
ABSTRACT
A long-term and huge challenge in nanomedicine is the substantial uptake and rapid clearance mediated by the mononuclear phagocyte system (MPS), which enormously hinders the development of nanodrugs. Inspired by the natural merits of extracellular vesicles, we therefore developed a combined “eat me/don’t eat me” strategy in an effort to achieve MPS escape and efficient drug delivery. Methodologically, cationized mannan-modified extracellular vesicles derived from DC2.4 cells were administered to saturate the MPS (eat me strategy). Then, nanocarriers fused to CD47-enriched exosomes originated from human serum were administered to evade phagocytosis by MPS (don’t eat me strategy). The nanocarriers were also loaded with antitumor drugs and functionalized with a novel homing peptide to promote the tumour tissue accumulation and cancer cell uptake (eat me strategy). The concept was proven in vitro as evidenced by the reduced endocytosis of macrophages and enhanced uptake by tumour cells, whereas prolonged circulation time and increased tumour accumulation were demonstrated in vivo. Specially, the strategy induced a 123.53% increase in tumour distribution compared to conventional nanocarrier. The study both shed light on the challenge overcoming of phagocytic evasion and provided a strategy for significantly improving therapeutic outcomes, potentially permitting active drug delivery via targeted nanomedicines.
Introduction
Nanoparticle-based drug delivery has emerged as a suitable strategy for treating and diagnosing cancer. An ideal nanocarrier must remain intact in the complex biological system, exhibit prolonged circulation time in the blood to maximize drug delivery to the intended site, and evade the mononuclear phagocyte system (MPS), which can remove it from the bloodstream [Citation1,Citation2]. Although much progress has been made in cancer-targeted drug delivery, the majority of the injected dose is cleared from the circulation by the MPS, preventing the achievement of proper therapeutic outcomes, impeding the delivery of a sufficient drug dose to the diseased site, and raising toxicity concerns [Citation3]. It has been reported that up to 99% of systemically administered nanoparticles of all shapes, sizes and chemical compositions are eliminated through the liver, whereas only 0.7% of the dose is delivered to the solid tumour [Citation4,Citation5]. The liver, as the largest solid organ in the body, contains the highest population of tissue-resident macrophages [Citation6], and the mouse liver contains 20–40 macrophages per 100 hepatocytes [Citation7]. Kupffer cells (KCs) comprise 80–90% of all tissue macrophages within the body, and they are responsible for the capture and clearance of foreign materials [Citation8,Citation9]. Therefore, understanding and controlling particle phagocytosis by macrophages are extremely critical for developing potential nanotherapeutics, but this remains a significant challenge.
In recent years, several strategies have been designed to reduce the non-specific accumulation of nanoparticles in the liver and spleen. The primary strategy includes optimization of the physicochemical properties of nanoparticles such as shape, size, charge and surface grafting, mostly commonly using the hydrophilic polymer poly(ethylene glycol) (PEG) [Citation10–Citation12]. However, such manipulation has limited ability to decrease MPS uptake because the protein corona layer considerably masks the biological identity of nanoparticles [Citation13]. Additionally, it has been demonstrated that stealth nanoparticles activate the immune system with loss of efficacy upon repeated administration, thereby lowering their binding/uptake by target cells [Citation14–Citation16]. Alternatively, surface modification of nanoparticles using CD47 or peptides derivative from this marker, termed the “don’t eat me” signal, has proven effective for enhancing drug delivery [Citation17]. A relatively new approach to extend circulation time involves the use of naturally derived cell membranes to shield nanoparticles from macrophages [Citation18–Citation22]. These biomimetic design strategies use different cell types such as red blood cells, platelets, leucocytes, cancer cells and stem cells. Nevertheless, a possible major issue is reorienting the targeted bio-inspired nanoparticles to a specific bodily location; for instance, delivering platelet membrane-coated nanoparticles away from sites of vascular injury may be a larger hurdle [Citation23]. Unlike optimizing nanoparticle design, direct blockade of the MPS has gained increasing attention. Because this strategy involves the use of phospholipid vesicles, its safety is assured, and the original physicochemical properties of the nanoparticles for drug delivery are maintained without any need for complicated surface functionalization. This approach is achieved by reversibly blocking MPS-mediated phagocytosis using empty liposomes or directly depleting MPS macrophages using chemical agents [Citation24–Citation27]. However, the rapid elimination of conventional liposomes from the MPS and the systematic toxicity induced by the chemical materials hamper the clinical usefulness of this strategy [Citation5,Citation28].
Extracellular vesicles (EVs) are cell-derived membrane-bound vesicles that have emerged as important messengers in intercellular communication and the regulation of various pathophysiological conditions [Citation29]. EVs are broadly categorized into three major subgroups based on their biogenesis and size: exosomes with a vesicle size of 30–200 nm, microvesicles of 200–1000 nm in diameter, and apoptotic bodies with diameters exceeding 1000 nm [Citation30]. Several studies demonstrated that EVs from various cell sources may adopt different innate homing capabilities in vivo [Citation31–Citation33]. It was found that EVs derived from DC2.4 cells more readily accumulated in the MPS than those isolated from muscle and melanoma cells [Citation34]. However, some EVs have been revealed to promote an immune privilege status, resulting in reduced MPS-mediated clearance [Citation35]. As an illustrative example, EVs isolated from immunocytes and primary fibroblast-like mesenchymal cells are positive for CD47 expression [Citation36,Citation37]. CD47 is known to bind to signal regulatory protein α, initiating the “don’t eat me” signal that blocks phagocytosis [Citation38].
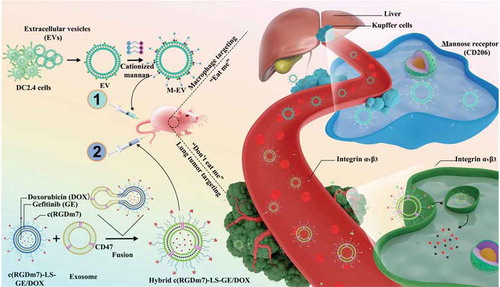
Accordingly, we have proposed a novel EV-based “eat me/don’t eat me” strategy with dual targeting effects to minimize nanoparticle sequestration by MPS organs (liver, spleen) and improve the lung tumour targeting efficiency. Given that EVs derived from DC2.4 cells are mainly captured by macrophages, we functionalized these vesicles with cationized mannan (termed M-EV) to enhance their macrophage targeting. The resulting M-EV are responsible for the “eat me” tactic. Conversely, the subsequent “don’t eat me” signal was achieved using CD47-expressing exosomes originating from human serum to further decrease nanocarrier clearance by the MPS. In this regard, a hybrid drug delivery system was designed by fusing exosomes harbouring CD47 with c(RGDm7)-modified nanocarriers co-loaded with doxorubicin (DOX) and gefitinib (GE), named Hybrid c(RGDm7)-LS-GE/DOX. This strategy was achieved first by injecting M-EV into mice via the tail vein to saturate the receptors of macrophages prior to the administration of Hybrid c(RGDm7)-LS-GE/DOX, thereby minimizing its sequestration by the MPS, improving its tumour accumulation, and enhancing its therapeutic efficacy against lung cancer (Schema 1). Interestingly, such a strategy could result in improved tumour targeting and enhanced therapeutic efficacy without negative effects on liver or spleen function.
Materials and methods
Surface plasmon resonance (SPR) of c(RGDm7)
The binding affinity of c(RGDm7) was investigated by SPR (BiacoreT200, GE Healthcare) at 25°C. Briefly, an amine-coupling kit (GE Healthcare, Buckinghamshire, UK) was used to immobilize integrin αvβ3 on a CM5 chip, with final integrin coupled levels of 16,000 RU. Afterwards, various concentrations of cycle RGD peptides were injected in a running buffer composed of HEPES (10 × 10−3 M, pH 7.4), NaCl (150 × 10−3 M), EDTA (3 × 10−3 M) and surfactant P20 (0.05%) at a flow rate of 30 μL/min. The contact time and dissociation time were both 1 min. The binding affinity KD value was measured as follows: KD = Kd/Ka.
Expression of integrin αvβ3
A549 and MCF-7 cells were grown on coverslips overnight. Then, the cells were washed with PBS, fixed by 4% paraformaldehyde, and blocked with 5% BSA for 1 h at room temperature. Next, we incubated cells with integrin αvβ3 (ab75872, Abcam, UK) rabbit monoclonal antibodies used as first antibodies. PBS was considered as negative control. The secondary antibody used was a Texas Red-conjugated Affinipure Goat Anti-rabbit IgG, which was incubated with cells at 37°C for 1 h. The cells were stained by Hoechst 33,258 for 15 min, and the receptor expression was analysed using a Leica TCS SP5 confocal laser-scanning microscope (CLSM, Heidelberg, Germany).
