupts vascular integrity and facilitates tumour metastasis via triggering endoplasmic reticulum (ER) stress in ECs and reduction of TJ-associated proteins like ZO-1 and CLDN5.
Materials and methods
Cell culture
Normal human umbilical cords were obtained with written consent from healthy donors with the approval of the Human Subjects Ethics Committee of Second Affiliated Hospital of Zhejiang University. Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cords by enzymatic detachment using collagenase. Briefly, umbilical cords were washed with phosphate buffer saline (PBS) three times and then digested using 1 mg/mL collagenase I (Gibco, USA) for 30 min. The enzymatic detachment was neutralized with M199 containing 10% foetal bovine serum (FBS) (Gibco, USA), and detached cells were collected and washed with PBS. Cells were cultured in EC medium (ECM, ScienCell, USA) supplemented with 10% FBS and EC growth supplement (ECGS, ScienCell, USA). All experiments were conducted with HUVECs in passages 3–6.
The human cervical squamous cancer cell line HeLa was identified by short tandem repeat profiling with a certificate provided by Sangon Biotech (Shanghai) Co. Ltd (China). HeLa cells were cultured in PRMI 1640 culture medium (GIBCO, USA), and human cervical epithelial cell (HCEC), mouse mammary carcinoma 4T1 cell as well as human cervical squamous cancer cell line Siha transfected with green fluorescent protein (GFP) were cultured in high-glucose Dulbecco’s modified Eagle’s medium (GIBCO, USA). Culture mediums were supplemented with 10% FBS, 100 U/mL penicillin sodium and 100 μg/mL streptomycin sulphate (Hyclone, USA), and cells were cultured at 37°C in humidified air containing 5% CO2.
Exosome isolation and characterization
Cells were grown at sub-confluence in growth media containing exosome-depleted FBS (prepared by overnight ultracentrifugation at 110,000 ×g at 4ºC) for 48 hrs. Conditioned medium was then collected and centrifuged at 300 ×g for 10 min, 2,000 ×g for 10 min and 10,000 ×g for 30 min to remove cells and cell debris. The supernatant was then concentrated with 10-kDa molecular weight cut-off hollow fibre membrane (Millipore, Billerica, MA, USA) at 2,500 ×g for 10 min. The supernatant was further filtered by a 0.22-μM filter (Millipore). The concentrated supernatant was then ultra-centrifuged at 110,000 ×g for 70 min on top of a 30% sucrose cushion (Optima L-90K; Beckman Coulter, USA). Exosomes were then collected and washed 1 time with PBS by centrifugation at 110,000 ×g for 70 min. Finally, exosomes were resuspended by 200 μL PBS. This exosome preparation was used for protein concentration measurement, transmission electron microscopy (TEM) analysis (Fig S1A), nanoparticle tracking analysis (NTA, )) and most of the experiments in this study. Exosomes from both HeLa cells and HCEC exhibited typical cup-shaped morphology (Fig S1A). The diameters of ExoHela and ExoHECE in TEM pictures are 61 ± 21 nm and 71 ± 27 nm, respectively.
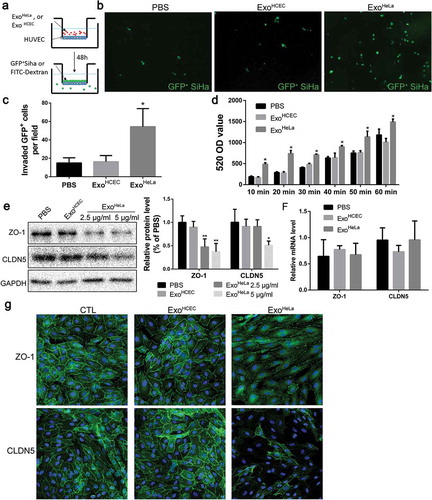
Figure 1. ExoHeLa down-regulated tight junctions and destroyed the barrier function of endothelial monolayers in vitro. (a) HUVECs were grown on the inserts of a dual well plate for 24 hrs to form endothelial monolayer. After treated with ExoHeLa or ExoHCEC for 24 hrs, GFP-labelled SiHa cells were loaded on the upper chamber and allowed for migration for 12 hrs. (b) GFP+ SiHa cells migrated through HUVEC monolayer were observed on the other side of the inserts under fluorescent microscopy. (c) The number of migrated cells in B were quantified (n = 6). (d) Similarly, after HUVECs monolayer on the inserts were treated with ExoHeLa or ExoHCEC for 24 hrs, FITC-Dextran (70kD) was added into the upper chamber. The fluorescence in the bottom wells was measured at different times. (e) Protein levels of ZO-1 and CLDN5 in HUVECs after treated with either 2.5 μg/mL or 5 μg/mL ExoHeLa were analysed by western blot. (f) mRNA levels of ZO-1 and CLDN5 in HUVEC were evaluated by real-time quantitative RT-PCR after the cells were treated with 5μg ExoHeLa or ExoHCEC or PBS for 48 hrs. (g) After treated with ExoHeLa or ExoHCEC for 48 hrs, HUVEC monolayers analysed by immunofluorescence for ZO-1 and CLDN5 (green). The nuclear were stained with Hoechst 33258 (blue). *P< 0.05, **P< 0.01.
The exosome preparation by ultra-centrifugation was further subjected to OptiPrep discontinuous gradient separation as described by Eichenberger et al. [Citation25] with minor modification. Briefly, OptiPrep (60% w/v) was diluted with 0.25 M sucrose/10 mM Tris, pH 7.4 to generate 40%, 35%, 30%, 25%, 20%, 10% and 5% w/v iodixanol solutions. Then, 1.5 mL of each concentration was sequentially (40% at bottom, 5% at top) layered with care to generate discontinuous iodixanol gradient, and the exosome suspension in 1 mL PBS was added to the top layer and ultracentrifuged at 110,000 ×g for 18 hrs at 4°C. All seven fractions (40%, 35%, 30%, 25%, 20%, 10% and 5% w/v iodixanol solutions) were collected separately and excess OptiPrep solution was removed by diluted to 12 mL in PBS and ultra-centrifuged at 110,000 ×g for 70 min. The resulting pellets were resuspended in 15μl PBS and subjected to western blot analysis for CD63 (Fig S1C). Fractions at 30%, 25%, 20% and 10% iodixanol were positive for CD63. Exosomes from these four fractions were collected and used for comparison with exosomes prepared by ultra-centrifugation for their effect on TJ proteins.
The protein content of the concentrated exosomes was determined using a bicinchoninic acid protein assay kit (Thermo, USA). Particle size of the purified exosomes was analysed using Zetasizer Nano series-Nano-ZS (Malvern, UK). Exosomes were diluted with PBS (1:50) and injected into the Zetasizer Nano instrument. Purified ExoHeLa showed a bell-shape distribution of diameter at 80–200 nm with a peak at 100 nm, and the diameter distribution of ExoHCEC ranges from 100–230 nm with a peak at 150 nm (Figure S1B). The concentrations of exosomal particle were determined using NTA with ZetaView instrument S/N 19–447 (Particle Metrix, Germany) which was operated by DKSH Shanghai Ltd (Shanghai, China). ExoHela and ExoHECE are in 6.4 × 106 particles/μg and 6.1 × 106 particles/μg, respectively.
