
ABSTRACT
Reimbursement decisions on new oncology drugs are now often made while uncertainty remains about a drug's risk–benefit profile. One consequence of this is a delay in patient access to valuable new medicines. We share our perspectives on strategies to mitigate sources of uncertainty in the health technology assessment process. These include flexible approaches for evaluating the additional benefit, such as better use of surrogate endpoints and health-related quality of life data, and renewed research efforts to define the optimal target population and generate real-world evidence post-authorisation.
Introduction
Reimbursement and price-setting for innovative cancer drugs is increasingly challenging. A major factor is the introduction of accelerated approval programmes. The Adaptive Pathways concept was introduced by the European Medicines Agency (EMA) in 2016 to enable early market authorisation of potentially beneficial new treatments for patient groups with unmet medical needs [Citation1]. The evidence for new oncology drugs approved through such pathways attracts public opinion, the media and policymakers, but can lack robustness [Citation2]. A further challenge is that target populations for some new drugs are not fully defined at the time of authorisation.
These ‘uncertainties’ about a drug’s risk–benefit profile present a challenge for payers. They already face a rapidly changing cancer treatment landscape and increasing drug volume, including personalised medicines with requirements for molecular testing and novel treatment strategies, which necessitate deviation from established pricing models. The consequences of ‘uncertainty’ for payers are to negotiate the drug price/budget impact through financial agreements, identify cost offsets or delay approval. This results in heterogeneity in the reimbursement and price-setting process across Europe. Delaying access to innovative cancer therapies due to uncertainty around therapeutic benefits might lead to unnecessary morbidity and mortality if older, less safe and less efficacious therapies are not replaced [Citation3], and counteracts the attempts of regulatory agencies to facilitate faster patient access, as part of a general effort to transform research into accepted clinical practice.
In this article, we share our perspectives on current challenges in the health technology assessment (HTA) of novel oncology drugs and strategies to mitigate sources of uncertainty.
Evaluating benefit: relevant endpoints from clinical trials
Overall survival and surrogate endpoints
Overall survival (OS) is regarded as the gold standard for demonstrating efficacy in oncology trials [Citation4] and is the preferred criterion for HTA [Citation5] (). Increasingly, there are situations where OS data are not available for HTA, even where drugs have been granted regular marketing authorisation without obligation to conduct further studies [Citation2]. Demonstrating an improvement in OS over standard care may not always be feasible in a clinical trial. For example, where studies in earlier lines of therapy allow patients to crossover on progression, OS results can be difficult to interpret due to the confounding effects of subsequent treatments. In many tumour types, survival has been extended substantially by innovative therapies (e.g., imatinib for chronic myeloid leukaemia [Citation6]; pertuzumab for first-line, metastatic HER2+ breast cancer [Citation7]; androgen receptor inhibitors and immunotherapy for metastatic prostate cancer [Citation8–Citation10]; proteasome inhibitors and immunomodulators for multiple myeloma [Citation11]) and median OS may not be reached within the timeframes of a pivotal trial and, most importantly, may be modified by later lines of therapies, which are neither well established by the literature nor by robust evidence. Similarly, in some haematological malignancies (e.g., indolent non-Hodgkin’s lymphomas, chronic lymphocytic leukaemia, multiple myeloma), a long follow-up period is required to demonstrate meaningful gain in OS due to the disease course [Citation12]. In trials of agents with targeted mechanisms of action and rare tumour types, recruiting sufficient eligible patients to provide the power to demonstrate an OS benefit is challenging.
Figure 1. Current and potential future use of endpoints in health technology assessment of oncology drugs.
HRQoL, health-related quality of life; HTA, health technology assessment; MCID, minimal clinically important difference; OS, overall survival; PFS, progression-free survival.
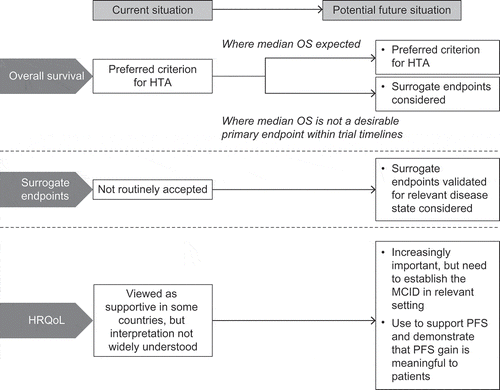
OS data will be required by HTA agencies for settings where a median OS can be realistically reached within the clinical study. However, OS may not always be a realistic endpoint and, in some contexts, payers may want to consider alternatives and may see the potential value of multi-criteria decision-making.