Synthesis and characterization of c(RGDm7)-PEG-DSPE
c(RGDm7)-PEG-DSPE was synthesized according to previously reported procedure [Citation39]. NHS-PEG2000-DSPE and c(RGDm7) at 2:1 molar ratio were dissolved in DMF. Then, drops of triethylamine were added to adjust the pH to 8.0. The liquid was gently stirred for 120 h at room temperature. The solution was then dialysed against deionized water for 48 h to remove the unconjugated c(RGDm7) and NHS-PEG2000-DSPE. The pure c(RGDm7)-PEG-DSPE was obtained after lyophilization. The conjugation product was verified by MALDI-TOF MS.
EV isolation and purification
Dendritic cell (DC2.4)-derived EV were isolated by differential centrifugation processes, as previously reported [Citation40–Citation42]. EV-free FBS was prepared by filtering FBS with a 100 nm filter, followed by ultracentrifugation at 100,000 × g for 16 h and subsequent filtration with a 100 nm filter. DC 2.4 cells were then cultured in media containing EV-depleted FBS for 48 h. The supernatant was centrifuged at 800 × g for 5 min, and 2000 × g for another 10 min to remove cells and debris, and then filtered using 0.2 μm filter (Millipore, Billerica, MA, USA). Next, the samples were subjected to ultracentrifugation at 100,000 × g (type SW32 Ti rotor) for 2 h using an ultracentrifuge (Optima L-100XP, Beckman Coulter, Brea, CA, USA). After discarding the supernatant, the pellet was resuspended in PBS, and ultracentrifuged again at 100,000 × g (type SW41 Ti rotor) for another 2 h. The sedimented vesicles were carefully suspended in PBS.
Isolation and purification of exosomes from human serum
Human serum purchased from ZOMANBIO Biotechnology Co. (Beijing, China) was diluted 8-fold in PBS and subjected to 15,000 × g centrifugation at 4°C for 30 min to pellet the cellular debris. After 2 h ultracentrifugation at 100,000 × g at 4°C, the pellet was resuspended in PBS. This resulting exosome-enriched sediment was subjected to second high speed centrifugation (100,000 × g for 2 h), and the collected exosomes were reconstituted and stored in PBS at – 80°C.
Synthesis and characterization of cationized mannan
The cationized mannan was synthesized by introducing spermine to the hydroxyl groups of mannan by means of a N,N-carbonyldiimidazole (CDI) activation method [Citation43]. Mannan (100 kDa, 20 mg) was suspended in 20 mL of anhydrous dimethyl sulphoxide (DMSO), and then spermine (748 mg) and CDI (105.2 mg) were added to the mixture. The reaction was kept under moderate stirring at 35°C for 20 h. Once the reaction was completed, the cationized mannan was obtained by dialysis and lyophilization. The yielded product was then analysed by proton nuclear magnetic resonance 1H-NMR, and infrared (IR) spectrophotometric analysis.
Preparation of M-EV
Extracellular vesicles modified with cationized mannan (M-EV) were engineered by mixing of EV isolated from DC2.4 cells with the cationized mannan. The reaction was then incubated at room temperature for 15 min. The protein concentrations of EV were determined using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL) according to the manufacturer’s instructions.
Characterization of M-EV and exosomes
M-EV and exosomes were characterized by nanoparticle tracking analysis (NTA) using a NanoSight LM14 instrument (Malvern Instruments, UK). The particle suspensions were diluted 50 times with PBS for optimal analysis. The morphology was visualized using a transmission electron microscope (TEM, H-9500, Hitachi). First, EV were dropped on copper grids coated with carbon film (Zhongjingkeyi Technology, Beijing, China) and allowed to diffuse onto the grid for 2 min at room temperature. Then the samples were stained for contrast with 2% uranyl acetate for 30 s. For western blot, M-EV and exosomes isolated from DC2.4 cells and human serum, respectively were lysed in RIPA buffer supplemented with complete protease inhibitor cocktail tablets (Roche). Samples were run on SDS-PAGE gels and transferred onto PVDF membranes (Bio-Rad). The blots were incubated with primary antibodies to CD47 rabbit monoclonal antibody (ab108415, Abcam, Cambridge, MA, USA), CD63 rabbit monoclonal antibody (ab134045, Abcam, Cambridge, MA, USA), or CD 81 mouse monoclonal antibody (ab79559, Abcam, Cambridge, MA, USA) overnight at 4°C. Secondary antibodies (1:5000; System Bioscience, Mountain View, CA) were incubated with the membranes for 30 min at 37°C. Signals were detected using chemiluminescent reagents from Pierce, according to the manufacturer’s instructions.
Preparation of hybrid vesicles modified with c(RGDm7)
The hybrid vesicles were prepared using a thin film hydration and extrusion method, followed by PEG-mediated fusion. In brief, a mixture of EPC/cholesterol/mPEG-DSPE/c(RGDm7)-PEG-DSPE (molar ratio of 55:40:3:2) for targeted vesicles or EPC/cholesterol/mPEG-DSPE (molar ratio of 55:40:5) for plain vesicles, and DiO in chloroform were rotary evaporated to produce a thin film, which was hydrated using normal saline solution. The suspensions were then extruded sequentially through 200 nm, 100 nm and 50 nm pore size polycarbonate membranes (Whatman PLC., UK) using an Avanti Mini-Extruder (Avanti Polar Lipids). Unilamellar vesicles were formed by gel filtration over a Sephadex G50 with normal saline solution (PBS). The second step is performed by mixing lipid vesicles and exosomes (molar ratio of 1/1) at 40°C for 2 h in a Thermomixer (Eppendorf). PEG 8000 (30%, w/w) was then added to the mixture to engineer the hybrid vesicles.
FRET-based lipid-mixing assay
Lipid vesicles-exosomes fusion was monitored by FRET-based lipid exchange assay as described previously [Citation44]. The FRET acceptor dye (DiI) and donor dye (DiO) were physically encapsulated into the lipid vesicles. A549 cells were then incubated with c(RGDm7)-LS and hybrid c(RGDm7)-LS for 4 h at 37°C. FRET images were visualized with a confocal laser scanning microscope (CLSM, TCS SP5, Leica, Germany). The FRET efficiency is defined as a difference of fluorescence intensity of the donor before and after photobleaching. The FRET efficiency is measured using this equation: FRETeff = (Dpost-Dpre)/Dpost, where Dpost and Dpre correspond to the fluorescence intensity of DiO after and before photobleaching, respectively. FRET occurs at DiO excitation wavelength (484 nm) and DiI emission wavelength (555–655 nm) [Citation45].
Preparation and characterization of Hybrid c(RGDm7)-LS-GE/DOX
c(RGDm7)-modified lipid vesicles co-loaded with doxorubicin (DOX) and gefitinib (GE), were also constructed using the ratio of components mentioned above. Then, GE with lipid in chloroform (weight ratio of 20:1) were rotary evaporated to produce a thin film. The dry lipid film was hydrated with (NH4)2SO4, and then extruded sequentially through 200 nm, 100 nm and 50 nm pore size polycarbonate membranes (Whatman PLC., UK) using an Avanti Mini-Extruder (Avanti Polar Lipids). Unilamellar vesicles were formed by gel filtration over a Sephadex G50 with phosphate buffered saline (PBS).The lipid vesicles were then actively loaded with DOX using conventional ammonium sulphate gradient method [Citation46]. Afterwards, c(RGDm7)-LS-GE/DOX were mixed with exosomes as previously to form Hybrid c(RGDm7)-LS-GE/DOX. DOX and GE release behaviours were investigated in PBS at pH 7.4 at 37°C using dialysis bags (MW, 14 kDa). At different time points, an equal volume of the dialysis medium was withdrawn to quantify the released drugs with HPLC analysis.
Cell selectivity of M-EV
To confirm the cell type specific uptake of M-EV, the cellular uptake study was conducted in different cell lines: RAW 264.7, DC2.4 and A549 cells. In brief, cells were seeded in 12-well plates overnight and then treated with 20 μg of M-EV-DiO for 4 h. The cells were analysed quantitatively by FACSAria II flow cytometer (BD Biosciences).
Cellular uptake in RAW 264.7
To evaluate the in vitro efficiency of M-EV, cellular uptake in murine RAW 264.7 macrophage cells was performed. M-EV and lipid vesicles were labelled with the lipophilic fluorescent probe DiO. RAW264.7 cells cultured in 12-well plates overnight, were preincubated with 20 μg of M-EV at 37°C for 4 h. After the pre-block with M-EV, macrophage cells were incubated with 5 μmol/L of LS-DiO, Hybrid LS-DiO, c(RGDm7)-LS-DiO or Hybrid c(RGDm7)-LS-DiO at 37°C for 4 h. The fluorescence intensity was determined using a Leica TCS SP5 confocal laser-scanning microscope and quantified by FACSAria II flow cytometer (BD Biosciences).