The exosomal markers Alix, CD63 and CD9, as well as Golgi-associated protein GM130, were analysed by western blot (Figure S1D) for HeLa cells and HCECs and exosomes purified from them. After exosomes were labelled with fluorescent PKH26, and then mixed with HUVECs for 12 hrs, uptaking of exosomes by HUVECs was observed (Figure S1E).
Cell proliferation assay
After HUVECs were plated on collagen-coated 96-well plates (2 ×103 cells/well) for overnight, and PBS, or ExoHCEC or ExoHeLa (5 μg/mL) was added to each well and cultured for 48 hrs. Then, 10 μL of Cell Counting Kit-8 (CCK-8, Dojindo, Japan) was added and incubated for 2 hrs at 37°C, and the absorbance was determined at a wavelength of 450 nm. HUVEC proliferation was also measured using the 5-ethynyl-2ʹ-deoxyuridine (EdU) Cell Proliferation Assay Kit (Ribobio) according to the manufacturer’s instructions. EdU-labelled cells were counted manually in five fields of view randomly selected from each well, and percentages were calculated.
Endothelial permeability and transendothelial migration analysis
For endothelial permeability analysis, HUVECs (2 × 104) were seeded on 0.3 cm2 polyethylene terephthalate transwell filters in a 24-well plate (0.4 μm pore size; BD Biosciences; Franklin Lakes, NJ) and allowed to reach confluence, followed by addition of PBS, or ExoHCEC or ExoHeLa (5 μg/mL) and continued culture for 48 hrs. FITC-dextran (10 mg/mL, average MW ~70,000; Sigma, USA) was added to the top well to reach final concentration at 10 mg/mL. The appearance of fluorescence in the bottom well representing the passage of FITC-dextran was monitored by taking 40 μL medium aliquots in a time course to measure fluorescence using a SpectraMax microplate reader (SpectraMax M5, Molecular Devices, USA) at 488 nm excitation and 520 nm emission.
For transendothelial migration assay, HUVECs were grown to form a monolayer in the upper well of transwells in a 24-well plate (8-μm pore size; BD Biosciences, USA), and PBS or ExoHeLa or ExoHCEC (100 μL, final 10 μg/mL) was added to the culture. After incubation for 24 hrs, Siha transfected with GFP (1 ×104) were added, and the transmigrated Siha cells in the bottom wells were counted under fluorescent microscope after other 12-hr culture. Each experiment was repeated three times, and each time three wells were tested for a sample.
Immunocytochemistry
Cells were washed with PBS containing 0.5% bovine serum albumin (BSA) and fixed with 4% paraformaldehyde for 15 min. The cells were then permeabilized with 0.5% Triton X-100 for 10 min, blocked with 5% BSA in PBS for 30 min at room temperature, and then incubated with primary antibody overnight at 4°C, followed by incubation with secondary antibodies for 1 hr at 37°C. Nuclei were stained with Hoechst 33258 for 5 min. Cells were then washed three times and viewed using a fluorescence microscope (Leica, Germany).
Real-time PCR analysis
Total RNA was extracted from HUVECs with TRIzol reagent (Invitrogen, USA). First-strand cDNA was synthesized from the total RNA (500 ng) using the TaKaRa 5X PrimeScript RT Master Mix Kit (TaKaRa, Japan) according to the manufacturer’s instruction. Subsequently, the quantitative real-time polymerase chain reaction (qRT-PCR) was performed using the TB Green Premix Ex Taq II Kit (TaKaRa, Japan) according to the manufacturer’s protocol. Each sample was performed in triplicated and all results were normalized to the expression of glyceraldehydes 3-phosphate dehydrogenase (GAPDH). Fold expression relative to the reference GAPDH gene was calculated using the comparative method 2−ΔCt. The primer sequences are listed in Supplemental Table S1.
Western blot analysis
Cell lysate was prepared using RIPA lysis buffer (Beyotime, China). The samples were separated by SDS-polyacrylamide gel (SDS-PAGE), transferred to a polyvinylidene fluoride membrane, and immunoblotted with the following antibodies: activating transcription factor-4 (ATF4; Abcam, ab184909), protein kinase RNA-like endoplasmic reticulum kinase (PERK; Cell Signalling Technology, 5683), Dicer (Cell Signalling Technology, 5362), mammalian target of rapamycin (mTOR, Cell Signalling Technology, 2972), sequestosome 1 (P62, Cell Signalling Technology, 88588), microtubule-associated protein 1 light chain 3B (LC3B, Cell Signalling Technology, 3868), CCAAT/enhancer-binding protein homologous protein (CHOP, Cell Signalling Technology, 2895), vascular endothelial growth factor receptor 2 (VEGFR2, Cell Signalling Technology, 9698), eukaryotic translation initiation factor 2 alpha (eIF2α; Abcam, ab169528), CD9 (Abcam, ab92726), phos-eIF2α (Abcam, ab32157), E2F1 (Abcam, ab179445), Alix (Abcam, ab117600), 78 kD glucose-regulated protein (GRP78; HuaBio, ER40402), Claudin-5 (CLDN5; Abcam, ab15106), ZO-1 (Proteintech, 21773-1-AP), matrix metalloproteinase 2 (MMP2, Huabio, ER40806), MMP9 (Huabio, ET1704-69), MMP14 (Huabio, ET1606-48), MMP15 (Huabio, ER1913-06) and GAPDH (Huabio, ET1702-66). After incubation of the membranes with peroxidase-conjugated secondary antibodies (Cell Signalling Technology), bands were visualized using enhanced chemiluminescence reagents (Bio-Rad).
Electron microscopy
For TEM analysis, the cells were collected and immediately fixed overnight in 3% glutaraldehyde. Then, the samples were rinsed three times with PBS and postfixed with 1% osmic acid for 2 hrs. After being rinsed three times with deionized water and serially dehydrated with 50%, 70%, 80%, 90% and 100% alcohol and 100% acetone, the samples were embedded in epoxy resin to form blocks of cells. Ultrathin sections (50 nm) were obtained by an ultramicrotome (Ultracut UCT, Leica, Germany). The sections were then stained with lead citrate and uranyl acetate and examined by TEM (T10, FEI, USA).
For exosome TEM analysis, exosomes were fixed with 4% paraformaldehyde, a drop of exosomes (20 mL) was pipetted onto a grid which was coated with formvar and carbon, standing for 5 min at room temperature. The excess fluid was removed with a piece of filter, and the sample was negatively stained with 3% (wt/vol) phosphotungstic acid (pH 6.8) for 5 min. After air-drying under an electric incandescent lamp, the sample was analysed by TEM (T10, FEI, USA).