Accordingly, drug developers will be required to demonstrate why OS is not a desirable endpoint. Where there is consensus that extending OS is not the primary objective, one solution is to demonstrate significant improvements in other aspects of treatment that are meaningful to all stakeholders, e.g., improving alternative patient-relevant endpoints; avoiding toxicities and harmful effects associated with standard treatments or procedures; delaying the need for more expensive or more aggressive treatments; or extending disease-free survival such that patients can benefit from the next generation of treatments [Citation13]. Interestingly, the German Institute for Quality and Efficiency in Healthcare (IQWiG), in its assessment of allogeneic stem cell transplant, made the comment that progression-free survival (PFS) might be a patient-relevant endpoint in cases where there is no further option, i.e., PFS means a sure progression to death [Citation14].
The majority of oncology drugs approved by the EMA and US Food and Drug Administration (FDA) without proven OS benefit at time of authorisation are conditionally approved on surrogate endpoints of PFS or objective response (OR) [Citation2,Citation15,Citation16]. Findings from clinical trials and meta-analyses on the relationship of surrogate endpoints to OS are heterogeneous and vary by cancer type and setting [Citation17–Citation26]. For these surrogate endpoints to be acceptable for HTA, there will need to be demonstration of a clear relationship with OS that has been validated in clinical studies in the relevant disease setting and target population. This will require effort by the developer and/or clinical societies in multiple clinical validation studies to confirm the reliability of surrogates to predict hard outcomes such as OS. Investigating other surrogate markers, such as biomarkers and minimal residual disease (haematological malignancies), should also be of value, particularly for targeted therapies.
Health-related quality of life
In cases where there is no OS improvement, and where PFS alone is not shown to be an appropriate surrogate, improvement in PFS with a corresponding improvement in health-related quality of life (HRQoL) may be acceptable for HTA (). HRQoL data could demonstrate whether delaying progression constitutes a clinically meaningful improvement for the patient, taking into account the impact of side effects as well as morbidity benefits. This is particularly important when the indicator of progression is radiographical rather than a symptomatic change in the patient’s health state. HRQoL data, measured at appropriate intervals before or during progression, may also demonstrate a benefit from delaying the need for subsequent treatment with more aggressive regimens that have a negative impact on patients’ QoL, e.g., chemotherapy.
HRQoL is an important element of the health gain from cancer drugs, but has not been widely accepted by payers across all jurisdictions. Many are unfamiliar with its use in healthcare decision-making, as HRQoL endpoints are not always routinely incorporated in pivotal trial designs [Citation27]. Variation between HTAs might be explained by differences in the level of understanding and interpretation of HRQoL data and how they translate into clinically meaningful gains [Citation27]. As patients take a more prominent role in decision-making [Citation28,Citation29], HRQoL data are likely to become of greater importance in reimbursement. However, there remains a need to demonstrate to payers that HRQoL is not an absolute measure and that its value to the patient varies in different conditions and disease states. Some patients might regard HRQoL as more important than OS [Citation30], e.g., patients with recurrence may value improvement in HRQoL more highly than those with early, asymptomatic disease [Citation31], and patients receiving chemotherapy may value improvement in fatigue [Citation32]. There is also a requirement for investment in studies to establish the minimum clinically-important difference in a patient-reported outcome instrument in the relevant patients and disease setting.
Several tools allow mapping HRQoL into utilities to allow Quality Adjusted Life Years (QALY) to be calculated. Indeed utilities and QALYs are commonplace in current oncology trial development, but they are only widely accepted in some jurisdictions, possibly because of the same concerns as HRQoL. Some authors [Citation33] have advocated for the usage of Q-TWIST (Quality-Adjusted Time Without Symptoms), or even ASCO (American Association for Clinical Oncology) has put forward a Value Framework [Citation34], but neither of these metrics are routinely being used by payers.