Cellular uptake in A549
To assess the in vitro targeting efficiency of hybrid vesicular delivery into lung tumour cells, the cellular uptake study was conducted on A549 cells. The cells were plated in a 12-well plate. After 24 h incubation, cells were subsequently incubated with DiO-loaded plain vesicles (LS), c(RGDm7)-LS, Hybrid LS or Hybrid c(RGDm7)-LS at the same final DiO concentration of 5 μmol/L, and incubated for a further 4 h. The cellular uptake was quantified by FACS analysis. Meanwhile, cells were viewed under a fluorescence microscope (Leica, Germany).
In vitro cytotoxicity study
The cytotoxicity of Hybrid c(RGDm7)-LS-GE/DOX against A549 cells was performed by MTT assay. A549 cells (3000 cells per 200 μL per well) were seeded into 96-well plates. After 24 h culture, free DOX and different nanoformulations at various concentrations were added to each well for 72 h and the cytotoxicity was monitored by MTT method.
In vivo targeting efficiency of eat me/don’t eat me strategy
Animal procedures were carried out in accordance with guidelines evaluated and approved by the Ethics Committee of Peking University. In order to explore the time interval between M-EV dose and the second dose of nanotherapeutics, the liver biodistributions of DiR-labelled EV and M-EV were evaluated at three predetermined time points (1, 2 and 4 h). Then, mice were autopsied to harvest the major organs, and their ex vivo images were recorded. We further investigated the blockade effect of M-EV on in vivo biodistribution of Hybrid c(RGDm7)-LS. The first dose of unlabelled M-EV and the second dose of different formulations were separated by an interval of 4 h (based on previous in vivo biodistribution study). Four hour after A549 tumour bearing mice were injected intravenously with DiR-loaded nanoformulations (the second dose), the nude mice were sacrificed, the main organs and tumours were dissected for ex vivo fluorescent intensity analysis.
Immunofluorescence analysis
The biodistribution of different nanoformulations in A549 tumour-bearing nude mice were evaluated under CLSM. DiO-loaded nanocarriers with or without M-EV pretreatment were injected into tumour-bearing nude mice. After 4 h, the tumour tissues were carefully removed, frozen in O.C.T. embedding medium, and sectioned at 3 μm thick. After that, the sections were dyed with Hoechst. For analysis, anti-CD31 antibody was used to detect the tumour angiogenesis.
Pharmacokinetics study
To evaluate effect of pretreatment with M-EV on the fate of secondly injected nanoformulations, pharmacokinetics study was conducted. Hybrid c(RGDm7)-LS-GE/DOX, Hybrid LS-GE/DOX, M-EV+Hybrid c(RGDm7)-LS-GE/DOX or M-EV+Hybrid LS-GE/DOX at a DOX dose of 5 mg/kg and a GE dose of 2 mg/kg, was injected intravenously into SD rats about 220 g, respectively. Next, 0.5 mL blood was collected at different time points (0.083, 0.167, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48 and 72 h) following injection. Samples were then centrifuged at 1000 g for 10 min and resultant supernatants were aspirated for analysis. Briefly, Daunorubicin hydrochloride (50 μL, 125 μg/mL) used as the internal standard was mixed with 200 μL of plasma samples and 1 mL of extraction solvent composed of chloroform and methanol (4:1, v/v). The mixture was vortexed for 1 min and centrifuged at 3000 rpm for 5 min. The lower organic layer was carefully collected and dried under nitrogen gas. The remaining residue at the bottom of tube was redissolved in 100 μL of methanol. For quantification of GE, 100 μL of Sorafenib as internal standard solution (100 μL, 25 μg/mL) was added to 200 μL of plasma and 500 μL of NaOH (0.1 N). After mixing, 1 mL of ethyl acetate was added, mixed for 1 min in the vortex mixer, and centrifuged 10 min at 4000 rpm. The supernatant collected was dried at 40°C under nitrogen gas and reconstituted in 100 μL of methanol. The plasma concentrations of DOX and GE were analysed by HPLC and the pharmacokinetic parameters were calculated by Kinetica 4.4 (Thermo, USA).
In vivo assessment of anti-tumour efficacy
A549 cells (5 × 106) were subcutaneously implanted into the axilla region of BALB/c nude mice. When the tumour reached an average size of 80 mm3, nude mice were randomly divided into six groups (n = 10) and intravenously injected with saline, free DOX, M-EV+Hybrid LS-GE, M-EV+Hybrid LS/DOX or M-EV+Hybrid LS-GE/DOX every two days for a total of five treatments. The dose of DOX was 2 mg/kg and that of GE was 0.8 mg/kg. Meanwhile, the control of free GE was given by oral gavage (at a dose of 20 mg/kg) successively for 6 days. The tumour volume ((length × width2)/2) and body weight of different groups were measured every two days during the entire study period. At the end of treatment, one mouse from each group was sacrificed, the tumours and major organs (heart, liver, spleen, lung and kidney) were fixed with 4% paraformaldehyde, sectioned and stained with haematoxylin and eosin (H&E) staining. Tumour cell apoptosis and angiogenesis were detected by TUNEL assay and CD31 staining according to the manufacturer’s protocol. The remaining mice were used for survival analysis. We defined overall survival when mice naturally died or tumour size exceeded 2000 mm3. To further evaluate the therapeutic efficacy of hybrid targeted vesicles containing both GE and DOX as well the pretreatment with M-EV, the models of A549 tumour-bearing nude mice were established as described above. Five groups of animals, 10 mice in each group, were intravenously administered with saline, Hybrid LS-GE/DOX, Hybrid c(RGDm7)-LS-GE/DOX, M-EV+Hybrid LS-GE/DOX or M-EV+Hybrid c(RGDm7)-LS-GE/DOX at a DOX dose of 2 mg/kg and a GE dose of 0.8 mg/kg every two days for a total of five doses. The tumour growth and body weight were also assessed every 2 days until the death of mice defined as previously. One mice in each group was randomly selected and then was euthanized for histopathology analyses with haematoxylin and eosin (H&E) staining, TUNEL and CD31 staining. The rest of the mice in each group were observed until death (n = 9).
Safety evaluation in animal
ICR mice (20–22 g) were randomly divided into six groups (n = 3) and received previous treatments. Three important hepatic indicators (ALT: alanine aminotransferase, AST: aspartate aminotransferase and ALP: alkaline phosphatase), two indicators for kidney functions (CRE: creatinine and BUN: blood urea nitrogen), and one heart indicator (CK: myocardial creatinine kinase) were analysed using a blood biochemical autoanalyzer (7080, HITACHI, Japan).
Results
Characterization of c(RGDm7), c(RGDm7)-PEG-DSPE and integrin expression
The affinity of c(RGDm7) for αvβ3 integrin was verified via surface plasmon resonance (SPR) (supplementary Figure 1). The SPR sensorgram illustrated that c(RGDm7) peptide exhibited a slow-on and slow-off model. The binding affinities of c(RGDm7), c(RGDyK) and c(RGDfK) were (6.47 ± 2.26)×10−6, (5.78 ± 1.5)×10−5, and (3.72 ± 0.93)×10−5 M, respectively, validating the increased affinity and selectivity of the cyclic RGD tripeptide upon the incorporation of aminocyclopentane as reported previously [Citation47]. Integrin αvβ3 has attracted substantial attention as a target in tumour cells and the tumour vasculature [Citation48,Citation49]. Recently, it was found that c(RGD) could facilitate the transcytosis of nanoparticles from the luminal to the abluminal side of tumour endothelial cells, facilitating the trafficking of nanoparticles through the tumour matrix to cancer cells [Citation50]. Based on these findings, c(RGDm7) was chosen as the candidate ligand to enhance the tumour targeting of our drug delivery system.
The expression of αvβ3 integrin was confirmed in A549 and MCF-7 cells via an immunofluorescence method. The results illustrated that A549 cells were positive for αvβ3 integrin, whereas MCF-7 cells lacked αvβ3 integrin expression (supplementary Figure 2).
c(RGDm7) was conjugated to NHS-PEG2000-DSPE by reacting the NHS group with the amino group. MALDI-TOF-MS of NHS-PEG2000-DSPE and c(RGDm7)-PEG-DSPE demonstrated an increase in mass from 2800 Da to 3367.62 Da after conjugation with c(RGDm7), indicating successful synthesis of the functional material c(RGDm7)-PEG-DSPE (supplementary Figure 3).
