
ABSTRACT
The transfer of microRNAs (miRs) via extracellular vesicles (EVs) is a functionally relevant mechanism of intercellular communication that regulates both organ homoeostasis and disease development. Little is known about the packaging of miRs into EVs. Previous studies have shown that certain miRs are exported by RNA-binding proteins into small EVs, while for other miRs and for large EVs, in general, the export mechanisms remain unclear. Therefore, a proteomic analysis of endothelial cell-derived large EVs was performed, which revealed that heterogeneous nuclear ribonucleoprotein U (hnRNPU) is abundantly present in EVs. EVs were characterized by electron microscopy, immunoblotting and nanoparticle tracking analysis. Taqman microRNA array and single qPCR experiments identified specific miR patterns to be exported into EVs in an hnRNPU-dependent way. The specific role of hnRNPU for vesicular miR-sorting was confirmed independently by gain- and loss-of-function experiments. In our study, miR-30c-5p was the miR whose export was most significantly regulated by hnRNPU. Mechanistically, in silico binding analysis showed that the export of miRs into EVs depends on the binding efficiency of the respective miRs to hnRNPU. Among the exported miRs, a significant enrichment of the sequence motif AAMRUGCU was detected as a potential sorting signal. Experimentally, binding of miR-30c-5p to hnRNPU was confirmed independently by RNA-immunoprecipitation, electrophoretic mobility shift assay and reciprocally by miR-pulldown. Nuclear binding of miR-30c-5p to hnRNPU and subsequent stabilization was associated with a lower cytoplasmatic abundance and consequently reduced availability for vesicular export. hnRNPU-dependent miR-30c-5p export reduced cellular migration as well as pro-angiogenic gene expression in EV-recipient cells. In summary, hnRNPU retains miR-30c-5p and other miRs and thereby prevents their export into large EVs. The data presented provide a novel and functionally relevant mechanism of vesicular miR export.
Introduction
The release of extracellular vesicles (EVs) is a well-conserved and ubiquitous mechanism in eukaryotic cells and organisms [Citation1,Citation2]. EVs transfer bioactive molecules between cells and, therefore, serve as a means of intercellular communication. This includes the transfer of nucleic acids, peptides, proteins and lipids [Citation3]. EVs are a heterogeneous group of membrane-coated bodies, which are traditionally grouped into two overlapping categories according to their size and their release mechanism. Small EVs (exosomes) are released by the multivesicular body and are 50 nm to 150 nm in size, large EVs (microvesicles) are released via direct budding from the cytoplasmic membrane and are between 50 nm and 1000 nm in size [Citation4]. Both EV subtypes have been shown to transfer functional microRNAs (miRs) into their recipient cells and to thereby contribute to organ homoeostasis and disease development [Citation5,Citation6]. Furthermore, miRs, which are encapsulated in small and large EVs, have been suggested as possible biomarkers for various disease entities, including cardiovascular, cancerous and inflammatory diseases [Citation7–Citation9]. Therefore, it is crucial to understand how the export of miRs into EVs is regulated in order to develop strategies to influence miR transfer therapeutically. Moreover, knowledge about vesicular export of miRs will help us to understand factors that can influence the presence and stability of circulating miRs and, consequently, affect the validity of miRs as biomarkers.
For small EVs, the sorting mechanisms for miRs have been shown to involve RNA-binding proteins [Citation10], ceramides [Citation11] and target mRNAs [Citation12]. In particular, the joint export of miRs and RNA-binding proteins, which can act as miR carriers through sequence-specific binding, is an important mechanism of miR sorting into small EVs. In this context, the heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) and the synaptotagmin-binding cytoplasmic RNA interacting protein (SYNCRIP) have been shown to be responsible for the export of a specific set of miRs into small EVs [Citation10,Citation13]. Furthermore, the Y box binding protein-1 (YBOX-1) as well as the human antigen R (HuR) have been implicated in the vesicular export of miRs into small EVs [Citation14,Citation15]. However, regarding the sorting mechanisms of miRs into large EVs, a possible mechanism has only very recently been attributed to caveolin-1 [Citation16]. In the present study, we characterize a novel role for heterogeneous nuclear ribonucleoprotein U (hnRNPU) in retaining miRs and thereby regulating their export into a distinct population of large EVs derived from endothelial cells.
Methods
Experimental model
Female HCAECs (Promocell, Cat. # C-12221) were cultured under standard conditions (37°C, 5% CO2, 100% relative humidity) until passage 7–8 using Endothelial Cell Growth Medium MV (Promocell Cat. # C-22020). HCAECs were used in this study, because endothelial cells are the only permanently resident and proliferating cells that are donors as well as recipients of blood-derived EVs. Furthermore, the phenotype of endothelial cells from human arteries has been shown to determine vascular health and disease development in patients. Prior to vesicle isolation the HCAECs at 90% confluency were subjected to Endothelial Cell Growth Medium MV without supplements for 24 h. Female Human cardiac fibroblasts (HCF, Promocell, Cat. # C-12375) were cultured under standard conditions (37°C, 5% CO2, 100% relative humidity) until passage 7–8 using fibroblast growth medium 3 (Promocell, Cat. # C-23025) Prior to vesicle isolation the HCFs at 90% confluency were subjected to basal fibroblast medium 3 (Promocell, Cat. # C-23230) without supplements for 24 h. THP-1 cells (Sigma-Aldrich, Cat. # 88081201–1VL) were cultured under standard conditions (37°C, 5% CO2, 100% relative humidity) using RPMI 1640 medium, GlutaMAX (Gibco, Cat. # 61870044) + 10% foetal bovine serum (Gibco, Cat. # A3160802) + 1% penicillin–streptomycin (Gibco, Cat. # 11548876). Prior to vesicle isolation the THP-1 cells were subjected RPMI 1640 Medium, GlutaMAX without supplements for 24 h. EV isolation was performed by use of a three-step centrifugation protocol in an Eppendorf Centrifuge 5430 with a FA-45-16-17 rotor as previously published by our group [Citation17]. Briefly, (i) centrifugation of the culture supernatant at 1500 g for 15 min, (ii) centrifugation of the supernatant at 20,000 g for 40 min, washing of the EV pellet in PBS, (iii) centrifugation at 20,000 g for 40 min ().
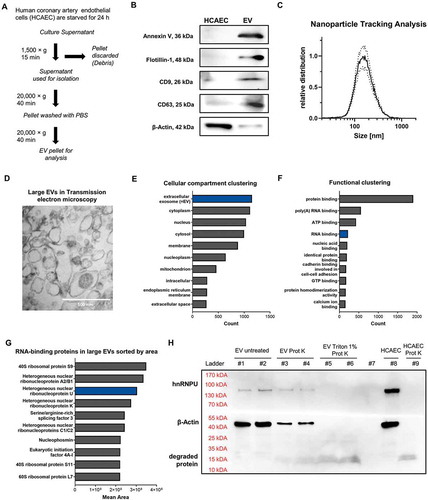
Figure 1. (A) Schematic diagram of the EV isolation protocol used. (B) Immunoblotting of Annexin V (36 kDa), Flotillin-1 (48 kDa), CD9 (26 kDa), CD63 (25 kDa) and β-Actin (42 kDa) in HCAECs and EVs. (C) Size distribution of EVs analysed by nanoparticle tracking analysis (size ~70–500 nm), dotted lines = SEM. (D) Transmission electron microscopic image (85,000×) of pelleted large EVs (size ~100–500 nm). (E) Proteomic analysis of large EVs via mass spectrometry: >3000 proteins the largest portion of which are annotated as vesicular proteins. (F) Functional annotation reveals a relevant portion of RNA-binding proteins. (G) Ranking of the top 10 RNA-binding proteins found in EVs, sorted by area. (H) Immunoblot of hnRNPU and β-Actin of EVs from HCAECs with and without prior degradation by proteinase K and 1% Triton X-100 confirms the presence of hnRNPU in EVs.
Nanoparticle tracking analysis
For nanoparticle tracking analysis, vesicles were isolated from 1.2 × 107 HCAECs as described above and resuspended in 1000 µL PBS and further diluted 1:5 for the analysis. Each sample was recorded three times with three cycles at 11 positions using a minimum brightness of 30 in a ZetaView BASIC NTA – Nanoparticle Tracking Video Microscope PMX-120 (Particle Metrix). The concentration was confirmed to be in the linear range of the ZetaView.
Protein isolation and immunoblotting
Immunoblotting of cells and EVs was performed after lysis in RIPA buffer (Sigma-Aldrich, Cat. # R0278) with 1:25 Protease Inhibitor Cocktail (Roche, Cat. # 4693132001). Lysates were ultrasonicated for 10 min and protein concentration was assessed with a Qubit-4 Fluorometer (Thermo Fisher Scientific) by use of a Qubit™ Protein Assay Kit (Thermo Fisher Scientific, Cat. # Q33211), according to the manufacturer’s instructions. For quantification of the EV protein markers as well as for the confirmation of the presence of hnRNPU in EVs 50 µg, for the confirmation of hnRNPU knockdown and overexpression 10 µg were diluted 2:1 in 3× Laemmli buffer and loaded onto an SDS-PAGE gel (Bio-Rad, Cat. # 456-1084 and 456–1024) by use of the Mini PROTEAN System (Bio-Rad). Subsequently, the protein was transferred onto a Roti-NC nitrocellulose membrane (Carl Roth GmbH, HP40.1) and blocked with 5% BSA (Sigma-Aldrich) for 1 h. Monoclonal mouse anti-hnRNP U antibody (Abcam, Cat. # ab10297, RRID:AB_297037) 1:2000, monoclonal mouse anti-Annexin V antibody (Abcam, Cat. # ab54775, RRID:AB_940268) 1:1000, mouse anti-flotillin-1 antibody (BD Bioscience, Cat. # 610820, RRID:AB_398139) 1:1000, mouse anti-CD63 antibody (Abcam, Cat. # ab59479, RRID:AB_940915) 1:1000, mouse Anti-CD9 antibody (Millipore, Cat. # CBL162, RRID:AB2075914) 1:1000 and mouse anti-β-Actin antibody (Sigma-Aldrich, Cat. # A1978, RRID:AB_476692) 1:2000 in BSA 5% were used to stain the membrane overnight at 4°C. After extensive washing with 0.1% TBST, the membrane was incubated with an HRP-conjugated rat monoclonal anti-mouse-IgG antibody (Sigma-Aldrich, Cat. # A9044, RRID: AB_258431) 1:3000 in 5% BSA for 1 h at RT. After washing again with 0.1% TBST, the membrane was developed with ECL primer western blotting detection reagent (Sigma-Aldrich) and imaged by use of a ChemoCam HR16-3200 Imager (INTAS). The images were analysed by the software ImageJ (RRID:SCR_003070).