RNA sequence
RNA was extracted from ExoHeLa and ExoHCEC, or HUVECs treated with or without ExoHeLa. For miRNA library construction and RNA sequencing, the complementary DNA (cDNA) libraries for single-end sequencing were prepared using Ion Total RNA-Seq Kit v2.0 (Life Technologies) according to the manufacturer’s instructions. The cDNA library had been size selected by PAGE Gel electrophoresis for miRNA sequencing. The cDNA libraries were then processed for the Proton Sequencing process according to the commercially available protocols. Samples were diluted and mixed, and the mixture was processed on a OneTouch 2 instrument (Life Technologies) and enriched on a OneTouch 2 ES station (Life Technologies) for preparing the template-positive Ion PI™ Ion Sphere™ Particles (Life Technologies) according to Ion PI™ Template OT2 200 Kit v2.0 (Life Technologies). After enrichment, the mixed template-positive Ion PI™ Ion Sphere™ Particles of samples were loaded on to 1 P1v2 Proton Chip (Life Technologies) and sequenced on Proton Sequencers according to Ion PI Sequencing 200 Kit v2.0 (Life Technologies). The results were presented as gene ontology (GO) analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis and miRNA-Gene-Network analysis. The Miranda, RNAhybrid and TargetScan were utilized as the tools for predicting differentially expressed miRNA target on mRNA.
The cDNA libraries were constructed for each pooled RNA sample using the VAHTSTM Total RNA-seq (Vazyme Biotech, Nanjing, China) according to the manufacturer’s instructions. Generally, the protocol consists of the following steps: depletion of rRNA and fragmented into 150–200 bp using divalent cations at 94°C for 8 min. The cleaved RNA fragments were reverse-transcribed into first-strand cDNA, then second-strand cDNA was synthesized, and fragments were end repaired, A-tailed and ligated with indexed adapters. Target bands were harvested through VAHTSTM DNA Clean Beads. The products were purified and enriched by PCR to create the final cDNA libraries and quantified by Agilent2200. The tagged cDNA libraries were pooled in equal ratio and used for 150 bp paired-end sequencing in a single lane of the Illumina Xten with 51 plus 7 cycles by NovelBio Corp. Laboratory (Shanghai, China). All mRNA sequencing data were uploaded to NCBI GEO repository with ID GSE128802 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc =GSE128802) and GSE128803 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc =GSE128803).
siRNA transfection
PERK small interfering (si) RNA (si-PERK) or DICER siRNA (si-DICER) and scrambled siRNA controls were synthesized by Ribobio (Guangzhou, China). Cells were seeded at 1 ×105 per well into 12-well plates 1 day before transfection. Lipofectamine 2000 (Invitrogen, USA) transfection reagent was employed to transfect cells with si-PERK or siDICER, or siRNA controls (all in 50 nM). Lipofectamine 2000/siRNA complexes were prepared in reduced serum medium, OptiMEM (Invitrogen, USA), at the recommended ratio of 1 μL of Lipofectamine 2000 per 20 pmol siRNA (1 μL). After adding the Lipofectamine 2000/siRNA complexes to the cells and cultured for 8 hrs, the cell growth medium was removed and the cells were incubated in the fresh medium containing 10% FBS for further treatment.
Liquid chromatograph–mass spectrometer/mass spectrometry analysis
The liquid chromatograph–mass spectrometer/mass spectrometry (LC-MS/MS) analysis of ExoHeLa, ExoShiha and triton-treated ExoHeLa was carried out in Capitbalbio Technology by using Q Exactive mass spectrometer (Thermo Scientific, USA). The peptide mixture was separated by reversed-phase chromatography on a DIONEX nano-UPLC system using an Acclaim C18 PepMap100 nano-Trap column (75 μm × 2 cm, 2 μm particle size) (Thermo Scientific, USA) connected to an Acclaim PepMap RSLC C18 analytical column (75 μm × 25 cm, 2 μm particle size) (Thermo Scientific, USA). Before loading, the sample was dissolved in sample buffer, containing 4% acetonitrile and 0.1% formic acid. A linear gradient of mobile phase B (0.1% formic acid in 99.9% acetonitrile) from 3% to 30% in 43 min followed by a steep increase to 80% mobile phase B in 1 min was used at a flow rate of 300 nL/min. The nano-LC was coupled online with the Q Exactive mass spectrometer using a stainless steel Emitter coupled to a nanospray ion source. MS analysis was performed in a data-dependent manner with full scans (350–1,600 m/z) acquired using an Orbitrap mass analyser at a mass resolution of 70,000 at 400 m/z in Q Exactive. Twenty most intense precursor ions from a survey scan were selected for MS/MS from each duty cycle and detected at a mass resolution of 35,000 at m/z of 400 in Orbitrap analyser. All the tandem mass spectra were produced by higher-energy collision dissociation (HCD) method. Dynamic exclusion was set for 18 s. All LC-MS/MS data were listed on Supplemental Online data.
Animal model and exosome delivery
The animal protocol was approved by Zhejiang University according to Chinese guidelines for laboratory animal care and use. Female 6-week-old BALB/c-nude mice were used for in vivo vascular permeability assay or neoplasms transplantation experiments. ExoHCEC, ExoHeLa or PBS (as control) were intravenously injected into the tail vein of mice (10 μg exosomes per injection; two injections per week).
In vivo vascular permeability assay
After 8 injections of exosomes, 100 mg/kg FITC-dextran (average MW ~70,000) was intravenously injected. One hour later, ear vessel bloodstream was examined. Mice were positioned on the stage of a laser confocal scanning microscope and their ear lobes were fixed beneath coverslips with a single drop of immersion oil. FITC-dextran in the bloodstream and its leakage out of vasculature were visualized. The mice were then sacrificed, and transcardiac perfusion with PBS was carried out to remove the excess dye. Lung and liver tissues were embedded in Tissue-Tek O.C.T. Compound (Sakura; Torrance, CA) to make frozen blocks for sectioning and immunofluorescent staining.
Tumour metastasis experiment
Luciferase-labelled 4T1 (2 × 105 cells) were injected into the No. 4 mammary fat pad of 6-week-old female BALB/c-nu mice. Immediately after implantation, mice were injected with 100 μL ExoHCEC, ExoHeLa (10 μg total per injection, two injections per week) or PBS via tail vein. When tumours became palpable, tumour volume was assessed by calliper measurements using the formula (width2 × length)/2 (mm3). Bioluminescence imaging was carried out using a Xenogen system (IVIS spectrum, Perkin Elmer, USA) on D0, D14 and D21. After six injections, mice were sacrificed. Lungs were paraformaldehyde-fixed and paraffin-embedded for haematoxylin and eosin (H&E) staining.
Statistical analysis
All data are presented as the mean ± standard deviation (SD). Statistical analyses were performed with Student’s t-test for comparisons between two groups and with ANOVA followed by Dunnett’s correction for more than two groups using SPSS version 17.0. P<0.05 was considered statistically significant.