Construction and characterization of cationized mannan
Mannan was cationized with spermine, a natural polyamine that displays superior properties over other types of amines, to promote its electrostatic interaction with the negative surface charge of EVs [Citation51]. In this study, spermine was grafted onto the hydroxyl groups of mannan after activation with N,N-carbonyldiimidazole (CDI). The cationic polysaccharides obtained in this manner were stable complexes. For instance, Thomsen et al. prepared cationic pullulan–spermine/DNA complexes that were successfully transfected into rat brain endothelial and human brain microvascular endothelial cells [Citation52]. The structure of cationized mannan was confirmed through IR and 1H-NMR analyses. The appearance of the carbonyl group peak of CDI and the cuspidal peak of spermine at 3300 cm−1 in the IR spectrogram of cationized mannan, confirmed the successful conjugation. 1H-NMR enabled further confirmation of the structure (supplementary Figure 4).
Preparation and characterization of M-EV and exosomes
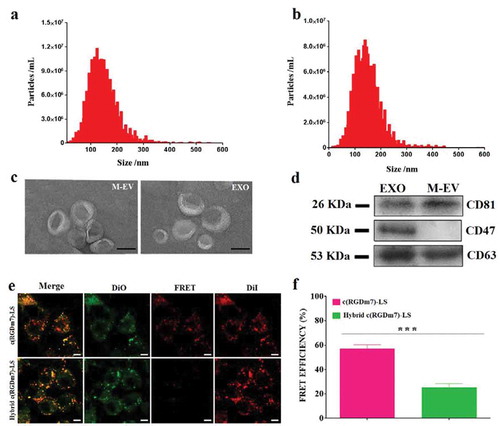
EVs and exosomes were isolated from DC2.4 cells and human serum, respectively, via sequential ultracentrifugation. Isolated EVs were characterized using different approaches including nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM), and Western blotting. As determined by NTA, the average diameters of M-EV and exosomes were approximately 160 and 119 nm, respectively ()). TEM revealed double-membrane vesicles within the diameter range of EVs ()). Protein markers are often used in the literature to characterize EVs, and CD63 and CD81 are the most frequently identified EV proteins. In this study, Western blotting revealed the presence of CD63 and CD81 on M-EV, whereas these two protein markers and CD47 were detected in the exosomes ()).
Figure 1. Characterization of M-EV and exosomes isolated from DC2.4 cells and human serum, respectively. (a, b) Nanoparticle tracking analysis (NTA) of M-EV and exosomes (EXO) isolated from the cell culture and human serum, respectively. (c) Transmission electron micrographs of M-EV and EXO. Scale bar: 50 nm. (d) Western blot of EV markers CD81 (~26 kDa), CD47 (~50 kDa), and CD63 (~53 kDa) on M-EV and EXO. (e) Confocal fluorescence imaging of DiO/DiI-loaded targeted vesicles incubated with A549 cells (green: DiO, red: DiD; scale bar: 10 μm). (f) FRET efficiency of hybrid and no-hybrid targeted vesicles.
Engineering and characterization of hybrid vesicles modified with c(RGDm7)
Different hybrid and functional nanocarriers named Hybrid c(RGDm7)-LS were first engineered using a thin film hydration and extrusion method, followed by PEG-mediated fusion. All of nanocarriers exhibited comparable mean particle sizes, polydispersity indexes and encapsulation efficiencies. The corresponding encapsulation efficiencies of DOX and GE in Hybrid c(RGDm7)-LS-GE/DOX were higher than 90 and 50%, respectively. The low encapsulation of GE was attributable to the bulk precipitates of GE during the vesicle preparation. Precipitation was ascribed to the crystalline nature of GE, which formed lower energy macroscopic crystals in aqueous solutions during the synthesis process [Citation53]. Conversely, the TEM images confirmed the uniform spherical particles of nanometre size, indicating that the ligand modification or fusion process did not influence the physical properties of vesicles (supplementary Figure 5).
The release profiles of DOX and GE from nanocarriers are presented in supplementary Figure 6. Recently, the therapeutic potential of sequentially drug-targeted delivery was demonstrated by several groups [Citation54]. In this study, we observed similarly slow drug release from both targeting and no-targeting hybrid nanocarriers within 72 h. However, GE was released much faster than DOX from all four delivery systems, probably because GE was encapsulated in the phospholipid bilayer whereas DOX was entrapped in the aqueous phase of the vesicles in the form of aggregated and gelatinous anthracycline sulphate salt. As shown in supplementary Figure 6, approximately 40% of DOX and 60% of GE were released from Hybrid c(RGDm7)-LS-GE/DOX in a controlled and sustained pattern within 3 days. These results revealed that Hybrid c(RGDm7)-LS-GE/DOX possessed differential release rates for DOX and GE.
Fusion efficiency with exosomes
To verify the occurrence of fusion between lipid vesicles (LS) and exosomes, the FRET assay was performed. FRET effects were obvious when A549 cells were incubated with DiO/DiI-loaded c(RGDm7)-LS. Because of the proximity of DiO and DiI, both the occurrence of FRET signals and an increase of DiI intensity were observed ()). When fluorescently labelled vesicles were fused with unlabelled exosomes, the distance between DiO and DiI dyes was increased, and consequently, the emission of DiI at 565 nm was decreased. As shown in ), FRET efficiency was decreased significantly after the fusion process, whereas prior to fusion, the FRET efficiency approached 60%, which was in agreement with the findings in ).
“Eat me/don’t eat me” strategy in macrophages in vitro
To evaluate the macrophage targeting efficiency of M-EV, the cell selectivity was performed in RAW 264.7, DC2.4, and A549 cells. Quantitative analysis by flow cytometry showed that M-EV were preferentially taken up by macrophages (supplementary Figure 7). While M-EV exhibited negligible cellular uptake in A549 cells, they were uptaken to some extent by DC2.4 cells. Furthermore, the uptake of M-EV in RAW264.7 cells was 2.20-, 5.78-fold higher than that of M-EV in DC2.4 and A549 cells, respectively. These results indicated the specificity of M-EV to macrophages, which were consistent with many other studies showing high expression level of mannose receptors on macrophages [Citation55–Citation57]. The uptake of various nanoformulations was also evaluated in murine RAW264.7 cells. According to the quantitative and qualitative data presented in ,c), uptake was significantly higher in the M-EV group than in the other groups even though all nanocarriers were internalized in macrophages to some extent. In fact, uptake was approximately 2.73-fold higher in the M-EV group than in the LS group. The sequence of cell uptake from high to low was as follows: M-EV, c(RGDm7)-LS, Hybrid c(RGDm7)-LS, LS, M-EV+Hybrid c(RGDm7)-LS, Hybrid LS and M-EV+Hybrid LS. Notably, the endocytosis of Hybrid c(RGDm7)-LS and Hybrid LS was significantly abrogated by pretreatment with M-EV. In particular, the cell uptake rates in the Hybrid c(RGDm7)-LS and Hybrid LS groups were approximately 1.51- and 1.32-fold of those in the M-EV+Hybrid c(RGDm7)-LS and M-EV+Hybrid LS groups, respectively. The mannose receptors on RAW264.7 cells might have been blocked by M-EV. Generally, the results demonstrated that the “eat me” strategy based on M-EV was successful and that M-EV might represent a promising modality for macrophage targeting.
Figure 2. In vitro cellular uptake of nanocarriers by macrophages and lung tumour cells. (a, b) Confocal microscopy images of nanoformulations internalized by RAW 264.7 and A549 cells, respectively. (Scale bar: 10 μm). (c, d) Quantification of cellular uptake of different formulations by flow cytometry analysis in RAW 264.7 and A549 cells, respectively. ***p < 0.001.
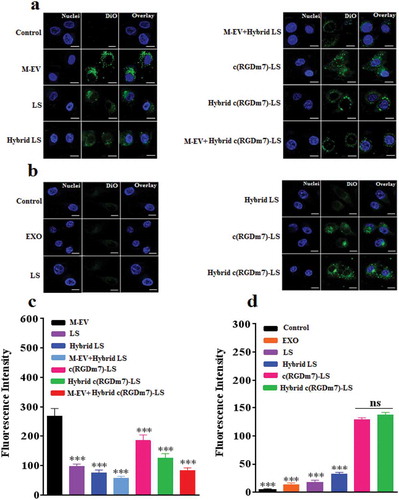
In addition, the results presented in ,c) illustrate that fusing vesicles with exosomes expressing CD47 on their surfaces further decreased the macrophage uptake of Hybrid c(RGDm7)-LS and Hybrid LS. Based on the comparison between nanocarriers with and without exosomes, the endocytosis of hybrid nanocarriers was decreased by 22–32%.
“Eat me” strategy in A549 cells in vitro
Furthermore, the cellular uptake of Hybrid c(RGDm7)-LS was examined in A549 cells as the tumour model. The result clearly illustrated that c(RGDm7)-LS, either alone or fused with exosomes, exhibited significantly higher uptake by A549 cells (6.52- or 6.10-fold increase, respectively) than unmodified vesicles (,d)). The effect of c(RGDm7) was in good accordance with its binding affinity for αvβ3 integrin, as shown previously in supplementary Figure 1, whereas LS only exhibited low-level cellular uptake.