Target populations
Identifying the optimal target populations for new drugs is becoming more complex due to use of multiple lines of treatment, resulting in defined groups of patients across the course of the disease. Yet, regulators often fail to make the label specific to the studied population, making scientific assumptions that are not acceptable to payers and approving indications for populations not explicitly covered by the clinical trial. Despite the potential economic benefits of such decisions, manufacturers and regulators should take into account the fact that such decisions are likely to delay and restrict patient access to therapy.
Precision medicine is providing the tools to discriminate populations and define those most likely to benefit from novel drugs. This can help with lowering of the number needed to treat to prevent one adverse outcome, which might increase efficiency of use as well as payers’ willingness to pay. Again, developers need to invest in translational research studies early in the development programme, and to engage and partner with clinical societies, regulators and HTAs to validate biomarkers; and understand their role in the biology of the disease in order to establish the medical need for that target population. Economic requirements for biomarker testing must be addressed early in the development programme [Citation35]. Defining an optimised target population will reduce post-approval uncertainty, and there should be a joint effort to determine future requirements for aligning access with the label, thus avoiding conditional approval, which may not be accepted across jurisdictions. Certainly, the potential of Joint Regulatory-HTA advice can help address some of the concerns, but still the recommendations are explicitly non-binding to national or sub-national payers. Interestingly, the EMA highlights itself a potential difference between ‘significant benefit of orphan medicines versus their added therapeutic value’ – and suggests ‘new areas of collaboration to explore possible synergies between HTA bodies and regulators [Citation36].
Addressing the changing cancer treatment landscape
Many cancers are now regarded as chronic diseases [Citation37], as the cancer treatment paradigm has evolved from front-line therapy and palliative care to use of multiple lines of therapy. This complicates healthcare requirements, with many patients needing long-term treatment and supportive/multidisciplinary care for managing side effects. The ‘structure–process–outcomes’ paradigm, used for evaluating the quality of healthcare, needs to be reassessed to ensure it reflects the current situation. Developers may need to better understand the acceptance and consequences of a new technology in a healthcare environment, and further invest to define the disease and ‘process’, so that the ‘outcomes’ assumptions remain relevant. The increasing role of molecular biology in cancer treatment also adds to the complexity of reimbursement due to the requirement for molecular companion diagnostics, single-gene tests or next-generation sequencing to identify eligible patients for targeted therapies.
It is important that we avoid unnecessary and obstructive complication of the HTA process. However, changes to the cancer treatment landscape, and the uncertainty about the risk–benefit profile of many new treatments, may necessitate alternative financing models, such as performance-linked, managed entry agreements [Citation38]. Implementing performance-related schemes will require a broad spectrum of evidence from randomised controlled trials and post-licensing registry studies. Establishing accessible registries and standardising how these data are used is a priority for enabling informed decision-making. As technology progresses, we can expect real-world data to become more accessible to policy makers and industry, to improve healthcare interventions and monitor the use of technologies once they have been introduced. Effort is required by all stakeholders to establish minimum quality standards on how prospectively gathered evidence (including real-world evidence) should be sourced, analysed and presented.
Concluding remarks
Reimbursement decisions on new oncology drugs are often being made, while uncertainty remains about a drug’s risk–benefit profile. Flexible approaches for evaluating the additional benefit are needed. Validated supportive endpoints, such as PFS and HRQoL, could have a more prominent role where OS is not a realistic endpoint. Research efforts should also focus on defining the target population and the disease early in the development programme, and generating evidence post-authorisation to define the risk–benefit profile.
Acknowledgments
This article was developed following a series of meetings held in Berlin, Germany between 2015 and 2017, organised and sponsored by Pfizer. We are grateful to Martin Buxton and Jean-Michel Hotton (RIP) for their valuable comments and discussion of the content.
Writing support was provided by Ashfield Healthcare, part of UDG Healthcare plc, funded by Pfizer Pharma GmbH.
Disclosure statement
Dr. Sola-Morales has consulted for most of the multinational pharmaceutical companies, including all ‘top 10’. He has received fees and honoraria for such consultancies, and is currently engaged in several projects with many of these companies. He holds ownership of several start-up companies in the healthcare field, and has had in the past stock ownership of ‘Top 10’ pharmaceutical companies. Dr. Mantovani has received research grants from Bayer, Biogen, Daiichi Sankyo, Sobi, Shire, Roche, Boehringer Ingelheim and has consulted for Bayer, Pfizer, Biogen, Roche.Dr. Timm Volmer consults for various pharmaceutical companies having products in HTA assessments. He also conducts research in areas of interest similar to the business interests of manufacturers such as Pfizer.