Electron microscopy of EVs
For transmission electron microscopic imaging, EVs from 1.2 × 107 HCAECs were pelleted as described above, fixed in 1.25% glutaraldehyde in 0.1 M cacodylate buffer overnight, dehydrated with ethanol and propylene oxide and embedded in Epon 812 (Serva, Cat. # 90529-77-4) again overnight. The samples were sliced with an Ultracut R microtome (Reichert) at 70 nm thickness. After double contrast staining by use of uranyl acetate and aqueous lead solution, images were taken with a CM 10 electron microscope (Philips).
Proteomic analysis of EVs
Peptide preparation
Proteins were stained with Coomassie Brilliant blue (Sigma-Aldrich). For protein identification, gel slices were subjected to in-gel digestion [Citation18,Citation19]. In brief, slices were washed consecutively with water, 50% acetonitrile (ACN, Sigma-Aldrich) and 100% ACN. Proteins were reduced with 20 mM dithiothreitol (Sigma-Aldrich) in 50 mM ammonium bicarbonate (Sigma-Aldrich) and alkylated with 40 mM iodoacetamide (Sigma-Aldrich) in the dark for 30 min. The slices were washed again and dehydrated with ACN. Dried slices were incubated with 330 ng sequencing grade trypsin (Promega) at 37°C overnight. The peptide extract was separated, and remaining peptides extracted with 50% ACN. Peptides were dried in a vacuum concentrator and stored at −20°C.
LC-MS measurements
Peptides were dissolved in 0.1% trifluoroacetic acid and 1/3 was injected onto a C18 trap column (20 mm length, 100 µm inner diameter, ReproSil-Pur 120 C18-AQ, 5 µm, Dr. Maisch GmbH) made in-house. Bound peptides were eluted onto a C18 analytical column (200 mm length, 75 µm inner diameter, ReproSil-Pur 120 C18-AQ, 3 µm, Dr. Maisch GmbH) with 0.1% formic acid (Sigma-Aldrich) as solvent A. Peptides were separated during a linear gradient from 2% to 35% solvent B (90% acetonitrile, 0.1% formic acid, both Sigma-Aldrich) within 80 min at 350 nl/min. The nanoHPLC was coupled online to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Peptide ions between 330 and 1600 m/z were scanned in the Orbitrap detector with a resolution of 30,000 (maximum fill time 400 ms, AGC target 106). The 25 most intense precursor ions (threshold intensity 5000, isolation width 1.0 Da) were subjected to collision induced dissociation (normalized energy 35) and analysed in the linear ion trap. Fragmented peptide ions were excluded from repeat analysis for 15 s.
Vesicular hnRNPU degradation assay
In order to confirm the presence of hnRNPU inside of EVs, EVs were isolated from 2.4 × 107 HCAECs per sample, as described above and then resuspended in 10 µL PBS or 1% Triton X-100. After 10 s of vortexing and incubation at RT for 5 min, half of the PBS-treated samples as well as the Triton X-treated samples were incubated with 5 µL proteinase K (20 mg/mL) solution (Thermo Fisher Scientific, Cat. # 25-530-049). 5 µL ddH2O were added to the PBS-treated samples in order to obtain three groups: untreated EVs, proteinase K-only treated EVs and Triton X + proteinase K-treated EVs. As an additional control, 20 µg of HCAEC lysate in 10 µL (prepared as described above) were treated with 5 µL proteinase K (20 mg/mL) solution or PBS. Subsequently, all samples were incubated 5 min at 65°C to allow the proteinase K to degrade the protein followed by 10 min at 95°C to deactivate the proteinase K. Next, 15 µL RIPA buffer (Sigma-Aldrich, Cat. # R0278) with 1:25 Protease Inhibitor Cocktail (Roche, Cat. # 4693132001) were added and the samples were prepared for blotting by ultrasonication, centrifugation and addition of 3× Laemmli buffer, as described above. The whole sample was then loaded onto am an SDS-PAGE gel and processed as described above for immunoblotting against hnRNPU and β-Actin.
hnRNPU knockdown
For siRNA transfection of HCAECs and HCFs, hnRNPU siRNA (Invitrogen, Cat. # AM16708, Assay ID 145413) as well as silencer negative control No. 1 siRNA (Invitrogen, Cat. # AM4611) were used at a final concentration of 10 nmol/L together with Lipofectamine RNAiMAX Transfection reagent (Invitrogen, Cat. # 13778150) at a final concentration of 3.75 µL/mL. Readouts were performed after 48 h of transfection. Negative control siRNA-transfected cells were used as a control group for all experiments on hnRNPU knockdown cells. For siRNA transfection of THP-1 cells 4 × 106 cells were used, following the same protocol as for HCAECS and HCFs. EV isolation from cells upon hnRNPU knockdown (EVshnRNPU kd) was performed as described above after 24 h of serum starvation.
Viability assay
HCAEC viability after hnRNPU knockdown or hnRNPU overexpression was assessed 48 h after transfection in a 96-well plate by use of the MTT Cell Growth Assay Kit (Sigma-Aldrich, Cat. # CT02). In brief, 0.01 mL of the AB Solution (MTT) were added to 0.1 mL of medium. The cells were incubated at 37°C for 4 h. Then 0.1 mL isopropanol with 0.04 N HCl were added to dissolve formazan and the absorbance was measured with a test wavelength of 570 nm and a reference wavelength of 630 nm in a Tecan Infinite M200 Plate Reader (Tecan).
Apoptosis assay
Apoptosis induction of hnRNPU knockdown or hnRNPU overexpression in HCAECs was quantified using the FITC Annexin V Apoptosis Detection Kit with 7-AAD (Biolegend, Cat. # 640922). The assay was performed in 12-well plates 48 h after transfection, strictly adhering to the manufacturer’s protocol. In brief, HCAECs were washed twice with PBS, detached and resuspended in 0.1 mL of Annexin V binding buffer. 5 µL of the Annexin staining solution and 5 µL of the 7-AAD solution were added and after 15 min of incubation another 0.6 mL of Annexin V binding buffer were added. The samples were analysed in a FACSCanto II (BD Bioscience). Compensation, gating and absolute quantification were performed with the software FlowJo V10 (BD Bioscience). Representative gating is displayed in Supplementary Figure S2G.
Proliferation assay
Proliferation after hnRNPU knockdown was quantified in a bromodeoxyuridine (BrdU)-incorporation experiment, as previously described. The assay was performed in a 24-well plate 48 h after the transfection. HCAECs were then incubated for 6 h in endothelial cell-growth medium MV with supplements and 10 µmol/L BrdU (Abcam, Cat. # ab142567). The cells were then washed with PBS, fixed in 4% paraformaldehyde (PFA) and washed three times with 1% Triton-X 100 in PBS. Antigens were exposed with 1 N HCL for 10 min on ice, 2 N HCL for 20 min at 37°C and 0.1 mol/L disodium tetraborate for 10 min at RT. Blocking was performed with 1% Triton-X 100, 1 mol/L glycine and 5% normal goat serum in PBS for 1 h at RT: For BrdU staining a rat monoclonal anti-BrdU antibody (Abcam, Cat. # ab6326) at 1:250 dilution (4°C overnight) and a Cy3-labelled goat anti-rat IgG antibody (Abcam, Cat. # ab98416) at 1:500 dilution (1 h at RT) were used. Subsequently, one drop of VECTASHIELD mounting medium with DAPI (Vector Laboratories, Cat. # H-1200) was added and the wells were sealed with a cover slip. Images were acquired by use of a Zeiss Axio Observer microscope and analysed with the ZEN 2.3 pro software.
hnRNPU overexpression
Plasmid expansion
The hnRNPU overexpression plasmid pcDNA3.1-hnRNPU-V5 was obtained from Susana Valente through Addgene (Addgene plasmid # 35974; RRID: Addgene_35974). This plasmid was transformed into the chemically competent E. coli strain, Zymo DH5α Mix, and spread on a selective kanamycin-containing bacterial agar plate (100 µg/mL). Surviving colonies were picked and transferred under aseptic conditions into 200 mL of starter culture. Bacteria were allowed to grow for 24 h under optimal bacterial growth conditions. Plasmid purification was performed by using the NucleoBond Xtra Maxi Kit (Macherey-Nagel, Cat. # 740414.10) according to the manufacturer’s recommendations. The presence of plasmid DNA was confirmed after restriction enzyme digestion by using the respective restriction endonucleases and Sanger sequencing (Microsynth Seqlab, Goettingen, Germany) prior to transfection into mammalian cells.
Plasmid transfection
The hnRNPU overexpression plasmid as well as a corresponding empty plasmid were transfected at a final concentration of 1 µg/mL by use of Lipofectamine 3000 (Invitrogen, Cat. # L3000008) at a final ratio of 3.75 µL/mL with 2.5 µL/mL P3000 reagent. Readouts were performed 48-h post-transfection. Overexpression was confirmed on the RNA and protein levels. For EV isolation, transfected cells were subjected to 24 h of serum-free culture.
RNA isolation
RNA isolation was performed by use of Trizol (Invitrogen, Cat. # 15596026) and chloroform as previously described [Citation9]. In brief, cells were washed with PBS and subsequently lysed in Trizol. RNA was isolated with chloroform, precipitated by use of isopropanol, washed twice with ethanol, dried and resuspended in water. Separate isolation of nuclear and cytoplasmic RNA was carried out with the cytoplasmic and nuclear RNA purification kit (Norgen Biotek, Cat. # 37400) strictly following the manufacturer’s protocol. RNA quality and concentration were assessed using a Nanodrop2000 (Thermo Fisher Scientific).