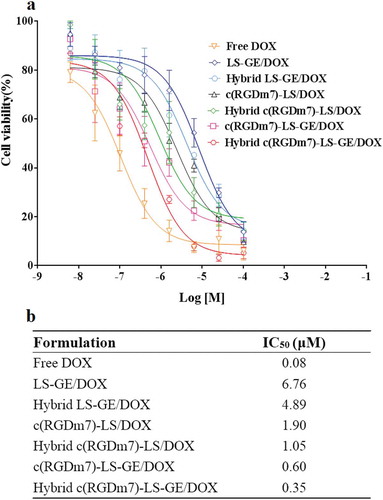
Next, we further evaluated the in vitro efficacy of drug-loaded Hybrid c(RGDm7)-LS in A549 cells. Among the nanocarriers, the IC50s of Hybrid c(RGDm7)-LS-GE/DOX, c(RGDm7)-LS-GE/DOX, Hybrid c(RGDm7)-LS/DOX, c(RGDm7)-LS/DOX, Hybrid LS-GE/DOX and LS-GE/DOX were 0.35, 0.60, 1.05, 1.90, 4.89, and 6.76 μM, respectively. Namely, Hybrid c(RGDm7)-LS-GE/DOX was approximately 19.3-fold more potent than LS-GE/DOX. Generally, the cytotoxicities of hybrid, targeting, and multi-drug–loaded nanocarriers were higher than those of non-hybrid, non-targeting, and single drug-loaded nanocarriers, respectively, confirming the necessity of exosome fusion, c(RGDm7) modification, and combination chemotherapy (). Such potent cytotoxicity might be attributable to the high cell uptake efficiency. Meanwhile, free DOX as a control exerted the strongest inhibitory effect as expected, likely because of the small molecule drug without release from LS, leading to rapid uptake by tumour cells. Of course, the in vivo fate of free DOX does not possess any advantage compared with nanocarriers [Citation58].
Figure 3. Antiproliferative effect of different co-loaded and single drug loaded nanocarriers on A549 cells (n = 3; mean ± SD). (a) Dose-response curves representing cell viability, and (b) IC50 values calculated for each group.
“Eat me/don’t eat me” strategy in vivo
To ascertain whether the proposed “eat me/don’t eat me” strategy could efficiently enhance the in vivo targeting efficiency, we explored the biodistribution of different nanoformulations by comparing whole-body and excised organ images after the pretreatment of mice with M-EV. Previous studies indicated that purified EVs are rapidly cleared from the circulation with a half-life ranging from 10 min to 5 h [Citation59,Citation60]. Therefore, intervals of 1, 2 and 4 h between M-EV administration and subsequent nanocarrier treatment were first tested. The fluorescent intensity of DiR-labelled M-EV in the liver was time dependent (supplementary Figure 8). As the time was extended to 4 h, more DiR-labelled M-EV migrated to the livers and spleens of mice, resulting in the strongest fluorescence intensity. Thus, we chose 4 h as the time interval between the dosing of M-EV and that of nanocarriers for the following study.
We then explored the in vivo blockade effect of M-EV on hybrid vesicles. ,b) shows in vivo whole-body fluorescence imaging and ex vivo fluorescence imaging of dissected organs from mice that were injected with different formulations, respectively. It was clear that M-EV mostly localized in the livers and spleens of tumour-bearing mice (,b)). The semiquantitative analysis of ex vivo images achieved the same conclusion ()), consistent with previous reports [Citation31,Citation34], as well as our macrophage uptake study.
Figure 4. In vivo biodistribution of Hybrid c(RGDm7)-LS. (a) In vivo whole-body imaging of subcutaneous A549 tumour-bearing nude mice injected intravenously with different DiR-labelled nanoformulations at 4 h with or without pre-block. (b) IVIS fluorescence image of tumours and main organs 4 h post-injection with or without pretreatment. (c, d) Quantification of the mean fluorescence signal intensities of major organs and tumours from model nude mice, respectively. Data were mean ± SD (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001).
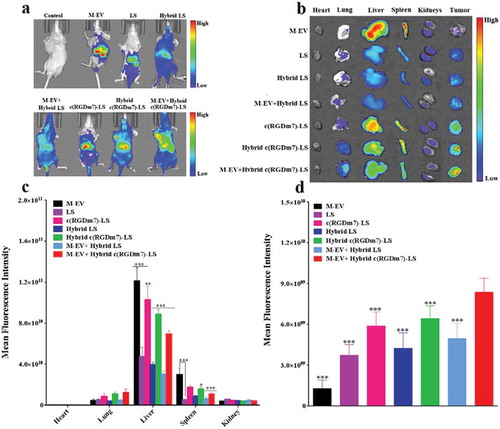
Next, we investigated the distribution of nanoformulations in tumour tissue. As indicated in ), the tumour accumulation of M-EV was the lowest, confirming the positive “eat me” strategy, together with the superior MPS targeting of M-EV as mentioned previously. In addition, the introduction of an exosome system and pretreatment with M-EV both enhanced the tumour accumulation of nanoformulations according to a comparison between LS and Hybrid LS, c(RGDm7)-LS and Hybrid c(RGDm7)-LS, as well as c(RGDm7)-LS and M-EV+c(RGDm7)-LS, Hybrid c(RGDm7)-LS and M-EV+Hybrid c(RGDm7)-LS. Thus, these two approaches did not impact the targeting ability of c(RGDm7)-LS in vivo, which was interesting and favourable because these two mechanisms were designed for MPS escape but not for targeting. However, it was noteworthy that c(RGDm7) resulted in a significantly higher distribution of targeted nanocarriers in tumour tissue than for non-targeted nanocarriers. Consequently, the M-EV+Hybrid c(RGDm7)-LS group exhibited the highest tumour distribution, as indicated by a 123.53% increase in fluorescence intensity within the tumour region compared with that of LS. This could be attributed to the blockade effect of M-EV, the MPS-escaping effect of hybrid nanocarriers, and ligand-mediated delivery due to c(RGDm7) conjugation. In total, this study confirmed the designed “eat me/don’t eat me” strategy at the animal level.
Numerous studies confirmed that RGD peptides specifically and strongly bind the integrin receptor αvβ3, which is overexpressed on tumour and angiogenic endothelial cells [Citation61], and RGD ligands constrained in a preferred cyclic conformation demonstrate an enhancement of integrin-binding affinity [Citation62]. In this study, an immunofluorescence analysis was conducted to investigate the co-localization of nanoparticles and A549 tumour tissue. Most of the administered Hybrid c(RGDm7)-LS co-localized with tumour vessels (CD31+) after M-EV pretreatment, whereas hybrid vesicles with or without pretreatment failed to accumulate in tumour vessels (supplementary Figure 9). These results suggest that M-EV+Hybrid c(RGDm7)-LS both targeted tumour cells and reached the tumour neovasculature.
Pharmacokinetics profiles
To further investigate the in vivo fate of the designed nanoformulations, a pharmacokinetic study was performed. The pharmacokinetic parameters were calculated as listed in supplementary Table 1 and 2 using the noncompartmental model based on the plasma drug concentrations. As shown in , all nanoformulations could maintain relatively high concentrations of DOX, which was still detectable at 72 h after injection, and a relatively low level of GE that was only detectable 24 h after injection. It was previously demonstrated that although EGFR inhibitors are more rapidly cleared from plasma than DOX, the antitumor efficacy of the combination therapy would not be affected [Citation63].
Figure 5. Plasma drug concentration versus time graphs of (a) DOX, and (b) GE following i.v. injection of various nanoformulations in rats (n = 6).
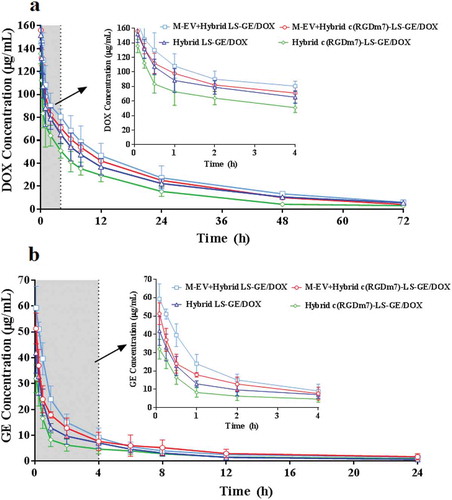
Compared with those of the Hybrid c(RGDm7)-LS-GE/DOX group, the area under the plasma concentration curve (AUC), plasma half-life (t1/2) and mean residence time (MRT) of the M-EV+Hybrid c(RGDm7)-LS-GE/DOX group were 1.54-, 1.18- and 1.15-fold higher, respectively, in the analysis of DOX and 1.91-, 1.55- and 1.36-fold higher, respectively, in the analysis of GE. Moreover, compared with the results of the Hybrid LS-GE/DOX group, these pharmacokinetic parameters were enhanced by 1.08-, 1.03- and 0.98-fold, respectively, in the analysis of DOX and by 1.41-, 1.05- and 1.13-fold, respectively, in the analysis of GE. The extended circulation time of the M-EV+Hybrid c(RGDm7)-LS-GE/DOX group might be attributable to the blockade effect of M-EV on liver and spleen accumulation and the MPS escape effect of exosomes in hybrid nanocarriers. Finally, further analysis is needed to determine whether these advantageous pharmacokinetic parameters result in enhanced antitumor efficacy.