Results
ExoHeLa deteriorated the barrier function of endothelial monolayer by disrupting EC TJs
To access the effect of ExoHeLa on the permeability of endothelial monolayer, transendothelial invasion of cancer cells (Siha/GFP) was examined (,)). There were significantly more GFP-labelled Siha cells migrated through ExoHeLa-treated HUVEC monolayer as compared with HUVEC treated with PBS or exosomes from normal HCECs (ExoHCEC) (,)). The changes in the permeability of endothelial monolayer after the exosome treatment were also detected by measuring the traversing of FITC-labelled dextran (70 KD) through HUVEC monolayers. Treatment of HUVEC with ExoHeLa significantly increased the pass of fluorescent probe through the EC layer as compared with those treated with ExoHCEC or PBS ()).
Since the permeability of the EC layer is associated with its TJ, TJ proteins in ECs were examined. TJ-associated ZO-1 protein level was down-regulated to 50% when treated with 2.5 μg/mL ExoHeLa for 48 hrs, while CLDN5 level remained unchanged. With higher ExoHeLa concentration (5 μg/mL), CLDN5 was also reduced by approximately 50% ()). However, the mRNA levels of ZO-1 and CLDN5 in ECs remained unchanged after exosome treatments ()). Immunofluorescence staining also revealed that there was a marked reduction in both ZO-1 and CLDN5 in HUVEC monolayers after they were treated with ExoHeLa, but not with PBS or ExoHCEC ()).
ExoHeLa increased vascular permeability in vivo
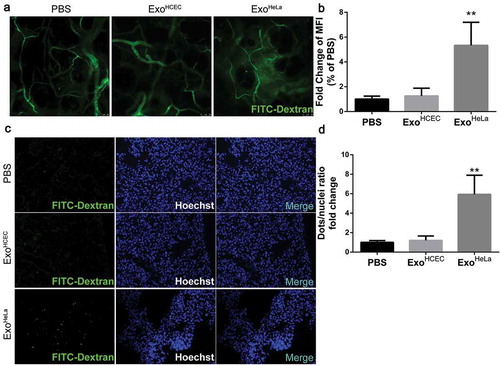
To further demonstrate the effect of ExoHeLa on endothelial barriers in vivo, mice were injected with ExoHeLa or ExoHCEC through tail veins followed with the injection of FITC-Dextran (Figure S2). Severe leakage of FITC-Dextran out of blood vessels in ear was detected in ExoHeLa-, but not in ExoHCEC- or PBS-treated mice (,)). Similar results were also observed in lung and liver as more fluorescence was detected in the sections of lung (,)) and liver (Figure S3A, S3B). Significantly less ZO-1 (Figure S3C) and CLDN5 (Figure S3D) were detected in pulmonary CD31 positive ECs. These results indicated that ExoHeLa treatment resulted in down-regulation of TJ proteins in ECs, which increased vascular permeability in different organs.
Figure 2. ExoHeLa increased vascular permeability in vivo. ExoHeLa or ExoHCEC or PBS were intravenously injected into the tail veins of Balb/c nude mice (n = 4 for PBS group, n = 3 for ExoHCEC group and n = 6 for ExoHeLa group) three times a week for ten times. (a) FITC-dextran (70kD) was intravenously injected and the leakage of FITC-dextran out of the ear vessel was examined alive under confocal microscope. (b) Fluorescent leaking out of vessels was quantified using image J as Mean Fluorescence Intensity (MFI). (c) Nude mice were sacrificed afterwards, and the appearance of injected FITC-dextran was examined in lung. (d) The number of FITC-dextran dots was quantified and data were presented as FITC-dextran dots/nuclei ratio. **P< 0.01.
ExoHeLa promoted tumour metastasis in vivo
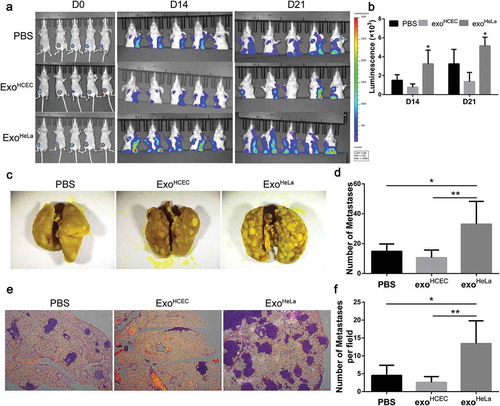
To determine whether ExoHeLa treatment can promote tumour metastasis, mouse mammary carcinoma 4T1 cells were transduced with luciferase gene and implanted into mice, followed with a consecutive daily injection of exosomes for 3 weeks as described in Materials and Methods section. Significantly more distant metastases were detected in ExoHeLa-treated mice by luminescence assay (,)), while the sizes of primary tumours were not significantly different among the three groups (Supplemental Figure S4). More than two-fold increased metastasis to lung was detected in the ExoHeLa-treated mice as compared to the ExoHCEC-treated mice (–)).
Figure 3. ExoHeLa promoted tumour metastasis in vivo. Luciferase-labelled 4T1 cells were injected into # 4 mammary fat pads of the nude mice. The mice simultaneously were injected with PBS, ExoHCEC, or ExoHeLa as indicated through tail vein (n = 5 for each group). (a) Bioluminescent imaging (BLI) at Day 0, 14 and 21 was examined. (b) Luminescence at Day 14 and 21 in A was quantified. (c) Mice were sacrificed on Day 21. Lungs were harvested and metastases were visualized after fixed with 4% paraformaldehyde containing 10% picric acid. White spots are the metastasized tumours. (d) Quantification of metastasis spots in C. (e). Representative images of lung sections showing tumour metastases in lungs after they were stained with haematoxylin and eosin. Metastasis tumours were stained into purple. (f) Quantification of metastasis spots in E. *P< 0.05, **P< 0.01.
Exosomal mircoRNA did not cause the down-regulation of TJs
To explore the mechanism of ExoHeLa-induced deterioration of endothelial integrity, exosomes were first treated with Triton X-100 to breakdown the integrity of exosomal membrane before they were added to HUVEC culture (Figure S5). The inhibitory effects of ExoHeLa on EC proliferation (Figure S5A, B, C) and TJ proteins along with other proteins (VEGFR2, E2F1) were reversed after Triton treatment (), Figure S5D), suggesting that components inside exosomes were responsible for the inhibitory effect.
Figure 4. Exosomal microRNAs were not responsible for the ExoHeLa-induced down-regulation of TJ proteins. ExoHeLa was treated with or without triton X100 before being co-incubated with HUVECs for 48 hrs. (a) Protein levels of ZO-1 and CLDN5 of HUVECs were evaluated by western blot. (b) RNA was extracted from exoHeLa and exoHCEC, small RNAs were analysed, and the most abundant small RNAs were listed. (c) Based on the small RNA sequencing results, HUVECs were treated with corresponding microRNA inhibitors along with exoHeLa, then analysed by western blot for protein level of ZO-1 and CLDN5. Quantification of ZO-1 and CLDN5 expression were presented as the percentage of that of PBS group, and the data are shown in a bar graph. (d) Quantification of DICER mRNA in HeLa cells using real-time quantitative PCR after the cells were treated with small interference RNA to knocked down DICER mRNA, (e) Protein level of DICER was examined by western blot. (f) Exosomes secreted by HeLa cells and DICER knocked down (DICER KD) HeLa cells were collected, exosomal RNA was extracted from exoHeLa and exoHeLa DICER KD. MiR1290 and miR3960 levels were analysed by Real-time quantitative PCR. (g) After treated with exoHeLa and exoHeLa DICER KD for indicated duration, HUVECs were subjected to western blot analysis for ZO-1 and CLDN5 proteins. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001.