Quantification of miR expression
MiRs were quantified with Taqman miR-Assays (all from Thermo Fisher Scientific: hsa-miR-30c Cat. # 4427975, Assay ID 000419; hsa-let-7d, Cat. # 4427975, Assay ID 002283; hsa-miR-20a, Cat. # 4427975, Assay ID 000580; hsa-miR-125a-3p, Cat. # 4427975, Assay ID 002199; RNU44, Cat. # 4427975, Assay ID 001094; snRNA U6 Cat. # 4440887, Assay ID 001973) in a 7500 HT Real-Time PCR instrument (Applied Biosystems). 10 ng of total RNA was reversely transcribed to cDNA by use of the TaqMan microRNA Reverse Transcription kit (Applied Biosystems, Cat. # 4366597), according to the manufacturer’s protocols. Then, quantitative real-time PCR was performed in triplicate using the TaqMan Universal Master Mix II (Applied Biosystems, Cat. # 4440040) and 1 µL of the cDNA solution after reverse transcription, which represents the equivalent of 0.67 ng RNA. Relative expression of miR-30c-5p, let-7d-5p and miR-20a-5p was calculated as ΔΔCT values, with RNU-44 as an internal control. Absolute miR expression was calculated by use of a concentration gradient of miR-30c-5p-mimic (Thermo Fisher Scientific, Cat. # 4464066 Assay ID MC11060) ranging from 6.67 × 10−10 to 6.67 × 10−17 g RNA and corresponding CT values from 6 to 39. MiR levels in EV-recipient cells were quantified after 24 h of incubation with EVshnRNPU kd/EVsControl.
MiR Array
miR Arrays were performed using TaqMan Array Human MicroRNA Card A v2.0 (Applied Biosystems Cat. # 4398965) in a 7900 HT fast real-time PCR instrument (Applied Biosystems). For cDNA preparation, 0.5 µg RNA was reversely transcribed by use of the TaqMan MicroRNA Reverse Transcription Kit and specific Megaplex RT Primers (Applied Biosystems, Cat. # 4399966). Subsequently, preamplification was performed using 2.5 µL of the RT product, Megaplex PreAmp Primers (Applied Biosystems, Cat. # 4399233), and the TaqMan PreAmp Master Mix (Applied Biosystems, Cat. # 4488593). 9 µL of the preamplification product was used for the final amplification with the MicroRNA Array Card and TaqMan Universal Master Mix II. The amplification curves were analysed with the Program RQ manager Version 1.2.1 (Applied Biosystems) to calculate CT values, using the same threshold across all respective HCAEC and EV samples. Only miRs that were stably expressed upon all three samples of control and hnRNPU knockdown HCAECs and EVs were included into the final analysis. ΔΔCT values were calculated by use of RNU-44, because RNU-44 was the most stably expressed endogenous control across the EV samples as assessed by CT value standard deviation: RNU-6b: SD 1.45, RNU-44: SD 0.79, RNU-48: SD 0.93.
Quantification of mRNA and pri-miR expression
For mRNA and pri-miR expression analysis, 0.5 μg RNA was reversely transcribed to cDNA by use of the Omniscript RT Kit (Qiagen, Cat. # 205111) with Primer “random” (Roche, Cat. # 11034731001), following the manufacturer’s protocol. The cDNA was amplified using 1 µL of the cDNA preparation, the Taqman Gene Expression Master Mix (Applied Biosystems, Cat. # 4440040), and Taqman probes for hnRNPU, c-myc, GAPDH, and 18S (all from Thermo Fisher Scientific: hnRNPU Cat. # 4331182, Assay ID Hs00244919_m1; GAPDH, Cat. # 4331182, Assay ID Hs02786624_g1; c-myc, Cat. # 4453320, Assay ID Hs00153408_m1; 18S, Cat. # 4331182, Assay ID Hs99999901_s1; pri-miR-30c-1, Cat. # 4427012, Assay ID Hs03303371_pri; pri-miR-30c-2 Cat. # 4427012, Assay ID Hs03302833_pri) in a 7500 HT real-time PCR instrument (Applied Biosystems). GAPDH was used as an internal control and relative expression was calculated as ΔΔCT values.
Prediction of RNA–protein interaction
A potential interaction of hnRNPU and miRs was first assessed in silico by use of the RNA-protein interaction prediction tool (RPISeq), which is freely available online: http://pridb.gdcb.iastate.edu/RPISeq/references.php [Citation20]. For the analysis, raw values predicted by the SVM classifier were used with a cut-off value of 0.5.
RNA-motif identification
In order to identify sequence motifs in the RNAs, which are particularly affected by hnRNPU-mediated sorting, we used the Multiple Em for Motif Elicitation tool (MEME), which is freely available online: http://meme-suite.org/tools/meme [Citation21]. As input sequences we used the 48 RNAs that showed hnRNPU-dependent export using the miR array data with a p < 0.05 in the unadjusted t-test (above the lower dotted line (p < 0.05) in ). A ZOOPS model was used in the classic mode of the software with a motif width of 4–8 bases.
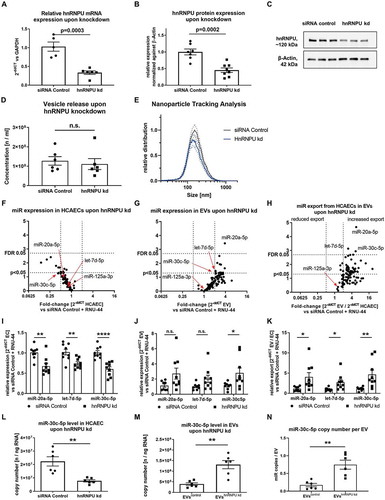
Figure 2. (A) Confirmation of hnRNPU knockdown (kd) in HCAECs on the mRNA level as 2−ΔΔCT relative to the siRNA control and GAPDH, n = 5/6. (B) Confirmation of hnRNPU knockdown on the protein level relative to β-Actin, n = 7/8. (C) Representative immunoblot: hnRNPU (~120 kDa), β-Actin (42 kDa). (D, E) Nanoparticle tracking analysis of EVs after hnRNPU knockdown (kd) in HCAECs vs siRNA control, n = 6; (D) total number of EVs released. (E) EV size distribution, dotted lines = SEM. (F–H) Volcano plots of Taqman microRNA array analyses of HCAECs (F), EVs (G), and relative export calculated as EVs/HCAECs (H) upon hnRNPU knockdown. Expression is calculated as 2−ΔΔCT relative to the siRNA control and RNU-44, n = 3, significance was tested with the two-stage step-up method of Benjamini, Krieger and Yektutieli, FDR level = 5%. (I–K) Validation of miR-20a-5p, let-7d-5p and miR-30c-5p expression in HCAECs and EVs upon hnRNPU knockdown as 2−ΔΔCT relative to siRNA control and RNU-44, n = 8/9. (I) HCAECs, (J) EVs, (K) relative export. All data are presented as the mean ± SEM, n.s. non-significant, unpaired t-test. (L, M) Validation of the regulation of miR-30c-5p export upon hnRNPU knockdown via quantification of absolute copy number of miR-30c-5p expression in HCAECs and EVs after hnRNPU knockdown, n = 6. N) Analysis of the miR copy number per EV particle after hnRNPU knockdown. All data are presented as the mean ± SEM, n.s. non-significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 unpaired t-test, except for F-H.
Crossed-linked RNA immunoprecipitation (RIP)
The Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Sigma-Aldrich, Cat. # 17–700) was used according to the manufacturer’s protocol. For each RIP reaction, 100 μL of the cellular pellet from HCAECs were fixed with 1% formaldehyde in PBS at RT for 10 min. The cross-linking reaction was stopped by adding 590 μL of 2.5 M glycine. Fixed cells were subsequently harvested and resuspended in RIP lysis buffer supplemented with protease/RNAse inhibitors. The lysate was obtained using a dounce homogenizer on ice (10 passes were performed to release the nuclei) followed by incubation on ice for 15 min. An equal volume of RIP lysis buffer was added to the cellular pellet. From the solution, 10 μL (10%) of the lysate was removed and stored as an “Input”. For each RIP reaction, 100 μL of lysate was mixed with 5 μg of mouse anti-IgG (negative control provided with the kit) and anti-hnRNPU antibody (Abcam, Cat. # ab10297), which was previously conjugated with Protein A/G magnetic beads (provided with the kit). Following overnight incubation at 4°C, the RNA-protein immunocomplex was extensively washed with RIP Wash Buffer (provided with the kit). Cross-linking was reversed by incubation with proteinase K. The immune-precipitated RNA was purified using a mixture of phenol:chloroform:isoamyl alcohol (125:24:25; Sigma-Aldrich, Cat. # P2069-400ML). 300 μL of the aqueous phase was separated from the upper portion of the tube after centrifugation and 50 μL salt solution-I, 15 μL salt solution-II, 5 μL precipitate enhancer, 850 μL ethanol, and 1 μL GlycoBlue dye was added to each tube, followed by 2-h incubation at −80°C to precipitate the RNA. The sample was then further centrifuged at 14,000 × g for 30 min and washed two times with 80% ethanol, followed by resuspension in RNAse-free water (10 μL). The entire sample was reverse transcribed into cDNA following treatment with DNase I and then the reverse transcriptase SuperScript VILO (Thermo Fisher Scientific, Cat. # 11756500) Master Mix. The final cDNA was diluted to 1 ng/μL for qRT-PCR analysis.