In vivo therapeutic efficacy against lung cancer
Lung cancer is the leading cause of cancer-related mortality worldwide, with non-small cell lung cancer (NSCLC) accounting for approximately 85% of all lung cancers [Citation64]. Many approved drugs targeting epidermal growth factor receptor (EGFR) such as cetuximab, panitumumab, gefitinib and erlotinib have exhibited only limited efficacy in both preclinical and clinical settings [Citation65]. It has been found that the suppression of EGFR signalling can remarkably sensitize NSCLC cells to the DNA-damaging agent DOX, leading to improved antitumor efficacy [Citation66,Citation67].
In this study, therapeutic efficacy was evaluated using a subcutaneous tumour model of A549 lung cancer. First, we compared antitumor efficacy between co-loaded and single drug-loaded nanocarriers using each of the free drugs as the controls. Although antitumor effects were observed in all test groups, the M-EV+Hybrid LS-GE/DOX group displayed the most significant antitumor effect, outperforming single drug-loaded nanoformulations and free drugs (supplementary Figures 10–13).
After confirming the superiority of combination therapy with GE and DOX, we further investigated the therapeutic efficacy of our designed nanoformulations, the targeted combinational hybrid nanosystem pretreated with M-EV, and their controls. Once palpable, tumour-bearing mice were treated with M-EV+Hybrid c(RGDm7)-LS-GE/DOX, Hybrid c(RGDm7)-LS-GE/DOX, M-EV+Hybrid LS-GE/DOX or Hybrid LS-GE/DOX every 2 days for 10 days. As shown in ), the tumour volume in the saline control group rapidly increased over 18 days, whereas the M-EV+Hybrid c(RGDm7)-LS-GE/DOX group exhibited the most remarkable tumour inhibition among the groups.
Figure 6. In vivo therapeutic efficacy of M-EV+Hybrid c(RGDm7)-LS-GE/DOX. (a) Tumour volume growth curves of A549 tumour-bearing nude mice injected with different treatments (n = 10). (b) Kaplan-Meier survival curve of nude mice after treatments (n= 9). (c) Photographs of mice bearing A549 subcutaneous tumours at the end of treatment. (d, e) TUNEL and CD31 staining of tumour sections. Scale bar: 200 μm.
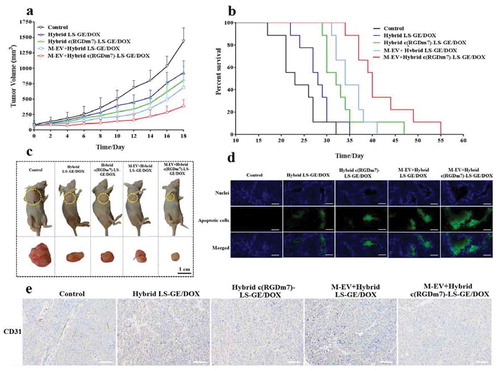
Among all groups, the median survival time of mice treated with M-EV+Hybrid c(RGDm7)-LS-GE/DOX was 40 days, which was significantly longer than those in the saline (23 days, p < 0.001), Hybrid LS-GE/DOX (28 days, p < 0.001), Hybrid c(RGDm7)-LS-GE/DOX (32 days, p < 0.01) and M-EV+Hybrid LS-GE/DOX groups (34 days, p < 0.01) ()).
We also studied tumour apoptosis and angiogenesis using TUNEL and CD31 staining, respectively. As shown in ,e), M-EV+Hybrid c(RGDm7)-LS-GE/DOX most strongly induced apoptosis and inhibited angiogenesis among the treatments. The obvious inhibitory effects of M-EV+Hybrid c(RGDm7)-LS-GE/DOX against angiogenesis was likely due to its better MPS-escape and effective drug delivery mediated by c(RGDm7) ligand that can bind to the integrin αvβ3 overexpressed on the tumour endothelial cells, thereby antagonizing its function as a key regulatory protein for tumour angiogenesis [Citation68,Citation69]. Moreover, both DOX and GE exerted additive anti-angiogenic effects via the modulation of fibroblast growth factor expression, and inhibition of epidermal growth factor receptor tyrosine kinase, respectively [Citation70,Citation71].
Safety study in animal
To design an effective drug delivery system, it is necessary to address the potential toxicity of nanocarriers. Thus, the in vivo toxicity of the nanoformulations was investigated via assessments of blood biochemical indices, haematoxylin and eosin (H&E) staining, and body weight loss. Clinically, free DOX is well known to induce chronic myocardial toxicity [Citation72]. In this study, free DOX caused substantial weight loss and noticeable cardiac tissue degeneration, presumably because of its cardiotoxicity (supplementary Figure 14). Typical biochemical markers were measured to assess liver, kidney, and heart function in the various treatment groups. As shown in supplementary Figure 15, the level of serum creatine kinase, a biomarker of cardiotoxicity, was significantly elevated in the free DOX group, again confirming its cardiotoxicity. Meanwhile, no nanoformulations induced any signs of cardiac muscle damage, hypertrophy, or adherent lobes. Moreover, no statistically significant difference in body weight was observed, indicating reduced systemic toxicity during the entire treatment period (supplementary Figure 16). Notably, the designed nanoformulation displayed no significant differences in terms of transaminase, aspartate aminotransferase, and alkaline phosphatase levels, in addition to heart and kidney function markers, relative to saline (supplementary Figure 17). Unlike other MRS depletion strategies exhibiting potent toxicity against macrophages [Citation25,Citation73], our strategy based on hybrid vesicles exhibited good biocompatibility and systemic safety.
Discussion
The mannose receptor (MR, CD206) is an endocytic pattern recognition receptor belonging to the C-type lectin family. It is well established that the MR is mostly expressed on the surface of most types of macrophages and dendritic cells (DCs), although they are also found in certain endothelial cells, smooth muscle cells, retinal epithelial cells, and so on [Citation55]. There are plenty of studies validating the high expression of the mannose receptor (CD206) on macrophages [Citation56,Citation57]. So, several studies have developed the mannose-modified nanoparticles to target macrophages overexpressing mannose receptors [Citation74,Citation75]. To target KCs, cationized mannan was modified onto the EVs surface to improve the targeting efficiency and macrophage uptake.
In the cell uptake study, a lipophilic fluorescent probe DiO was used. It could be only loaded into the lipid vesicles, since almost no DiO could dissociate in the aqueous medium or biological environments [Citation76]. Namely, the efficiency of the labelling fluorescence to various lipid nanocarriers can be the same as long as we load the same amount of DiO into these nanocarriers. In fact, the same final concentration of the labelling probe was used for all nancarriers in the uptake study, in order to provide the comparability among different nanocarriers.
The specific content of EVs might influence their in vivo fate. It is reported that the clearance of EVs from the circulation is closely related to their uptake in specific organs, and their biodistribution most probably is dependent on their cellular origin, in addition to the presence of various surface markers [Citation34]. In addition, exosomes derived from organotropic human breast cells that metastasized primarily to the lungs were more efficiently accumulated in the lungs, whereas those isolated from pancreatic cancer cells that tend to metastasize to the liver were mainly distributed in the liver [Citation77]. Another example is the “don’t eat me” signal, including CD47 on the surface of vesicles, which inhibits phagocytosis by macrophages [Citation37]. Our experiments demonstrated this. Conversely, PEGylation has been utilized to provide “stealth” properties for the uptake of ligand-modified nanocarriers by macrophages during systemic circulation [Citation12]. However, this strategy is rather limited because the ligand modified on the surface of nanoparticles could be screened by the formed protein corona, which masks the targeting capability and influences the biological effects of nanoparticles [Citation78,Citation79]. This might partially explain why several actively targeted nanomedicines have not been approved for clinical practice [Citation80]. In this study, we successfully developed an integrative approach to actualize such an absconding effect.