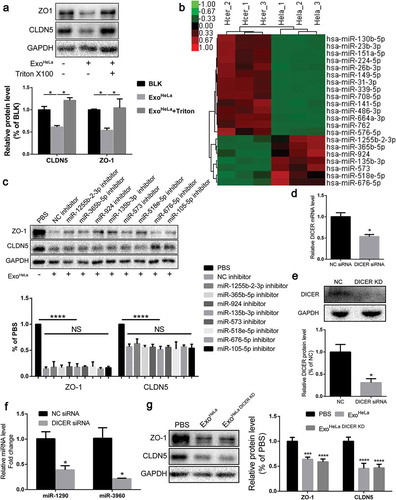
Since microRNAs (miRNAs) from exosomes play important roles in exosome-mediated effects, all miRNAs in the exosomes were sequenced and profiled. ExoHeLa and ExoHCEC exhibited different miRNA compositions (Figure S6, S7, S8). The miRNAs having regulatory potential on ZO-1 or CLDN5 and being more than two-fold difference between ExoHeLa and ExoHCEC are listed in ), and the miRNA-Gene-Network is presented in Figure S6. The inhibitors for these miRNAs along with the ExoHeLa were used to treat HUVEC. Strikingly, preconditioning of HUVECs with the miRNA inhibitors could not reverse the down-regulation of ZO-1 or CLDN5 in HUVECs by ExoHeLa ()).
To further confirm the possibility that miRNAs were not involved in the inhibition of TJ proteins in HUVECs, DICER mRNA was knocked down by DICER siRNA ()). As a consequence, DICER protein was down-regulated by ~70% in HeLa cells ()). The amount of exosomal miRNAs, represented by two highly expressed miRNAs (miR-1290 and miR-3960) in exosomes, was reduced by 60% and 75%, respectively, after the DICER knock-down ()). However, DICER knock-down did not attenuate the down-regulating effect of HeLa exosomes on TJ proteins in HUVECs ()), indicating that miRNAs may not be involved in the inhibitory effect of HeLa exosomes on TJ proteins.
Inhibition of either ubiquitination or autophagy cannot reverse the protein level of ZO-1 and CLDN5
Degradation of cellular proteins could be through lysosome or proteasome pathways. It has been shown that ubiquitination can trigger the proteasome-dependent degradation of CLDN5 [Citation26]. To examine whether ExoHeLa-induced down-regulation of TJ proteins is through the enhanced ubiquitination pathways, proteasome inhibitor MG132 was added to the HUVEC culture along with ExoHeLa. As shown in ), MG132 did not reverse the inhibitory effect of ExoHeLa on the expressions of CLDN5 and ZO-1, indicating that ubiquitination pathway may not be involved in the down-regulation of TJ proteins.
Figure 5. Reduction of TJ proteins in ExoHeLa treated HUVECs was not through ubiquitination or autophagy. (a) HUVECs were preconditioned with PBS or MG132 followed by treatment with ExoHeLa as indicated. Protein levels of ZO-1 and CLDN5 were analysed by western blot and quantified. (b) After treated with either PBS or ExoHeLa, HUVECs were visualized under transmission electron microscopy. Autophagosomes were indicated using black arrows. (c) HUVECs were treated with ExoHeLa for specified times, and the protein level of mTOR, P62 and LC3B I/II was analysed by western blot. (d) HUVECs were preconditioned with PBS or CQ followed by treatment with ExoHeLa. ZO-1 and P62 proteins were evaluated by western blot. Quantification of the proteins was shown in the bar graph. (e) Similarly, after HUVECs were treated with 3MA along with ExoHeLa, ZO-1 and CLDN5 proteins were evaluated by western blot. Quantification of the proteins was shown in the bar graph. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001.
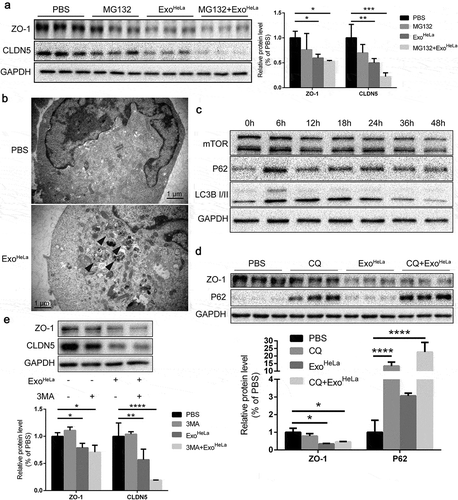
Next, we evaluated whether autophagy was induced by ExoHeLa. Electron microscopy unveiled the formation of autophagosomes in HUVECs after treated with ExoHeLa ()). Autophagosome marker LC3-II was significantly increased and reached a plateau at 12 h after HUVECs were treated with ExoHeLa ()). Autophagy receptor P62 in HUVEC was also increased after the ExoHeLa treatment. These data demonstrated that ExoHeLa could induce autophagy in ECs. Then, we pretreated HUVECs with 3-Methyladenine (3-MA) or chloroquine (CQ), both of which can interrupt autophagy flux, before treating the cells with ExoHeLa. Inhibition of the autophagy flux by 3-MA or CQ could not reverse the down-regulation of ZO-1 or CLDN5 expression by ExoHeLa (,)), indicating that the reduction of TJ proteins was not via ExoHeLa induced-autophagy.
ExoHeLa induced ER stress in HUVECs
To determine the effects of ExoHeLa on ECs, alteration of gene expression in HUVECs was analysed by comparing mRNA sequences in HUVECs before and after ExoHeLa treatment. GO analysis as well as pathway analysis identified major changes in cellular functions such as cell survival, migration and adhesion, proliferation, RNA expression and inflammation (Figure S9, S10, S11), among which the intrinsic apoptotic signalling pathway in response to ER stress showed a relatively significant change in rich factor (Figure S10). Based on these findings, we analysed the mRNAs of ER stress-related genes in HUVECs, including eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3), activating transcription factor-4 (ATF4), endoplasmic reticulum to nucleus signalling 1 (ERN1), endoplasmic reticulum oxidoreductase 1 alpha (ERO1A), heat shock protein family A member 5 (HSPA5), inositol 1,4,5-trisphosphate receptor 1 (ITPR1), protein phosphatase 1 regulatory subunit 15A (PPP1R15A) and DNA-damage inducible transcript 3 (DDIT3). All of them were significantly elevated in the ExoHeLa-treated HUVECs as compared with the ExoHCEC-treated ()). Besides, the phosphorylation of protein kinase RNA-like ER kinase (PERK) and eukaryotic translation initiation factor 2 alpha (eIF2α), as well as the protein level of ATF4, 78 kD glucose-regulated protein (GRP78) and CCAAT/enhancer-binding protein homologous protein (CHOP) were dramatically elevated in a time-dependent manner after ExoHeLa-treatment ()). These data indicate that ER stress was induced after HUVECs were treated with ExoHeLa.