Biotinylated microRNA pull-down
Biotinylation of miRs was performed by use of the Pierce™ RNA 3ʹ-End Desthiobiotinylation Kit (Thermo Fisher Scientific, Cat. # 20163) according to the manufacturer’s protocol. Biotinylated miRs were purified with an miRNeasy Mini Kit (Qiagen, Cat. # 217004). 50 pmol of biotinylated miRs were conjugated with 50 μL of Dynabeads M-280 Streptavidin by using 1× RNA Capture Buffer (provided with Pierce™ Magnetic RNA-Protein Pull-Down Kit, Cat. # 20164) according to the manufacturer’s protocol. Total cellular extracts were collected with Pierce IP Lysis Buffer (Life Technologies Cat. # 87787). Then, 1 mg of cellular extract was pre-cleared with 20 μL of Dynabeads M-280 Streptavidin (Thermo Fisher Scientific, Cat. # 11205D). The biotin-RNA-streptavidin complex was incubated with 1 mg of pre-cleared cellular extract at 4°C for 1 h. After washing five times with washing buffer (150 mM KCl; 25 mM TRIS-HCl at pH = 7.4; 5 mM EDTA; 0.5 mM DTT; 0.5% NP40; and cOmplete Protease Inhibitor Cocktail (Roche, Cat. # 4693132001), the bound proteins were eluted with 50 μL of Biotin Elution Buffer after incubation for 30 min at 37°C with agitation. The entire elution volume was loaded onto a 4–15% polyacrylamide precast protein gel (Bio-Rad, Cat. # 456-1084). Subsequently, immunoblotting for hnRNPU using an anti-hnRNPU antibody (Abcam, Cat. # ab10297) was performed as described above.
EMSA
The electrophoretic mobility shift assay was conducted by use of the LightShift Chemiluminescent RNA EMSA Kit (Thermo Fisher Scientific, Cat. # 2158). The assay was carried out according to the manufacturer’s instructions. In brief, 0.96 µg purified human hnRNPU (OriGene Technologies, Cat. # TP301627) were incubated with 1 pmol 3ʹ-biotinylated miRs (custom ordered from Sigma-Aldrich: hsa-miR-30c-5p: UGUAAACAUCCUACACUCUCAGC-[Btn], hsa-miR-30c-5p-mut1 UGUAUUGUAGGAACACUCUCAGC-[Btn], has-miR-30c-5p-mut2 UGUACCUCGAAGACACUCUCAGC-[Btn], hsa-miR-125a-3p ACAGGUGAGGUUCUUGGGAGCC-[Btn]), in 20 µL of the provided binding buffer with 4% glycerol for 30 min at room temperature. Subsequently, the miR-protein complex was loaded on a 5% polyacrylamide gel (Bio-Rad Cat. # 4565014) and transferred to a positively charged 0.45 µm nylon membrane (Thermo Fisher Scientific, Cat. # 77016). Blocking, washing, equilibration and luminol/peroxide-staining of the membrane was carried out as described in the manufacturer’s protocol. Images of the membrane were obtained using a ChemoCam HR16-3200 Imager (INTAS).
miR-stability assay
Stability of miR-30c-5p and miR-125a-3p upon hnRNPU knockdown were assessed after transcriptional inhibition by Actinomycin D (Sigma-Aldrich, Cat. # A1410) 5 μg/mL for 8 h, 12 h and 16 h [Citation22]. General inhibition of genetic transcription was assessed by quantification of the unstable mRNA of c-myc as previously described [Citation22].
HnRNPU immunostaining
Immunocytochemistry of endothelial cells was performed by using anti-hnRNPU antibody (Abcam) and Atto 565 phalloidin (Sigma-Aldrich Cat. # 94072). 3 × 104 HCAECs per well were grown in a 4-well chamber slide. 24 h after seeding, cells were rinsed with PBS and fixed with 4% paraformaldehyde in PBS for 10 min at RT. Fixed cells were washed with PBS and incubated with 0.25% Triton X-100 in PBS for 10 min at RT for permeabilization of the cell membranes. After three washing steps with PBS, cells were incubated with the blocking solution (0.25% Triton X-100; 1% bovine serum albumin [BSA] in PBS) for 1 h at RT. Subsequently, cells were incubated with anti-hnRNPU antibody 1:500 in the blocking solution overnight at 4°C. After extensive washing with PBS, the cells were incubated with Alexa Fluor-488 conjugated secondary antibody (Thermo Fisher Scientific, Cat. # A28175, 1:1000) and Atto 565 phalloidin (Sigma-Aldrich) for 60 min at RT. After washing with PBS, DAPI staining was applied and the chamber slide was mounted by using ProLong Gold Antifade (Invitrogen, Cat. # 502081). Images were taken with a Zeiss Axio Observer inverted microscope and analysed with the ZEN 2.3 pro software.
MicroRNA fluorescence in situ hybridization
For miR fluorescence in situ hybridization (miR FISH) in HCAECs, the View RNA Cell Plus Assay (Invitrogen, Cat. # 88–19000) was used together with the hsa-miR-30c-5p ViewRNA Cell Plus Probe Set, Alexa Fluor 546 (Thermo Fisher Scientific, Cat. # VM-06, Assay ID VM1-10339-VCP) and the hsa-miR-125a-3p, ViewRNA Cell Plus Probe Set (Thermo Fisher Scientific, Cat. # VM-06, Assay ID VM1-40405-VCP). The assay was carried out on cover slips in a 24-well plate and was performed in triplicate. As recommended by the manufacturer, samples with the target probe omitted were used as a negative control. The protocol was carried out as described by the manufacturers with all buffers and solutions included in the kit. In brief, the HCAECs were fixed and permeabilized with Fixation/Permeabilization Solution for 30 min at room temperature, washed three times with PBS with RNase Inhibitor, fixed again with Fixation Solution for 1 h at RT and washed again with PBS with RNase Inhibitor. The hybridization was performed for 2 h at 40°C in an Incucell V 55 Incubator (MMM Group) with the respective probes diluted 1:100 in ViewRNA Cell Plus Probe Set Diluent. With intermittent washing steps, hybridization of the preamplifier for 1 h at 40°C, hybridization of the amplifier for 1 h at 40°C, and hybridization of the label probe for 1 h at 40°C were carried out as recommended by the manufacturer. Finally, the slides were washed in PBS and then mounted with Vectashield mounting medium with DAPI (Vector Laboratories, Cat. # H-1200). Imaging was performed with a Zeiss Axiovert 200 M microscope and AxioVision software.
Scratch-wound assay
To assess the cellular migration capacity, a scratch-wound assay was used as previously published by our group [Citation5]. In brief, HCAECs were grown to confluence in a 6-well plate (approx. 1.8 × 106 cells). After performing a scratching with a sterile 200 µL pipet tip, the HCAECs were incubated with EVs from an equal number of cells, which were resuspended in the same volume as was used for EV generation (EVshnRNPU kd or EVssiRNA Control). The cells were photographed in a marked position at 0, 6 and 10 h and the remaining cell-free area was measured and correlated (in percentage) to the initially scratched area, images were taken with a Zeiss Axiovert 200 M microscope and AxioVision software.
Transwell migration assay
To confirm the effects of EVs on HCAEC migration, a transwell migration assay was performed as previously described [Citation5]. In brief, 105 HCAECs were seeded onto the upper chamber of a transwell polycarbonate insert with 8.0 µm pore size. After 1 h, EVhnRNPU kd or EVsiRNA Control were inserted into to the lower chamber followed by 6 h of incubation to allow the cells to migrate. The insert was then removed and cells on the upper side of the membrane were removed with a cell scraper. The membranes were fixed with 4% fresh paraformaldehyde, washed with PBS and stained with Vectashield mounting medium with DAPI (Vector Laboratories, Cat. # H-1200). Migration of HCAECS was assessed by counting DAPI-positive cells in five random microscopic fields (10×) of each membrane, images were taken with a Zeiss Axiovert 200 M microscope and AxioVision software.
Angiogenesis-related gene expression array
For the analysis of the angiogenic effect of EVhnRNPU kd compared to EVsiRNA Control, RT2 Profiler PCR Array Human Angiogenesis (Qiagen Cat. # PAHS-024Z) was used. Reverse transcription was conducted by use of 1 µg RNA and the RT2 First Strand Kit (Qiagen Cat. # 330404) following the manufacturer’s instructions. The equivalent of 8.5 ng RNA was used per well, together with RT2 SYBR Green qPCR Mastermix (Qiagen Cat. # 330500). GAPDH was used as an endogenous control to calculate fold-change expression (2−ΔΔCT).
Transfection of miR-30c-5p and miR-122-3p mimics/inhibitors in HCAECs
In order to generate HCAECs that exhibit increased or reduced miR-30c-5p expression, HCAECs were transfected with miR-30c-5p mimic, miR-30c-5p inhibitor, or control RNA (all Invitrogen: miR-30c-5p miRVana miRNA mimic, Cat. # 4464066, Assay ID MC11060; miR-30c-5p miRVana miRNA inhibitor, Cat. # 4464084, Assay ID MH11060; miRVana miRNA mimic Negative Control 1, Cat. # 4464060) at 10 nM final concentration using Lipofectamine 2000 (Invitrogen, Cat. # 11668019) for 24 h. The transfection efficiency was confirmed by qPCR, showing an approximately 30-fold upregulation and 2-fold downregulation upon miR-30c-5p mimic and inhibitor transfection, respectively. Functional assays were started 24 h after the transfection and performed within 48 h from the start of the transfection. The migration assay was carried out as described above. To generate EVmiR-mimic and EVmiR-inhibitor miR-30c-5p mimic, miR-30c-5p inhibitor, siRNA negative control and miR-122-3p miRVana miRNA mimic (Invitrogen, Cat. # 4464066, Assay ID MC13109) as well as miR-122-3p miRVana miRNA inhibitor (Invitrogen Cat. # 4464084,Assay ID MH13109) were transfected as described above. Subsequently the cells were cultured in serum free medium for 24 h, EVs were isolated after 24 h by the protocol described above, and used for co-culture and functional experiments with HCAECs, exactly as described for EVhnRNPU kd.