As demonstrated above, the retention of CD47 on hybrid vesicles might ensure a macrophage escape effect. Importantly, when the “eat me” and “don’t eat me” strategies were utilized simultaneously, their repressive effect on macrophage decrease was most evident. For example, M-EV+Hybrid c(RGDm7)-LS exhibited 120.96% lower cellular endocytosis than c(RGDm7)-LS. This result was likely attributable to the combination of both M-EV pretreatment and the functional contribution of CD47 as the “don’t eat me” signal of exosomes in hybrid nanocarriers. In short, this investigation confirmed the efficiency of the “don’t eat me” strategy in macrophages.
Moreover, the cellular uptake in A549 cells confirmed that the conjugation of c(RGDm7) significantly enhanced the in vitro targeting ability of nanocarriers to αvβ3-expressing tumour cells. In other words, c(RGDm7) functionalization provided an effective “eat me” signal for tumour cells. Additionally, the presence of CD47 on hybrid nanocarriers did not attenuate the targeting ability of c(RGDm7)-LS, although it did not enhance such effect significantly, as evidenced by the comparison between LS and Hybrid LS, as well as between c(RGDm7)-LS and Hybrid c(RGDm7)-LS.
In vivo and ex vivo fluorescence observations clearly revealed superior phagocytosis escape of hybrid vesicles compared with that of non-hybrid vesicles. Collectively, with the synergistic effects of CD47 expression as a “don’t eat me” signal and the macrophage-targeting ability, M-EV pretreatment significantly reduced the distribution of Hybrid LS and Hybrid c(RGDm7)-LS in the liver and spleen. It has been reported that macrophages efficiently accumulate EVs through the recognition of phosphatidylserine on the outer leaflet of EVs [Citation81]. These previous studies might partially explain our findings.
Interestingly, although the injected dose was the same, the sum of the fluorescence intensity was different in each mouse group at each time point. The reason seems that the different types of nanocarriers display different body distribution patterns, which result in different detection efficacy. Briefly, the lipid vesicles in blood are invisible because the blood strongly absorbs and scatters light from the visible part of the spectrum [Citation82] and only these in organs and tissues are detectable by this living imaging technique. For the mouse group with less sum of fluorescence intensity, the rest of labelled nanocarriers may stay in blood system or was already eliminated from the body.
Indeed, the in vivo and ex vivo imaging cannot detect the fluorescent nanocarriers in the bloodstream. Therefore, we have conducted a direct determination of the blood drug concentration for encapsulated GE and DOX at different time points (). Importantly, the pharmacokinetic profiles of the designed nanoformulations were improved by the introduction of our “eat me/don’t eat me” strategy. To our knowledge, no active targeted nanomedicine has advanced into phase III clinical trials [Citation83]. It has been reported that ligands functionalized on the surfaces of nanoparticles can undermine the long-circulating property of PEGylation [Citation84]. As verified by our study, however, M-EV pretreatment could improve the blood circulation of hybrid targeted vesicles while preserving their targeting ability. The circulation kinetics of nanocarriers are largely determined by their interaction with MPS. Consequently, the lipid vesicles with low MPS clearance circulated in the blood stream for a prolonged time. Thus, the prolonged blood circulation of our “eat me/don’t eat me” strategy is of considerable significance, which further supports the enhanced tumour accumulation observed in this study.
Finally, it was demonstrated that both c(RGDm7) functionalization and M-EV played crucial roles in terms of inhibiting tumour growth, prolonging the survival of test animals, promoting apoptosis, and suppressing angiogenesis. Together, these studies strongly supported that combination chemotherapy, pretreatment with M-EV, presence of “don’t eat me” signal and c(RGDm7) modification bestowed the nanoformulation with significantly better therapeutic effects, suggesting its utility as a potential treatment regimen.
In summary, we designed an EV-based “eat me/don’t eat me” strategy for active anticancer therapy. To address the issue of MPS-induced nanoparticle elimination, DC2.4-derived EVs modified with cationized mannan were utilized to saturate macrophages, and a type of lipid vesicles was fused with exosomes expressing CD47 (don’t eat me signal) to further minimize MPS uptake. Meanwhile, a type of cyclic homing peptide as an “eat me” approach was used to modify the hybrid vesicles loaded with both a cytotoxic agent and EGFR inhibitor. Via systematic in vitro and in vivo investigations, this strategy was demonstrated to be favourable in terms of reducing endocytosis by macrophages, enhancing the uptake of tumour cells, prolonging the in vivo circulation time, lowering the distribution to the liver and spleen, increasing tumour accumulation, suppressing tumour growth, improving survival rates, inducing tumour apoptosis, inhibiting angiogenesis and reducing the side effects of free drugs. Because MPS-mediated clearance is a long-term and huge challenge in the nanomedicine field that enormously hinders the development of nanodrugs, the current proof-of-concept study may be of considerable significance.
Scheme 1. “Eat me/don’t eat me” strategy to achieve macrophage escape and improve the tumour targeting efficiency. The cationized mannan-modified extracellular vesicles derived from DC2.4 cells (M-EV) are i.v. injected into the mouse via the tail vein to saturate the receptors of macrophages, prior the administration of a subsequent dose of exosome-hybrid targeted vesicles co-loaded with doxorubicin (DOX) and gefitinib (GE), namely “Hybrid c(RGDm7)-LS-GE/DOX”, thereby minimizing their sequestration by the liver, improving the tumour accumulation and enhancing the therapeutic efficacy on lung cancer.
Supplemental Material
Download MS Word (15.7 MB)Acknowledgements
We thank Dr. Jing Wang in the State Key Laboratory of Natural and Biomimetic Drug, Peking University for the experimental assistance of surface plasmon resonance technology.
Disclosure statement
The authors declare no conflict of interest.
Supplementary material
Supplemental data of this article can be accessed here.
Additional information
Funding
References
- Dobrovolskaia MA, Aggarwal P, Hall JB, et al. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5:487–16.
- Rao L, Bu LL, Xu JH, et al. Red blood cell membrane as a biomimetic nanocoating for prolonged circulation time and reduced accelerated blood clearance. Small. 2015;11:6225–6236.
- Tsoi KM, MacParland SA, Ma XZ, et al. Mechanism of hard-nanomaterial clearance by the liver. Nat Mater. 2016;15:1212.
- Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014.
- Zhang YN, Poon W, Tavares AJ, et al. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348.
- Guilliams M, Dutertre CA, Scott CL, et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity. 2016;45:669–684.
- Lopez BG, Tsai MS, Baratta JL, et al. Characterization of Kupffer cells in livers of developing mice. Comp Hepatol. 2011;10:2.
- Gregory SH, Wing EJ. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J Leukoc Biol. 2002;72:239–248.
- Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14:996–1006.
- Yu M, Zheng J. Clearance pathways and tumor targeting of imaging nanoparticles. ACS Nano. 2015;9:6655–6674.
- Walkey CD, Olsen JB, Guo H, et al. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134:2139–2147.
- Dai Q, Walkey C, Chan WC. Polyethylene glycol backfilling mitigates the negative impact of the protein corona on nanoparticle cell targeting. Angew Chem Int Ed Engl. 2014;53:5093–5096.
- Salvati A, Pitek AS, Monopoli MP, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8:137–143.
- Ishida T, Maeda R, Ichihara M, et al. Accelerated clearance of PEGylated liposomes in rats after repeated injections. J Control Release. 2003;88:35–42.
- Ishida T, Masuda K, Ichikawa T, et al. Accelerated clearance of a second injection of PEGylated liposomes in mice. Int J Pharm. 2003;255:167–174.
- Ishida T, Ichihara M, Wang X, et al. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J Control Release. 2006;112:15–25.
- Rodriguez PL, Harada T, Christian DA, et al. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339:971–975.
- Thamphiwatana S, Angsantikul P, Escajadillo T, et al. Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc Natl Acad Sci U S A. 2017;114:11488–11493.
- Yoo JW, Irvine DJ, Discher DE, et al. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521–535.
- Parodi A, Quattrocchi N, van de Ven AL, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8:61–68.
- He H, Guo C, Wang J, et al. Leutusome: A biomimetic nanoplatform integrating plasma membrane components of leukocytes and tumor cells for remarkably enhanced solid tumor homing. Nano Lett. 2018;18:6164–6174.
- Fang RH, Hu CM, Luk BT, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181–2188.
- Gulati NM, Stewart PL, Steinmetz NF. Bioinspired shielding strategies for nanoparticle drug delivery applications. Mol Pharm. 2018;15:2900–2909.
- Tavares AJ, Poon W, Zhang YN, et al. Effect of removing Kupffer cells on nanoparticle tumor delivery. Proc Natl Acad Sci U S A. 2017;114:10871–10880.
- Diagaradjane P, Deorukhkar A, Gelovani JG, et al. Gadolinium chloride augments tumor-specific imaging of targeted quantum dots in vivo. ACS Nano. 2010;4:4131–4141.
- Liu T, Choi H, Zhou R, et al. RES blockade: A strategy for boosting efficiency of nanoparticle drug. Nano Today. 2015;10:11–21.