Figure 6. ExoHeLa triggered ER stress in HUVECs. (a) The mRNAs of ER stress-related genes in HUVECs were analysed by Real-time quantitative PCR. The relative mRNA levels were presented in comparing with those in PBS-treated HUVECs. (b) The expressions of ER stress-related proteins in ExoHeLa treated HUVECs were analysed by western blot. (c) ExoHeLa was labelled with fluorescent dye PKH26 (red) and then mixed with HUVECs for the specified times. After HUVECS were fixed in 4% paraformaldehyde and subjected to immunofluorescence staining with Abs against either Calreticulin for ER (green) or GM130 for Golgi (green) or LAMP1 for lysosome (green). Cell nuclear was labelled with Hoechst 33258 (blue). Cellular distribution of the uptaken ExoHeLa in HUVECs was observed under laser confocal microscope. (d) PERK gene in HUVECs was knocked down by small interference RNA and confirmed by Real-time quantitative PCR. (e) Morphological change between normal HUVECs and PERK KD HUVECs treated with exoHeLa. Magnification: origin×40, inserter×100; (f) Cell proliferation measured by CCK8 and (g) ZO-1 and CLDN5 proteins by western blot were analysed for HUVECs after treated with the specified siRNA and ExoHeLa. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001.
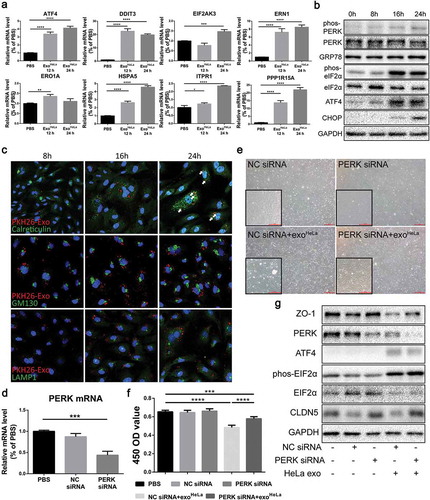
To explore the possible destination of exosomes uptook by HUVECs, fluorescent-labelled ExoHeLa were colocalized with ER, Golgi and lysosome with antibodies against Calreticulin, GM130 and LAMP1, respectively. ExoHeLa were colocalized with ER after mixed with HUVECs for 16 hrs ()). Interestingly, puncta of Calreticulin and LAMP1, but not GM130, increased with the progression of exosomes treatment, indicating the alternation of homoeostasis within ER and lysosomes.
ER stress increases the kinase activity of PERK to promote eIF2α phosphorylation, which resulted in the down-regulation of protein synthesis and cell growth. We further used RNA interference to knock down PERK in HUVEC ()) and reassessed the effect of ExoHeLa on PERK-down-regulated HUVECs. The proliferation of HUVECs was improved (,)), and the protein expression of ZO-1 and CLDN5 was also restored ()), indicating that ER stress was responsible for the down-regulation of TJ proteins in HUVECs.
In addition, the protein components of ExoHeLa and triton-treated ExoHeLa were compared using LC-MS/MS analysis (See Online Supplemental data). Unique peptides that can only be detected in ExoHeLa at least two times were listed in Table S2, and grouped according to their functions (Figure S12), which include 60S ribosomal protein L28 isoform 5 (RPL28), syntaxin-7 isoform b (STX-7), charged multivesicular body protein 4b (CHMP4B). Since numerous MMPs have been reported to be presented in different cancer cell lines [Citation27], MMPs were examined by both LC-MS/MS analysis and western blot. Although MMPs were detected in HeLa and Shiha cells, no MMPs were detectable in ExoHeLa (Figure S13, Table S2, Online Supplemental Excel data). MMP14 and MMP15, but not MMP2 and MMP9, were detected in ExoSiha.
Discussion
Penetration of dissociated cancer cells through the vascular endothelium is a key step for cancer metastases [Citation28]. The permeability of endothelial and epithelial cells is mainly governed by TJs [Citation29,Citation30], which are therefore the first barrier for cancer cells to overcome in order to metastasize [Citation31]. Our study shows that ExoHeLa could suppress the expression of several TJ proteins in ECs, including ZO-1 and CLDN5 both in vitro and in vivo. Along with the down-regulation of TJ proteins, increased endothelium permeability was observed. We employed an intravital evaluation system [Citation32] to determine the dynamic kinetics of vascular permeability in vivo. Our results demonstrated that i.v. injection of ExoHeLa resulted in the reduction of ZO-1 and CLDN5 expression in vessels and increased vascular permeability in vital organs such as liver and lung. Tumour metastasis was also significantly increased by ExoHeLa treatment. We further found that the ExoHeLa-induced inhibitory effect on ZO-1 and CLDN5 in ECs is not associated with exosomal microRNA, but rather with the ER stress induced by ExoHeLa. Our results suggested that exosomes secreted from tumour cell can trigger ER stress in ECs and break down endothelial integrity to allow tumour cells penetrate through vascular wall to achieve tumour metastasis.
An increasing number of studies have found that tumour-derived exosomes play an important role in the occurrence and development of cancer [Citation33,Citation34]. In our study, the inhibitory effect of ExoHeLa on TJ proteins was totally abrogated by triton X-100 treatment, indicating that the components inside exosomes, instead of exosome membrane or other ingredients co-deposited with exosomes during ultracentrifugation, caused the down-regulation of TJs. It is possible that such effect could be caused by amorphous materials aggregated on the exosomes that were prepared by ultra-centrifugation. To exclude the possibility, exosomes were purified further through density gradient ultracentrifugation (Fig S1C). Such prepared exosomes were used to treat HUVEC, and the expression of ZO-1 and CLDN5 in HUVECs was also inhibited (Fig S14), confirming such effect of ExoHeLa on down-regulation of TJ proteins is indeed caused by ExoHeLa. MMPs could also be a candidate responsible for the effect of ExoHeLa on TJ proteins in ECs since various MMPs were presented in different cancer cell lines [Citation27]. However, this possibility was dimmed as we did not detect MMPs in ExoHeLa (Fig S13, Table S2, Online Supplemental Excel data).