Quantification and statistical analysis
Data are presented as the mean ± standard error of the mean (SEM) throughout the manuscript. The number of independent replications of the respective experiment is reported as n in the figure legends. All statistical analyses were performed with the software Prism8 (GraphPad, RRID:SCR_002798). Statistical details are displayed in the figure legends. Means of two groups were compared with an unpaired t-test. Means of more than two groups were compared by a one-way ANOVA and a Tukey post-hoc test (applies for miR-stability assay and miR mimic/inhibitor transfection experiments). Correlation of the binding prediction score (SVM classifier) and significance of the hnRNPU-dependent miR-export increase was assessed by use of Pearson’s correlation coefficient. Statistical analysis of the miR array cards was performed using a multiple t-test tool with an FDR level of 5% according to the two-stage step-up method of Benjamini, Krieger and Yektutieli. All reported p-values are two-sided.
Mass spectrometric raw data processing and analysis of database searches were performed with Proteome Discoverer software 2.1.0.81 (Thermo Fisher Scientific). Peptide identification was done with an in-house Mascot server version 2.5 (Matrix Science Ltd., London, UK). MS2 data were searched against human sequences in SwissProt (release 2016_02) and common contaminants. Precursor ion m/z tolerance was 8 ppm, fragment ion tolerance 0.5 Da. Tryptic peptides with up to two missed cleavages were searched. Carbamidomethylation of cysteines was set as a static modification. Oxidation of methionine and N-terminal protein acetylation were allowed as dynamic modifications. Mascot results were assigned q-values by the percolator algorithm version 2.05 as implemented in Proteome Discoverer [Citation23]. Spectra of peptide spectrum matches (PSMs) with q > 0.01 were sent to a second round of database search with semitryptic enzyme specificity (one missed cleavage allowed) where carbamidomethylation was searched as a dynamic modification. Proteins were included if at least two peptides were identified with q ≤ 0.01. False positive rates were estimated to be 0.8%, 1.3% and 1.0% on PSM, peptide and protein levels, respectively.
Data availability
The raw proteomic data for the RNA binding proteins are shown for this manuscript in Supplementary Table S1. qPCR raw data from all experiments in this manuscript including raw CT values and concentration curves for copy number experiments are displayed in Supplementary Table S2. MiR array raw data are shown for this manuscript in Supplementary Table S3. All further data that support the findings of this manuscript are available on request from the corresponding authors.
Results
RNA-binding proteins are highly expressed in large EVs
In order to identify proteins that could be involved in miR sorting and export into large EVs, we isolated large EVs from human coronary artery endothelial cells (HCAECs) by differential centrifugation (). Large EVs were characterized according to the current recommendations of the International Society for Extracellular Vesicles (ISEV) [Citation24]. To this end, we used immunoblotting ( and Supplementary Figure S1), nanoparticle tracking analysis (), and transmission electron microscopy (), which showed that the isolated EVs had a diameter of ~70 to 500 nm and expressed surface markers that are typical for large EVs (microvesicles), such as Annexin V, Flotillin-1 as well as CD9 and CD63. In contrast, the cytoplasmic protein β-Actin was underrepresented in the EV lysate. To investigate which proteins are expressed within large EVs, a proteomic analysis was performed, using mass spectrometry to detect more than 3000 different proteins within large EVs. Cellular compartment (GOTERM_CC_DIRECT) clustering by use of DAVID revealed that vesicular proteins were highly enriched in the lysate (). A relevant portion of the identified proteins were annotated as RNA-binding proteins, according to the (GOTERM_MF_DIRECT) (). Among them, various types of heterogeneous nuclear ribonucleoproteins were prominently expressed in the isolated EVs, including hnRNPA2B1 and hnRNPU (, Supplementary Table S1). Vesicular encapsulation of hnRNPU was confirmed by a degradation experiment with proteinase K, with and without previous treatment with 1% Triton-X 100 (). Based on its high relative abundance in large EVs among RNA-binding proteins in the proteomic analysis and its unknown role in sorting miRs into large EVs, we focused our studies on hnRNPU to further understand the regulation of miR export into large EVs.
Downregulation of hnRNPU decreases miR levels intracellularly and increases miR levels in large EVs
In order to study the function of hnRNPU on vesicular miR export, siRNA-mediated knockdown of hnRNPU was performed in HCAECs and the knockdown efficiency was confirmed on both the mRNA (33.51 ± 4.65% remaining) and protein (45.20 ± 6.74% remaining) level (–C) as well as by Immunocytochemistry (Supplementary Figure S2A). hnRNPU knockdown led to a small, yet significant increase in HCAEC viability but had no impact on apoptosis or proliferation (Supplementary Figure S2B–D,G). Neither the release of large EVs nor the size distribution was significantly affected by hnRNPU knockdown (,E). In order to investigate a role of hnRNPU in vesicular miR export regulation, Taqman miR arrays were performed in EVs and HCAECs with or without hnRNPU knockdown (Supplementary Table S3). Interestingly, hnRNPU knockdown caused a trend towards an overall reduction of intracellular miR-expression (), whereas the miR-content of various miRs in large EVs tended to be increased upon hnRNPU knockdown (). In order to estimate the cellular export of miRs into large EVs, a miR expression ratio between HCAECs and EVs was calculated, showing a tendency towards increased vesicular export of all miRs (). Statistical analysis using a two-step approach with a false discovery rate (FDR)-level of 0.05 revealed a robustly and significantly increased export of 3 miRs upon hnRNPU knockdown: miR-20a-5p, let-7d-5p and miR-30c-5p. MiR array results were validated by using two independent approaches: relative (single qPCR) and absolute (copy number analysis) quantification. Single qPCR analysis revealed that miR-30c-5p showed the most significant increase in export into large EVs upon hnRNPU knockdown (–K). To exclude that the differentially regulated export of miR-30c-5p after hnRNPU knockdown could be an artefact of the normalization against RNU-44, the absolute expression level of miR-30c-5p was quantified as copy number in HCAECs and large EVs after hnRNPU knockdown and the corresponding controls. In line with the relative qPCR results, the absolute copy number of miR-30c-5p was significantly decreased after hnRNPU knockdown in HCAECs, whereas its copy number was increased in large EVs compared to control-siRNA transfection (,M and Supplementary Table S2). MiR-30c-5p copy number per particle was significantly increased from 0.13 ± 0.03 to 0.68 ± 0.19 copies per particle ().
Overexpression of hnRNPU reduces miR-30-5p export into large EVs
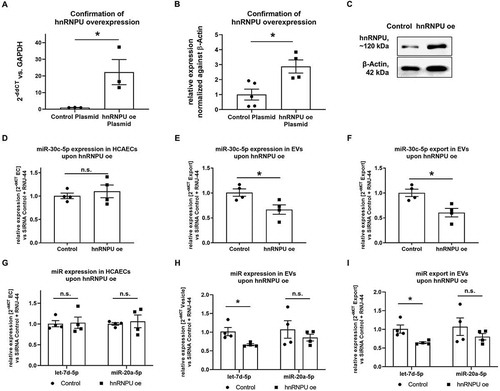
In order to confirm the role of hnRNPU in the regulation of miR export, hnRNPU was overexpressed in HCAECs through plasmid transfection. Stable overexpression of hnRNPU in HCAECs was confirmed 48 h after the transfection at both the mRNA and protein levels (–C). No significant difference was observed in HCAEC viability after hnRNPU overexpression, while early apoptosis was slightly, yet significantly increased after overexpression of hnRNPU (Supplementary Figure S2E,F). In contrast to when hnRNPU was downregulated, overexpression of hnRNPU reduced the export of miR-30c-5p into EVs (–F). A similar, although less pronounced, effect was observed for let-7d-5p and for miR-20a-5p miRNAs (–I).
Figure 3. (A) Confirmation of effective overexpression (oe) of hnRNPU mRNA after plasmid transfection as 2−ΔΔCT relative to the empty vector control and GAPDH, n = 3. (B) Confirmation of effective oe of hnRNPU protein after plasmid transfection relative to β-Actin, n = 4/5. (C) Representative immunoblot: hnRNPU (~120 kDa), β-Actin (42 kDa). (D–F) Quantification of miR-30c-5p in HCAECs and EVs upon hnRNPU overexpression as 2−ΔΔCT relative to the empty vector control and RNU-44. Shown are HCAECs (D), EVs (E), and relative export calculated as EVs/HCAECs (F), n = 4. (G–I) Quantification of let-7d-5p and miR-20a-5p expression as 2−ΔΔCT relative to the siRNA control and RNU-44 in HCAECs (G), EVs (H), and of relative miR export into EVs (I) upon hnRNPU overexpression, n = 4. All data are presented as the mean ± SEM, n.s. non-significant, *p < 0.05, unpaired t-test.
Downregulation of hnRNPU increases vesicular miR-30c-5p export in fibroblasts but not in monocytes
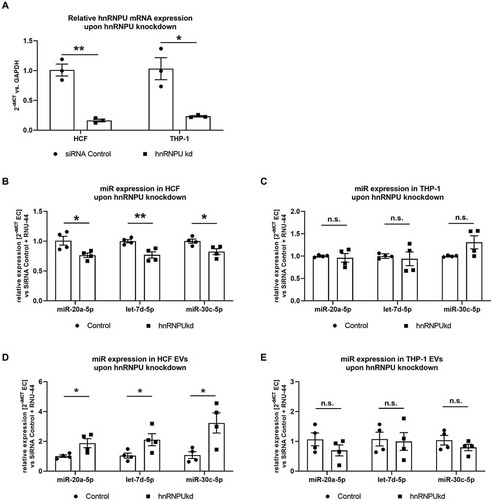
Large EV-encapsulated miRs are released by various types of cells that are relevant to the pathophysiology of atherosclerosis. We therefore investigated, if the aforementioned mechanism is limited to HCAECs, or if it also applies to different cell types, such as fibroblasts or immune cells. To this end, we conducted a knockdown of hnRNPU in human cardiac fibroblasts (HCF) as well as in THP-1 cells (a monocytic cell line, ) and quantified miR-20a-5p, let-7d-5p and miR-30c-5p both in cells and EVs using the same approach as for HCAECs. We found that knockdown of hnRNPU leads to a similar pattern of cellular and vesicular distribution of miR-20a-5p, let-7d-5p and miR-30c-5p in HCFs, as was shown before in HCAECs (,D). In THP-1 cells, however, no significant differences were observed in the cellular expression or EV content of miR-20a-5p, let-7d-5p and miR-30c-5p upon hnRNPU knockdown (,E).