- Sun X, Yan X, Jacobson O, et al. Improved tumor uptake by optimizing liposome based RES bockade strategy. Theranostics. 2017;7:319–328.
- Tang Y, Wang X, Li J, et al. Overcoming the reticuloendothelial system barrier to drug delivery with a “don’t-eat-us” strategy. ACS Nano. 2019;13:13015–13026.
- van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–228.
- Soo CY, Song Y, Zheng Y, et al. Nanoparticle tracking analysis monitors microvesicle and exosome secretion from immune cells. Immunology. 2012;136:192–197.
- Wiklander OP, Nordin JZ, O’Loughlin A, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316.
- Charoenviriyakul C, Takahashi Y, Morishita M, et al. Cell type-specific and common characteristics of exosomes derived from mouse cell lines: yield, physicochemical properties, and pharmacokinetics. Eur J Pharm Sci. 2017;96:316–322.
- Wen SW, Sceneay J, Lima LG, et al. The biodistribution and immune suppressive effects of breast cancer-derived exosomes. Cancer Res. 2016;76:6816–6827.
- Wei G, Jie Y, Haibo L, et al. Dendritic cells derived exosomes migration to spleen and induction of inflammation are regulated by CCR7. Sci Rep. 2017;7:42996.
- Batrakova EV, Kim MS. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Control Release. 2015;219:96–405.
- Kaur S, Singh SP, Elkahloun AG, et al. CD47-dependent immunomodulatory and angiogenic activities of extracellular vesicles produced by T cells. Matrix Biol. 2014;37:49–59.
- Kamerkar S, LeBleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503.
- Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24:225–232.
- Du W, Fan Y, Zheng N, et al. Transferrin receptor specific nanocarriers conjugated with functional 7peptide for oral drug delivery. Biomaterials. 2013;34:794–806.
- Melo SA, Luecke LB, Kahlert C, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–182.
- Melo SA, Sugimoto H, O’Connell JT, et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26:707–721.
- El-Andaloussi S, Lee Y, Lakhal-Littleton S, et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012;7:2112–2126.
- Jo J, Ikai T, Okazaki A, et al. Expression profile of plasmid DNA by spermine derivatives of pullulan with different extents of spermine introduced. J Control Release. 2007;118:389–398.
- Scott BL, Van Komen JS, Liu S, et al. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 2003;372:274–300.
- Chen H, Kim S, Li L, et al. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Förster resonance energy transfer imaging. Proc Natl Acad Sci U S A. 2008;105:6596–6601.
- Haran G, Cohen R, Bar LK, et al. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151:201–215.
- Auzzas L, Zanardi F, Battistini L, et al. Targeting alphavbeta3 integrin: design and applications of mono- and multifunctional RGD-based peptides and semipeptides. Curr Med Chem. 2010;17:1255–1299.
- Choi N, Kim SM, Hong KS, et al. The use of the fusion protein RGD-HSA-TIMP2 as a tumor targeting imaging probe for SPECT and PET. Biomaterials. 2011;32:7151–7158.
- Belhadj Z, Ying M, Cao X, et al. Design of Y-shaped targeting material for liposome-based multifunctional glioblastoma-targeted drug delivery. J Control Release. 2017;255:132–141.
- Nel A, Ruoslahti E, Meng H. New insights into “Permeability” as in the enhanced permeability and retention effect of cancer nanotherapeutics. ACS Nano. 2017;11:9567–9569.
- Jo J, Okazaki A, Nagane K, et al. Preparation of cationized polysaccharides as gene transfection carrier for bone marrow-derived mesenchymal stem cells. J Biomater Sci Polym Ed. 2010;21:185–204.
- Thomsen LB, Lichota J, Kim KS, et al. Gene delivery by pullulan derivatives in brain capillary endothelial cells for protein secretion. J Control Release. 2011;151:45–50.
- Pinkerton NM, Grandeury A, Fisch A, et al. Formation of stable nanocarriers by in situ ion pairing during block-copolymer-directed rapid precipitation. Mol Pharm. 2013;10:319–328.
- Chen H, Wang Y, Yao Y, et al. Sequential delivery of cyclopeptide RA-V and doxorubicin for combination therapy on resistant tumor and in situ monitoring of cytochrome c release. Theranostics. 2017;7:3781–3793.
- Taylor PR, Martinez-Pomares L, Stacey M, et al. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944.
- Shibaguchi K, Tamura A, Terauchi M, et al. Mannosylated polyrotaxanes for increasing cellular uptake efficiency in macrophages through receptor-mediated endocytosis. Molecules. 2019;24:439.
- Zhang J, Li H, Wu Q, et al. Tumoral NOX4 recruits M2 tumor-associated macrophages via ROS/PI3K signaling-dependent various cytokine production to promote NSCLC growth. Redox Biol. 2019;22:101116.
- Shi C, Guo D, Xiao K, et al. A drug-specific nanocarrier design for efficient anticancer therapy. Nat Commun. 2015;6:7449.
- Takahashi Y, Nishikawa M, Shinotsuka H, et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol. 2013;165:77–84.
- Saunderson SC, Dunn AC, Crocker PR, et al. CD169 mediates the capture of exosomes in spleen and lymph node. Blood. 2014;123:208–216.
- Cai W, Chen X. Anti-angiogenic cancer therapy based on integrin alphavbeta3 antagonism. Anticancer Agents Med Chem. 2006;6:407–428.
- Dechantsreiter MA, Planker E, Matha B, et al. N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem. 1999;42:3033–3040.
- He Y, Su Z, Xue L, et al. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J Control Release. 2016;229:80–92.
- Sher T, Dy GK, Adjei AA. Small cell lung cancer. Mayo Clin Proc. 2008;83:355–367.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388.
- Mok TS, Wu YL, Yu CJ, et al. Randomized, placebo-controlled, phase II study of sequential erlotinib and chemotherapy as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2009;27:5080–5087.
- Lee MJ, Ye AS, Gardino AK, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–794.
- Zhang C, Jugold M, Woenne EC, et al. Specific targeting of tumor angiogenesis by RGD-conjugated ultrasmall superparamagnetic iron oxide particles using a clinical 1.5-T magnetic resonance scanner. Cancer Res. 2007;67:1555–1562.
- Nieberler M, Reuning U, Reichart F, et al. Exploring the role of RGD-recognizing integrins in cancer. Cancers (Basel). 2017;9:116.
- Vitale DL, Spinelli FM, Del Dago D, et al. Co-treatment of tumor cells with hyaluronan plus doxorubicin affects endothelial cell behavior independently of VEGF expression. Oncotarget. 2018;9:36585–36602.
- Hirata A, Ogawa S, Kometani T, et al. ZD1839 (Iressa) induces antiangiogenic effects through inhibition of epidermal growth factor receptor tyrosine kinase. Cancer Res. 2002;62:2554–2560.
- Chatterjee K, Zhang J, Honbo N, et al. Doxorubicin cardiomyopathy. Cardiology. 2010;115:155–162.
- Ohara Y, Oda T, Yamada K, et al. Effective delivery of chemotherapeutic nanoparticles by depleting host Kupffer cells. Int J Cancer. 2012;131:2402–2410.
- Zhu S, Niu M, O’Mary H, et al. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Mol Pharm. 2013;10:3525–3530.
- Gao J, Chen P, Singh Y, et al. Novel monodisperse PEGtide dendrons: design, fabrication, and evaluation of mannose receptor-mediated macrophage targeting. Bioconjug Chem. 2013;24:1332–1344.
- Münter R, Kristensen K, Pedersbæk D, et al. Dissociation of fluorescently labeled lipids from liposomes in biological environments challenges the interpretation of uptake studies. Nanoscale. 2018;10:22720–22724.
- Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335.
- Tenzer S, Docter D, Kuharev J, et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol. 2013;8:772–781.
- Corbo C, Molinaro R, Taraballi F, et al. Unveiling the in vivo protein corona of circulating leukocyte-like carriers. ACS Nano. 2017;11:3262–3273.
- Hadjidemetriou M, Al-Ahmady Z, Mazza M, et al. In vivo biomolecule corona around blood-circulating, clinically used and antibody-targeted lipid bilayer nanoscale vesicles. ACS Nano. 2015;9:8142–8156.
- Matsumoto A, Takahashi Y, Nishikawa M, et al. Role of phosphatidylserine-derived negative surface charges in the recognition and uptake of intravenously injected B16BL6-derived exosomes by macrophages. J Pharm Sci. 2017;106:168–175.
- Negwer I, Best A, Schinnerer M, et al. Monitoring drug nanocarriers in human blood by near-infrared fluorescence correlation spectroscopy. Nat Commun. 2018;9:5306.
- Rosenblum D, Joshi N, Tao W, et al. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9:1410.
- Suk JS, Xu Q, Kim N, et al. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28–51.