Growing evidence indicated that exosomal components, especially microRNAs, can suppress the expression of TJ proteins in ECs [Citation14,Citation19]. Our exosomal microRNA sequencing data show that there are several exosomal miRNA(s) in ExoHeLa that may directly or indirectly modulate the expression of TJ proteins. However, we found that the corresponding miRNA inhibitors cannot neutralize the down-regulation effect of ExoHeLa on TJ proteins ()), indicating that exosomal miRNAs may not be responsible for the decrease of TJ proteins. This was further approved by our Dicer knock-down experiment. It has been approved that RNase III protein, Dicer, is required for the process of microRNA [Citation35]. When we knocked down Dicer in HeLa cells by siRNA, the miRNAs in exosomes were dramatically reduced ()). However, when these exosomes (ExoHeLa Dicer KD) were used to treat ECs, the TJ proteins in ECs were still decreased ()). All these results suggested that exosomal miRNAs are not responsible for the ExoHeLa-induced reduction of TJ proteins. Indeed, this finding is discordant with those previous results. It possibly due to the global protein synthesize arrest caused by eIF2α phosphorylation, which may abrogate the microRNA regulation effects on TJ protein expression.
Protein degradation via lysosome and ubiquitin/proteasome system are two routine pathways for cellular protein degradation. Interestingly, autophagy was observed in ECs after treated with ExoHeLa. Several studied have demonstrated that TJ proteins can be degraded by autophagy/lysosome and ubiquitin/proteasome [Citation36–Citation39]. For example, Wang et al. showed that particulate matter (PM) exposure could induce ZO-1 relocation from the cell periphery into lysosome, accompanied by significant reductions in ZO-1 protein levels, which required calpain activation [Citation40]. Human immunodeficiency virus (HIV)-1 protein gp120 is also implicated as a cause of the breakdown of TJs between ECs due to increased degradation of ZO-1 and ZO-2 by proteasome [Citation41]. In our study, we tried to clarify whether ExoHeLa down-regulates the TJs due to the enhanced protein degradation via autophagy/lysosome and ubiquitination/proteasome pathway. It has been reported that MG-132, a proteasome inhibitor effective for the lysosomal proteases [Citation42], can inhibit the ubiquitination of CLDN5 [Citation26]. However, our results indicated that inhibition of proteasome by MG132 cannot stop the reduction of TJ proteins in ExoHeLa-treated HUVECs. Furthermore, neither PI3K-type III inhibitor 3-MA, which inhibits the initiation of autophagy [Citation43] nor CQ, an autophagy inhibitor via inhibition of the acidification of lysosomes [Citation44], could restore the TJ proteins. These findings imply that neither autophagy/lysosome nor ubiquitination/proteasome pathway is responsible for the down-regulation of TJs in ExoHeLa-treated ECs.
Through the mRNA expression analysis of ExoHeLa-treated HUVECs, we found a significant upregulation in “intrinsic apoptotic signalling pathway in response to ER stress” pathway in the ExoHeLa-treated ECs relative to the control cells (Figure S10). ER plays essential roles in the physiologic regulation of many cellular processes. Alterations in ER homoeostasis caused by the accumulation of misfolded proteins or alteration in the calcium or redox balance of the ER lead to a condition called ER stress [Citation45]. Recent studies have also provided evidence that ER stress is capable of inducing autophagy in mammalian cells [Citation46]. We confirmed that ER stress in ECs is provoked by ExoHeLa as the ER stress-related genes and proteins were dramatically elevated in the ExoHeLa-treated ECs ()). ER stress increases the kinase activity of PERK to promote eIF2α phosphorylation, which inhibited massive protein synthesis and cell growth [Citation47,Citation48]. This is also in accordance with our findings that multiprotein synthesis arrest was observed (Figure S5C). Additionally, knocking down of PERK by RNA interference restored the protein level of ZO-1 and CLDN5, indicating the activation of PERK/eIF2α/ATF4 pathway causes TJ down-regulation in ECs.
Recently, Pommier et al. reported a paradoxical phenomenon that disseminated cancer cells (DCCs) from pancreatic ductal adenocarcinoma exhibit an unresolved ER stress response, which enables DCCs to escape immunity and establish latent metastases [Citation49]. In addition, Wu et al. [Citation50] recently reported that bladder cancer EVs could induce unfolded protein response in human SV-HUC urothelial cells, and result in the neoplastic transformation. Inhibition of EV uptake prevented this transformation. It is perceivable that the rapid growth of cancer cells requires supper fast synthesis of proteins, which could result in many misfolded proteins, and consequently lead to ER stress. In response, certain proteins are down-regulated in cancer cells, which could decrease the antigenicity of cancer cells and impair their immunogenicity. Therefore, ER stress could be a cancer cell-intrinsic mechanism to achieve an immune-privileged status [Citation51].
In conclusion, our finding that ExoHeLa trigger ER stress in HUVECs, together with the findings by Pommier et al. [Citation49] and Wu et al. [Citation50], may provide a novel conception that exosomal component from cancer cells, such as HeLa cell, could trigger ER stress in the recipient cells. We postulate that unfolded proteins in cancer cells are packaged into exosomes and delivered to normal cells and then cause ER stress in the recipient cells. Although the components in exosomes that triggers ER stress in ECs are still unknown, some unique peptides that can be detected only in ExoHeLa (Table S2), including RPL28, STX-7, CHMP4B, etc., could be potential targets. This study may provide a new mechanism of tumour metastasis and may become a potential new therapeutic target. Further study is required to identify the details of the exosomal components that trigger ER stress.
Limitation in this study
The present study has a number of limitations. First, our investigation focused on HeLa cell only, and further investigations are required to verify whether this phenomenon is common for other tumour cells. Second, there are differences about exosomes between an experimental and a clinical situation because exosomes are concentrated in laboratory. Third, the exact component in HeLa exosomes causing ER stress in ECs remained unclear, which needs to be further investigated.
High Lights
This study identified that the exosomes derived from HeLa cells inhibit the TJ expression in ECs, which in turn, increase the vascular permeability in vitro.
HeLa exosomes also break down the vascular integrity and increase tumour metastasis in vivo.
Further investigations indicate that ER stress is induced in HeLa exosomes treated ECs.
This investigation unveiled a novel mechanism for HeLa exosome-induced tumour metastasis.
Author contributions
Yinuo Lin: conception and design, performed experiments, data analysis and interpretation, manuscript writing.
Chi Zhang: collection and assembly of data, data analysis.
Pingping Xiang: collection and/or assembly of data.
Jian Shen: collection and/or assembly of data.
Weijian Sun: financial support.
Hong Yu: overall supervision of the project, conception and design, data analysis and interpretation, manuscript writing and revision, final approval of the manuscript and financial support.
Declaration of interests
No potential conflicts of interest were disclosed by all authors.
Supplemental Material
Download MS Word (4.6 MB)Acknowledgments
We thank Drs. Rongzheng Xu, Xun Hu, Yi Sun and Yuqi Wang from Zhejiang University for their advice on the project, Dr. Beibei Wang in the Center of Cryo-Electron Microscopy (CCEM), Zhejiang University for her technical assistance on transmission electron microscopy, and Mr. Zexin Chen in the Second Affiliated Hospital of Zhejiang University for his assistance on statistical analysis.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Egeblad M, Nakasone E, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18(6):884–16.
- Dutta S, Warshall C, Bandyopadhyay C, et al. Interactions between exosomes from breast cancer cells and primary mammary epithelial cells leads to generation of reactive oxygen species which induce DNA damage response, stabilization of p53 and autophagy in epithelial cells. PLoS One. 2014;9(5):e97580.