Figure 4. (A) Confirmation of hnRNPU knockdown (kd) in HCFs and THP-1 cells on the mRNA level as 2−ΔΔCT relative to the siRNA control and GAPDH, n = 3. (B–E) Quantification of miR-30c-5p, let-7d-5p and miR-20a-5p in HCF (B) and THP-1 cells (C) and their respective EVs (D, E) upon hnRNPU knockdown as 2−ΔΔCT relative to the siRNA control and RNU-44, n = 4. All data are presented as the mean ± SEM, n.s. non-significant, *p < 0.05, **p < 0.01, unpaired t-test.
HnRNPU binds miR-30c-5p in a sequence specific manner but not miR-125a-3p
As hnRNPU is known to be an RNA-binding protein and because cellular and vesicular miR-30c-5p levels were regulated depending on hnRNPU expression, we explored if miR-30c-5p binds to hnRNPU. To this end, we used an RNA–protein interaction prediction tool (RPISeq), which uses only sequence information [Citation20]. The three miRs whose vesicular levels were most significantly increased upon knockdown of hnRNPU were also predicted by RPISeq to bind hnRNPU: SVM classifier: miR-20a-5p: 0.65; let-7d-5p: 0.689; miR-30c: 0.6, cut-off 0.5 (). However, miR-125a-3p, which did not exhibit increased export upon hnRNPU knockdown, was predicted to not bind hnRNPU: miR-125a-3p: 0.047. To investigate if the binding prediction for the miRs that were analysed correlates with the extent of vesicular export regulation, we performed a correlation analysis of the binding score (SVM classifier) with the -LOG(p) value of export regulation (x-axis of ). We found a significant correlation between the predicted binding and the significance of the export regulation (). Analysis of the sequences of the miRs whose export was significantly regulated by hnRNPU (simple p < 0.05 x-axis of ) revealed the motif AAMRUGCU to be significantly enriched within the regulated miRs (motif present in 11 out of 48 miRs, e-value: 0.0021) ( and Supplementary Figure S3A). The predicted binding of miR-30c-5p, miR-20a-5p and let-7d-5p to hnRNPU was confirmed via cross-linked RNA immunoprecipitation (RIP) of hnRNPU with magnetic beads, followed by qPCR to quantify miRs in the precipitate. In contrast to miR-125a-3p and RNU44, miR-30c-5p and also miR-20a-5p and let-7d-5p were highly enriched in the precipitate (, Supplementary Figure S4). In order to confirm that the identified motif was in fact the binding region of miR-30c-5p and hnRNPU, mutated versions of miR-30c-5p were tested for binding to hnRNPU in an electrophoretic mobility shift assay. The first mutation (miR-30c-5p-mut1) was only partial, as it preserved the purine and pyrimidine pattern of the motif and used bases that were also predicted in positions 3, 5 and 6. The second mutation (miR-30c-5p-mut2) was complete, as the purines and pyrimidines were exchanged throughout the motif (, left panel). In the electrophoretic mobility shift assay with pure recombinant hnRNPU, native miR-30c-5p and the partially mutated miR-30c-5p-mut1 were shown to bind to hnRNPU, whereas binding was abolished almost completely for miR-30c-5p-mut2 and for miR-125a-3p, as a negative control (). Additionally, binding of hnRNPU and miR-30c-5p was confirmed by use of miR-mediated pulldown of hnRNPU ().
Figure 5. (A) RNA-protein interaction prediction via the RPISeq-Platform (SVM classifier). Values of 0.5 or greater indicate a likely RNA-protein interaction. (B) Correlation analysis of predicted RNA-protein interactions and the significance level of the hnRNPU-dependent vesicular miR export. (C) Motif analysis of the significantly regulated miRs (simple p < 0.05) by use of a ZOOPS model in the MEME software. AAMRUGCU is present in 11 of 48 analysed miRs sequences, e-value: 0.0021. (D) Cross-linking immunoprecipitation of hnRNPU and quantification of miR-30c-5p, RNU-44 and miR-125a-3p expression by qPCR as a percentage of 10% input, n = 3. (E) Electrophoretic mobility shift assay of hnRNPU, native miR-30c-5p, miR-30c-5p-mut1 (with partially mutated motif = purine and pyridines preserved), miR-30c-5p-mut2 (with complete mutation of the motif = purines and pyrimidines exchanged), and miR-125a-3p. (F) hnRNPU pulldown with miR-30c-5p and miR-125a-3p and quantification of bound hnRNPU by immunoblotting. All data are presented as the mean ± SEM, ****p < 0.0001, two-way-ANOVA + Tukey post-hoc test.
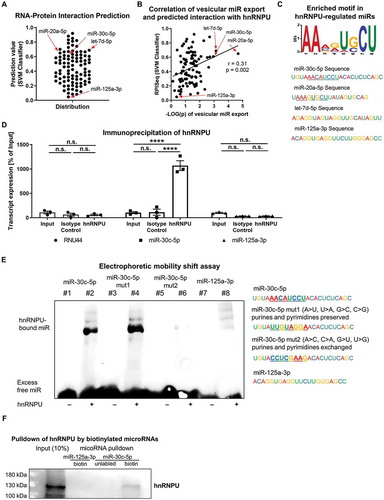
hnRNPU stabilizes miR-30c-5p and retains miR-30c-5p in the nucleus
To further investigate how hnRNPU regulates the cellular fate of miR-30c-5p, we sought to elucidate how hnRNPU affects miR-stability, nuclear/cytoplasmic distribution and pri-miR expression of miR-30c-5p. To explore if a loss of hnRNPU function affects the stability of miR-30c-5p and thereby reduces the overall intracellular copy number of miR-30c-5p, we performed a miR-stability assay upon hnRNPU knockdown. First, we confirmed by qPCR that transcription was completely inhibited by treatment with Actinomycin D (an inhibitor of transcription) for 8 h via quantification of c-myc mRNA, which is known to be rapidly degraded (Supplementary Figure S5C). hnRNPU protein stability was not affected by Actinomycin D treatment and RNU-44 was neither affected by hnRNPU knockdown nor by Actinomycin D treatment (Supplementary Figure S5A,B,D). However, stability of miR-30c-5p was significantly impaired by downregulation of hnRNPU after 8 h, 12 h and 16 h of Actinomycin D treatment while miR-125a-3p stability was much less affected by hnRNPU knockdown (,B) This is indicative of a stabilizing effect of hnRNPU on miR-30c-5p. In contrast to mature miR expression, expression levels of pri-miR-30c-1 and pri-miR-30c-2 remained unchanged following hnRNPU knockdown (Supplementary Figure S6). Subsequently, we further characterized the function of hnRNPU by assessing the cellular distribution of hnRNPU protein via immunocytochemistry. Direct staining of hnRNPU revealed an unbalanced distribution of protein between the nucleus and cytoplasm, with an abundant nuclear localization of hnRNPU (). To investigate the influence of hnRNPU on the distribution of miR-30c-5p in different cellular compartments, cytoplasmic and nuclear RNA was isolated separately upon hnRNPU knockdown. Separation of nuclear and cytoplasmic RNA was confirmed by quantification of the nuclear snRNA U6 and the cytoplasmic 18 S in the lysates (Supplementary Figure S7A) [Citation25,Citation26]. Relative cytoplasmic miR-30c-5p expression was significantly increased after hnRNPU knockdown compared to nuclear expression ( and Supplementary S7D), while RNU-44 distribution was not significantly affected by knockdown of hnRNPU (Supplementary Figure S7C). These results were confirmed by miR fluorescence in situ hybridization (miR-FISH) in combination with knockdown of hnRNPU. miR-FISH showed a nuclear enrichment of miR-30c-5p in the presence of hnRNPU, whereas a redistribution towards the cytoplasm and an overall signal reduction was observed upon knockdown of hnRNPU (, left panel). MiR-125a-3p exhibited a more even distribution between the nucleus and cytoplasm in miR-FISH, which was not visibly altered upon knockdown of hnRNPU (, middle panel). Of note, the applied miR-FISH probes recognize both mature miR-30c-5p and precursor molecules of miR-30c-5p. As pri-miR-30c-1/2 expression was unchanged after hnRNPU knockdown, as opposed to mature miR-30c-5p expression ( and , Supplementary Figure S6), the changes observed in the fluorescence signal can be attributed to mature miR-30c-5p. In summary, these findings indicate that hnRNPU binds and stabilizes miR-30c-5p and retains it in the nucleus of HCAECs. Loss of hnRNPU leads to a more cytoplasmic distribution of miR-30c-5p and can thereby facilitate the encapsulation and subsequent export of miR-30c-5p into large EVs. In line with the absence of a significant increase in vesicular miR-30c-5p export in THP-1 cells (), there was no redistribution from the nucleus to the cytoplasm upon knockdown of hnRNPU in THP-1 cells (Supplementary Figure S7B).
Figure 6. (A,B) MiR stability assay upon hnRNPU knockdown and 0–16 h Actinomycin D treatment, miR-30c-5p and miR-125a-3p were quantified as 2−ΔΔCT relative to the siRNA control and RNU-44 (A) and miR-30c-5p was additionally measured as absolute quantification as copy number (B), n = 3. (C) Immunocytochemical staining of hnRNPU (lower left panel, green), nuclear staining with DAPI (upper left panel, blue), and F-actin staining with Phalloidin (upper right panel, red) in HCAECs confirms the predominantly nuclear expression of hnRNPU, lower right panel = merge. (D) Relative quantification of nuclear and cytoplasmic miR-30c-5p expression as 2−ΔΔCT relative to nuclear expression and RNU-44, n = 3. (E) MicroRNA fluorescence in situ hybridization for miR-30c-5p (in the left panel), miR-125a-3p (middle panel) without probe (right panel) in control siRNA-treated HCAECs (upper row) and after hnRNPU kd (lower row). All data are presented as the mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, unpaired t-test, ANOVA + Tukey post-hoc test for A, B.
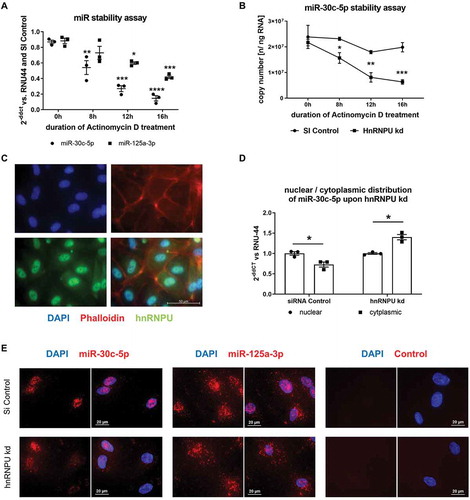
Increased hnRNPU-dependent miR-30c-5p export into large EVs leads to increased miR-30c-5p levels and reduced migration in EV-recipient cells
In order to evaluate the biological effects of increased miR-30c-5p export into large EVs on EV-recipient cells, we quantified miR levels of miR-30c-5p after 24 h incubation with EVs from hnRNPU-knockdown HCAECs (EVhnRNPU kd) and from corresponding control cells (EVcontrol). miR-30c-5p levels were significantly increased in HCAECs treated with EVhnRNPU kd (). Furthermore, we performed functional analyses of HCAECs after incubation with EVhnRNPU kd and EVcontrol. Target endothelial cell migration was significantly impaired upon treatment with EVhnRNPU kd in the scratch-wound assay and the transwell migration assay (,C). To elucidate the effects of EVshnRNPU kd on cellular angiogenesis, we performed a qPCR-based angiogenesis array after stimulation of recipient HCAECs with EVshnRNPU kd. Treatment with EVhnRNPU kd led to the upregulation of anti-angiogenic genes, such as THBS2, SERPINF1, CCL11, TIMP3 and CXCL9, while pro-angiogenic genes, such as HGF, MMP9 and TNF, were downregulated compared to treatment with EVsiRNA Control (, Supplementary Figure S8).
Figure 7. (A) Absolute quantification of miR-30c-5p levels as copy number in HCAECs after incubation with EVhnRNPU kd compared to EVsiRNA Control, n = 6. (B) HCAEC migration upon treatment with EVhnRNPU kd in a scratch-wound assay with representative images, n = 9/12. (C) HCAEC migration upon treatment with EVhnRNPU kd in a transwell migration assay with representative images, n = 6. (D) qPCR-based analysis of 84 angiogenesis-related genes upon treatment with EVhnRNPU kd compared to EVsiRNA Control. Top 10 upregulated and downregulated genes are displayed as 2−ΔΔCT relative to EVsiRNA Control and GAPDH, n = 3. All data are presented as the mean ± SEM, *p < 0.05, **p < 0.01, ANOVA + Tukey post-hoc test for A, B, C, unpaired t-test for D.
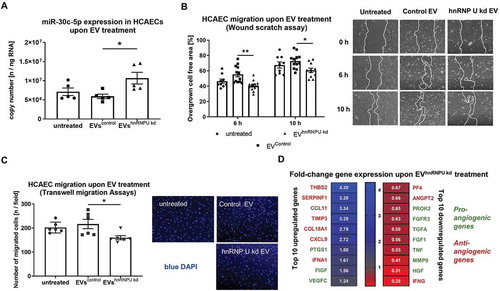
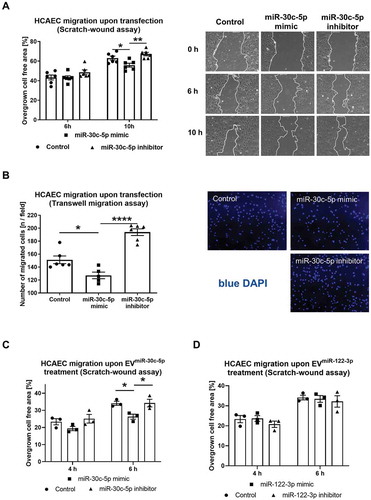
External upregulation and intercellular transfer by EVs of miR-30c-5p both lead to reduced HCAEC migration
To evaluate if the observed effect of EVhnRNPU kd treatment is potentially due to increased vesicular miR-30c-5p export and transfer, we transfected a miR-30c-5p mimic and an inhibitor into HCAECs. HCAEC migration was significantly reduced following transfection of the miR-30c-5p mimic, in both the scratch-wound and the transwell migration assays (,B). Additionally, EVs from miR-30c-5p (or miR-122-3p as a negative control without any known function in cell migration) mimic and inhibitor transfected HCAECs were co-incubated with native HCAECs and cellular migration was assessed in a scratch-wound assay. Confirming the anti-angiogenic effect of vesicular miR-30c-5p transfer, treatment with EVmiR−30c−5p-mimic lead to a similar reduction in endothelial migration as treatment with EVhnRNPU kd (,D).
Figure 8. (A) HCAEC migration upon miR-30c-5p mimic/inhibitor transfection at 10 nM final concentration for 24 h in a scratch-wound assay compared to control RNA transfection with representative images, n = 6. (B) HCAEC migration upon miR-30c-5p mimic/inhibitor transfection at 10 nM final concentration for 24 h in a transwell migration assay compared to control RNA transfection with representative images, n = 6. C, D) HCAEC migration upon treatment with EVmiR−30c−5p-mimic and EVmiR−30c−5p-inhibitor (C) EVmiR−122−3p-mimic and EVmiR−122−3p-inhibitor compared to EVcontrol RNA (D) in a scratch-wound assay, n = 3. All data are presented as the mean ± SEM, *p < 0.05, **p < 0.01, ****p < 0.0001, one-way ANOVA + Tukey post-hoc test.
Discussion
Despite the increasing application of EV-incorporated miRs as biomarkers and therapeutic agents, the mechanisms which regulate miR export and sorting on a cellular level have been insufficiently explored. While miR export into small EVs is mainly carried out through joint export with specific RNA-binding carrier proteins, the regulation of miR export into large EVs is poorly studied. In the present manuscript, we describe a new mechanism for the regulation of miR export into large EVs. We show, for the first time, that hnRNPU limits the export of miR-30c-5p, let-7d-5p and miR-20a-5p into large EVs and has an impact on EV function and the miR levels and function of EV-recipient cells ().
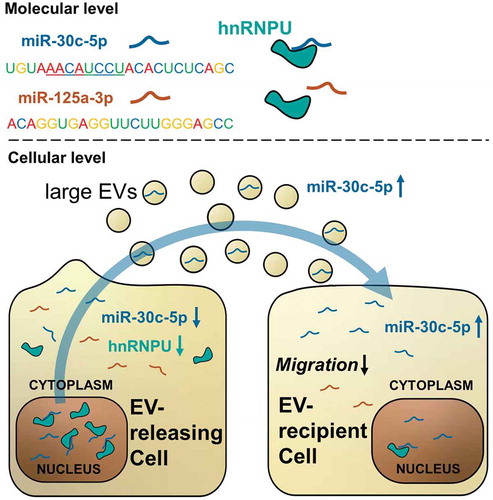
Figure 9. Graphical summary of the study results: hnRNPU binds a limited set of miRs in a sequence specific manner, including miR-30c-5p and excluding miR-125a-3p. Downregulation of hnRNPU triggers a relocation of previously bound miR-30c-5p from the nucleus to the cytoplasm, from where miR-30c-5p is available for export into large EVs. In this way downregulation of hnRNPU leads to increased vesicular levels of miR-30c-5p and causes effective transfer of miR-30c-5p to EV recipient cells. Here, increased levels of miR-30c-5p translate into reduced migratory potential and anti-angiogenic gene expression.
The previously described vesicular miR sorting mechanisms by RNA binding proteins, such as hnRNPA2B1, Y-BOX1, SYNCRIP and Argonaute protein 2 (AGO2), promote miR export, in part, by binding to specific sequence motifs of the exported miRs [Citation10,Citation13,Citation15,Citation27,Citation28]. Like the aforementioned RNA binding proteins, hnRNPU is present in EVs. However, the regulatory capacity of hnRNPU in vesicular miR export depends on its nuclear abundance. Our data suggest that hnRNPU binds miR-30c-5p in the nucleus, protects it from degradation, and thereby enhances the relative nuclear expression of miR-30c-5p. Downregulation of hnRNPU increases the relative concentration of miR-30c-5p in the cytoplasm and consequently facilitates miR-30c-5p export into EVs. Of note, miR-30c-5p exhibits none of the previously described exosomal sorting motifs (hnRNPA2B1: GGAG, CCCU; SYNCRIP: GGCU) and has not been described to be exported via hnRNPA2B1or SYNCRIP (Supplementary Figure S3B) [Citation10,Citation13]. Therefore, the vesicular export of miR-30c-5p is unlikely to be mediated by hnRNPA2B1 or SYNCRIP. Besides the identified carrier proteins hnRNPA2B1, SYNCRP, YBOX-1 and AGO2, the HuR also plays a distinct role in miR sorting into EVs, acting as a positive modulator of miR export. HuR is not exported into EVs for miR sorting, but acts in proximity to the exosomal sorting machinery, competitively to AGO2 [Citation14]. hnRNPU may instead reduce miR export by remote binding of the miR in the nucleus.
Interestingly, Squadrito et al. discovered a mechanism of secondary, remote regulation of vesicular miR export through binding of miRs to target mRNAs. The relative abundance of the target mRNAs was found to be inversely correlated to vesicular miR export, indicating that mRNAs can act like miR sponges and thereby reduce miR export [Citation12]. The mechanism we discovered in our study extends this result by showing that not only the presence of target mRNAs reduces the export of miRs, but also RNA-binding proteins, such as hnRNPU can influences miR export in a similar way.