- Chowdhury R, Webber JP, Gurney M, et al. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget. 2015;6(2):715–731.
- Li L, Li C, Wang S, et al. Exosomes derived from hypoxic oral squamous cell carcinoma cells deliver miR-21 to normoxic cells to elicit a prometastatic phenotype. Cancer Res. 2016;76(7):1770–1780.
- Skog J, Würdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–1476.
- Yuan A, Farber E, Rapoport A, et al. Transfer of microRNAs by embryonic stem cell microvesicles. PLoS ONE. 2009;4(3):e4722.
- Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659.
- Koppers-Lalic D, Hackenberg M, Bijnsdorp I, et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014;8(6):1649–1658.
- Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980.
- Zhu J, Lu K, Zhang N, et al. Myocardial reparative functions of exosomes from mesenchymal stem cells are enhanced by hypoxia treatment of the cells via transferring microRNA-210 in an nSMase2-dependent way. Artif Cells Nanomed Biotechnol. 2017;1–12. DOI:10.1080/21691401.2017.1388249
- Peinado H, Aleckovic M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(6):883–891.
- Szajnik M, Czystowska M, Szczepanski M, et al. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE. 2010;5(7):e11469.
- Plebanek MP, Angeloni NL, Vinokour E, et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat Commun. 2017;8(1):1319.
- Hsu YL, Hung JY, Chang WA, et al. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene. 2017;36(34):4929–4942.
- Umezu T, Tadokoro H, Azuma K, et al. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014;124(25):3748–3757.
- Tadokoro H, Umezu T, Ohyashiki K, et al. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J Biol Chem. 2013;288(48):34343–34351.
- Gesierich S, Berezovskiy I, Ryschich E, et al. Systemic induction of the angiogenesis switch by the tetraspanin D6.1A/CO-029. Cancer Res. 2006;66(14):7083–7094.
- Strilic B, Yang L, Albarrán-Juárez J, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536(7615):215–218.
- Zhou W, Fong MY, Min Y, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25(4):501–515.
- Li J, Li Z, Jiang P, et al. Circular RNA IARS (circ-IARS) secreted by pancreatic cancer cells and located within exosomes regulates endothelial monolayer permeability to promote tumor metastasis. J Exp Clin Cancer Res. 2018;37(1):177.
- Martin T, Jiang W. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. 2009;1788(4):872–891.
- Brennan K, Offiah G, McSherry E, et al. Tight junctions: a barrier to the initiation and progression of breast cancer? J Biomed Biotechnol. 2010;2010:460607.
- Martin T, Watkins G, Mansel R, et al. Loss of tight junction plaque molecules in breast cancer tissues is associated with a poor prognosis in patients with breast cancer. Eur J Cancer. 2004;40(18):2717–2725.
- Jia W, Lu R, Martin TA, et al. The role of claudin-5 in blood-brain barrier (BBB) and brain metastases (review). Mol Med Rep. 2014;9(3):779–785.
- Eichenberger RM, Talukder MH, Field MA, et al. Characterization of secreted proteins and extracellular vesicles provides new insights into host-parasite communication. J Extracell Vesicles. 2018;7(1):1428004.
- Mandel I, Paperna T, Volkowich A, et al. The ubiquitin-proteasome pathway regulates claudin 5 degradation. J Cell Biochem. 2012;113(7):2415–2423.
- Schröpfer A, Kammerer U, Kapp M, et al. Expression pattern of matrix metalloproteinases in human gynecological cancer cell lines. BMC Cancer. 2010;10(undefined):553.
- Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13(12):858–870.
- Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9(7):268–273.
- Jiang WG, Martin TA, Matsumoto K, et al. Hepatocyte growth factor/scatter factor decreases the expression of occludin and transendothelial resistance (TER) and increases paracellular permeability in human vascular endothelial cells. J Cell Physiol. 1999;181(2):319–329.
- Martin T, Mansel R, Jiang W. Antagonistic effect of NK4 on HGF/SF induced changes in the transendothelial resistance (TER) and paracellular permeability of human vascular endothelial cells. J Cell Physiol. 2002;192(3):268–275.
- Egawa G, Nakamizo S, Natsuaki Y, et al. Intravital analysis of vascular permeability in mice using two-photon microscopy. Sci Rep. 2013;3:1932.
- Ruivo C, Adem B, Silva M, et al. The biology of cancer exosomes: insights and new perspectives. Cancer Res. 2017;77(23):6480–6488.
- Becker A, Thakur B, Weiss J, et al. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30(6):836–848.
- Ha M, Kim V. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–524.
- Zhang C, Yan J, Xiao Y, et al. Inhibition of autophagic degradation process contributes to claudin-2 EXPRESSION increase and epithelial tight junction dysfunction in TNF-α treated cell monolayers. Int J Mol Sci. 2017;18(1):e157.
- Nighot P, Hu C, Ma T. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem. 2015;290(11):7234–7246.
- Stamatovic S, Johnson A, Sladojevic N, et al. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann N Y Acad Sci. 2017;1397(1):54–65.
- Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284(31):21036–21046.
- Wang T, Wang L, Moreno-Vinasco L, et al. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part Fibre Toxicol. 2012;9:35.
- Nakamuta S, Endo H, Higashi Y, et al. Human immunodeficiency virus type 1 gp120-mediated disruption of tight junction proteins by induction of proteasome-mediated degradation of zonula occludens-1 and −2 in human brain microvascular endothelial cells. J Neurovirol. 2008;14(3):186–195.
- Elliott PJ, Zollner TM, Boehncke WH. Proteasome inhibition: a new anti-inflammatory strategy. J Mol Med. 2003;81(4):235–245.
- Wu YT, Tan HL, Shui G, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–10861.
- Rubinsztein DC, Gestwicki JE, Murphy LO, et al. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6(4):304–312.
- Lenna S, Han R, Trojanowska M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life. 2014;66(8):530–537.
- Chandrika BB, Yang C, Ou Y, et al. Endoplasmic reticulum stress-induced autophagy provides cytoprotection from chemical hypoxia and oxidant injury and ameliorates renal ischemia-reperfusion injury. PLoS One. 2015;10(10):e0140025.
- Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A. 2000;97(23):12625–12630.
- Liang G, Yang J, Wang Z, et al. Polycystin-2 down-regulates cell proliferation via promoting PERK-dependent phosphorylation of eIF2alpha. Hum Mol Genet. 2008;17(20):3254–3262.
- Pommier A, Anaparthy N, Memos N, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science. 2018;360:6394.
- Wu CH, Silvers CR, Messing EM, et al. Bladder cancer extracellular vesicles drive tumorigenesis by inducing the unfolded protein response in endoplasmic reticulum of nonmalignant cells. J Biol Chem. 2019;294(9):3207–3218.
- Kabacaoglu D, Ciecielski KJ, Ruess DA, et al. Immune checkpoint inhibition for pancreatic ductal adenocarcinoma: current limitations and future options. Front Immunol. 2018;9:1878.