Importantly, the EV population we investigated in our study differs from the EVs which were used in the above-mentioned studies of hnRNPA2B1, SYNCRIP, Y-BOX1 and HuR, which all used small EVs (exosomes). In our study, we used relatively large EVs which express typical markers of larger EVs (e.g. microvesicles, ). The high abundance of hnRNPA2B1 in the isolated large EVs in our proteomic data suggests that hnRNPA2B1 may generally also be involved in miR sorting into large EVs, which has very recently been confirmed [Citation16]. , the aforementioned mechanisms are unlikely to apply to miR-30c-5p, because miR-30c-5p (and many other miRs) does not contain the required exosomal sorting motifs (Supplementary Figure S3B). It has been argued that protein sorting into large EVs is less strict in comparison to small EVs, where it is tightly regulated by the ESCRT machineries [Citation4,Citation29]. Remote regulation of miR sorting by mRNA sponges, as shown by Squadrito et al. [Citation12], or by RNA-binding proteins like hnRNPU, may therefore be particularly relevant for large EVs, because it does n not require any dedicated protein-sorting machinery.
In the miR array data after hnRNPU knockdown, we saw that vesicular miR content besides just miR-30c-5p is altered by hnRNPU downregulation. Although the expression of other miRs was changed less significantly, a trend towards increased export of miRs was observed for various miRs. While the extent of the regulation of export through hnRNPU varies from mild to strong, depending on the miR, none of the miRs were exported less upon hnRNPU knockdown. Moreover, we show that the extent of regulation of miR export correlates with the predicted binding to hnRNPU and that the sequence motif AAMRUGCU is enriched in regulated miRs (). This suggests that the described miR sorting mechanism is particularly pronounced for miR-30c-5p but is not exclusive. Of note, the role of hnRNPU for miR-30c-5p export regulation has been shown to be relevant beyond endothelial cells. In our experiments, silencing of hnRNPU in fibroblasts lead to a similar cellular as well as vesicular distribution pattern of miR-30c-5p as observed in endothelial cells. In a monocytic cell line, however, this effect was not visible, which may be due to the non-adherent nature of monocytes or to the relatively small cytoplasmatic volume, because we showed that redistribution of miRs from the nucleus to the cytoplasm plays a key role in the hnRNPU-dependent miR sorting mechanism.
Although we have tested the influence of downregulation and overexpression of hnRNPU on vesicular miR-export, we were thus far unable to test the effects of a complete silencing of hnRNPU. In mice, complete loss of hnRNPU function leads to an early embryonic lethality [Citation30]. This may be due to the broad range of functions that hnRNPU is exerting in cellular RNA processing. In previous work, hnRNPU has been shown to bind various RNA species and to regulate transcription, splicing and stability of mRNAs [Citation31–Citation34]. Due to its broad role in RNA processing, dysfunction of hnRNPU has pathophysiological implications for various organ systems and diseases. Mutations of the hnRNPU gene have been linked to the development of epileptic encephalopathy [Citation35]. HnRNPU expression has been suggested as a prognostic marker for colorectal cancer [Citation36]. Loss of hnRNPU function in the myocardium leads to development of cardiomyopathy [Citation34]. In the present study, we show that downregulation of hnRNPU causes reduced migration and a predominantly anti-angiogenic phenotype in otherwise untreated endothelial EV-recipient cells. This effect appears to be mediated by an increased transfer of miR-30c-5p, as shown in and –C. Nevertheless, we have shown that miR-30c-5p is not the only miR whose vesicular export is affected by hnRNPU. Therefore, it seems likely that there are also other miRs that are increasingly transferred to recipient cells via EVhnRNPU kd and that the combined signal from of all these miRs leads to the observed anti-angiogenic phenotype. Until now, a specific role for hnRNPU in the regulation of stabilization, compartmental distribution and intercellular trafficking of miRs has not been reported. Altered vesicular miR stability, distribution and transfer may therefore be an important pathophysiological mechanisms in disorders that are caused by dysfunction of hnRNPU.
EV-encapsulated miRs are used as diagnostic and prognostic biomarkers for multiple diseases, in particular for cancer and cardiovascular disease [Citation7,Citation37]. The release of miRs into EVs, and subsequently into bodily fluids, depends on the biological state of the cell releasing the EVs. Therefore, mechanisms which directly regulate vesicular miR export can influence the level of circulating miRs. Insufficient understanding of miR export mechanisms in different EV subsets cases misinterpretation of miR levels and leads to erroneous conclusions.
The new mechanism for regulating miR export into EVs that is described in this manuscript may contribute to a better understanding of miR export into large EVs, which are largely understudied in comparison to small EVs (exosomes), but functionally not less relevant. Our study provides new insights into the regulation of vesicular miR export, which can help to better interpret the levels of circulating miR biomarkers and may help to develop new treatment strategies based on horizontal transfer of EV-associated miRs.
Disclosure of interest
The authors report no conflicts of interest.
Supplemental Material
Download Zip (2.5 MB)Acknowledgments
We thank Anna Flender and Paula Levermann for excellent technical support. We thank Dr. Meghan Campbell for her critical reading and valuable suggestions to improve the manuscript.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article
Additional information
Funding
References
- Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun. 2012;80(6):1948–21.
- Yáñez-Mó M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066.
- Hergenreider E, Heydt S, Tréguer K, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14(3):249–256.
- van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–228.
- Jansen F, Yang X, Hoelscher M, et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation. 2013;128(18):2026–2038.
- Singh R, Pochampally R, Watabe K, et al. Exosome-mediated transfer of miR-10b promotes cell invasion in breast cancer. Mol Cancer. 2014;13:256.
- Thind A, Wilson C. Exosomal miRNAs as cancer biomarkers and therapeutic targets. J Extracell Vesicles. 2016;5:31292.
- Mi S, Zhang J, Zhang W, et al. Circulating microRNAs as biomarkers for inflammatory diseases. Microrna. 2013;2(1):63–71.
- Liu Y, Li Q, Hosen MR, et al. Atherosclerotic conditions promote the packaging of functional microRNA-92a-3p into endothelial microvesicles. Circ Res. 2019;124(4):575–587.
- Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980.
- Kosaka N, Iguchi H, Yoshioka Y, et al. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285(23):17442–17452.
- Squadrito ML, Baer C, Burdet F, et al. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 2014;8(5):1432–1446.
- Santangelo L, Giurato G, Cicchini C, et al. The RNA-binding protein SYNCRIP is a component of the hepatocyte exosomal machinery controlling microRNA sorting. Cell Rep. 2016;17(3):799–808.
- Mukherjee K, Ghoshal B, Ghosh S, et al. Reversible HuR-microRNA binding controls extracellular export of miR-122 and augments stress response. EMBO Rep. 2016;17(8):1184–1203.
- Shurtleff MJ, Yao J, Qin Y, et al. Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc Natl Acad Sci U S A. 2017;114(43):E8987–E8995.
- Lee H, Li C, Zhang Y, et al. Caveolin-1 selectively regulates microRNA sorting into microvesicles after noxious stimuli. J Exp Med. 2019;216(9):2202–2220.
- Jansen F, Zietzer A, Stumpf T, et al. Endothelial microparticle-promoted inhibition of vascular remodeling is abrogated under hyperglycaemic conditions. J Mol Cell Cardiol. 2017;112:91–94.
- Rosenfeld J, Capdevielle J, Guillemot JC, et al. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal Biochem. 1992;203(1):173–179.
- Jenö P, Mini T, Moes S, et al. Internal sequences from proteins digested in polyacrylamide gels. Anal Biochem. 1995;224(1):75–82.
- Muppirala UK, Honavar VG, Dobbs D. Predicting RNA-protein interactions using only sequence information. BMC Bioinformatics. 2011;12:489.
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36.
- Bail S, Swerdel M, Liu H, et al. Differential regulation of microRNA stability. RNA. 2010;16(5):1032–1039.
- Käll L, Storey JD, MacCoss MJ, et al. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res. 2008;7(1):29–34.
- Théry C, Witwer KW. Minimal information for studies of extracellular vesicles 2018 (MISEV 2018): a position statement of the international society for extracellular vesicles and update of the MISEV 2014 guidelines. International Society of Extracellular Vesicles; 2018, Mount Royal, New Yersy, USA .
- Turunen TA, Roberts TC, Laitinen P, et al. Changes in nuclear and cytoplasmic microRNA distribution in response to hypoxic stress. Sci Rep. 2019;9(1):10332.
- Rouquette J, Choesmel V, Gleizes PE. Nuclear export and cytoplasmic processing of precursors to the 40S ribosomal subunits in mammalian cells. Embo J. 2005;24(16):2862–2872.
- Guduric-Fuchs J, O’Connor A, Camp B, et al. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genomics. 2012;13:357.
- Shurtleff MJ, Temoche-Diaz MM, Karfilis KV, et al. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife. 2016;5. DOI:10.7554/eLife.19276
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383.
- Roshon MJ, Ruley HE. Hypomorphic mutation in hnRNP U results in post-implantation lethality. Transgenic Res. 2005;14(2):179–192.
- Kiledjian M, Dreyfuss G. Primary structure and binding activity of the hnRNP U protein: binding RNA through RGG box. Embo J. 1992;11(7):2655–2664.
- Xiao R, Tang P, Yang B, et al. Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol Cell. 2012;45(5):656–668.
- Yugami M, Kabe Y, Yamaguchi Y, et al. hnRNP-U enhances the expression of specific genes by stabilizing mRNA. FEBS Lett. 2007;581(1):1–7.
- Ye J, Beetz N, O’Keeffe S, et al. hnRNP U protein is required for normal pre-mRNA splicing and postnatal heart development and function. Proc Natl Acad Sci U S A. 2015;112(23):E3020–3029.
- de Kovel CG, Brilstra EH, van Kempen MJ, et al. Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol Genet Genomic Med. 2016;4(5):568–580.
- Hope NR, Murray GI. The expression profile of RNA-binding proteins in primary and metastatic colorectal cancer: relationship of heterogeneous nuclear ribonucleoproteins with prognosis. Hum Pathol. 2011;42(3):393–402.
- Jansen F, Nickenig G, Werner N. Extracellular vesicles in cardiovascular disease: potential applications in diagnosis, prognosis, and epidemiology. Circ Res. 2017;120(10):1649–1657.