References
- European Medicines Agency. Final report on the adaptive pathways pilot. 2016. [cited 2017 Nov 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Report/2016/08/WC500211526.pdf
- Davis C, Naci H, Gurpinar E, et al. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European medicines agency: retrospective cohort study of drug approvals 2009-13. Bmj. 2017;359:j4530.
- Jönsson B, Wilking N. The effect of cancer drug vintage on cancer survival and mortality. Ann Oncol. 2007;18(Suppl3):iii67–5.
- Blumenthal GD, Kluetz PG, Schneider J, et al. Oncology drug approvals: evaluating endpoints and evidence in an era of breakthrough therapies. Oncologist. 2017;22(7):726–767.
- Jönsson B. Bringing in health technology assessment and cost-effectiveness considerations at an early stage of drug development. Mol Oncol. 2015;9(5):1025–1033.
- Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376:917–927.
- Swain SM, Kim SB, Cortés J, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013;14(6):461–471.
- Fizazi K, Scher HI, Molina A, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13(10):983–992.
- Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422.
- Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–1197.
- Anderson KC. The 39th David A. Karnofsky lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012;30(4):445–452.
- Smith BD, DeZern AE, Bastian AW, et al. Meaningful endpoints for therapies approved for hematological malignancies. Cancer. 2017;123(10):1689–1694.
- Thornton Snider J, Batt K, Wu Y, et al. The option value of innovative treatments for non-small cell lung cancer and renal cell carcinoma. Am J Manag Care. 2017;23(10):e340–e346.
- Institute for Quality and Efficiency in Healthcare (IQWiG). Report N17-02: Allogeneic stem cell transplantation in aggressive B-cell non-Hodgkin lymphoma and in T-cell non-Hodgkin lymphoma. Available from: https://www.iqwig.de/en/projects-results/projects/non-drug-interventions/n-projekte/n17-02-allogeneic-stem-cell-transplantation-in-aggressive-b-cell-non-hodgkin-lymphoma-and-in-t-cell-non-hodgkin-lymphoma.7810.html
- Aggarwal C, Borghaei H. Treatment paradigms for advanced non-small cell lung cancer at academic medical centers: involvement in clinical trial endpoint design. Oncologist. 2017;22(6):700–708.
- Kim C, Prasad V. Cancer drugs approved on the basis of a surrogate end point and subsequent overall survival: an analysis of 5 years of US food and drug administration approvals. JAMA Intern Med. 2015;175(12):1992–1994.
- Hasan B, Greillier L, Pallis A, et al. Progression free survival rate at 9 and 18 weeks predict overall survival in patients with malignant pleural mesothelioma: an individual patient pooled analysis of 10 European organisation for research and treatment of cancer lung cancer group studies and an independent study validation. Eur J Cancer. 2014;50(16):2771–2782.
- Beauchemin C, Cooper D, Lapierre MÈ, et al. Progression-free survival as a potential surrogate for overall survival in metastatic breast cancer. Onco Targets Ther. 2014;7:1101–1110.
- Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–172.
- Foster NR, Renfro LA, Schild SE, et al. Multitrial evaluation of progression-free survival as a surrogate end point for overall survival in first-line extensive-stage small-cell lung cancer. J Thorac Oncol. 2015;10(7):1099–1106.
- Tan A, Porcher R, Crequit P, et al. Differences in treatment effect size between overall survival and progression-free survival in immunotherapy trials: a meta-epidemiologic study of trials with results posted at ClinicalTrials.gov. J Clin Oncol. 2017;35(15):1686–1694.
- Ciani O, Davis S, Tappenden P, et al. Validation of surrogate endpoints in advanced solid tumors: systematic review of statistical methods, results, and implications for policy makers. Int J Technol Assess Health Care. 2014;30(3):312–324.
- Fiteni F, Westeel V, Bonnetain F. Surrogate endpoints for overall survival in lung cancer trials: a review. Expert Rev Anticancer Ther. 2017;17(5):447–454.
- Hellman MD, Chaft JE, William WN Jr, et al. Pathological response after neoadjuvant chemotherapy in resectable non-small-cell lung cancers: proposal for the use of major pathological response as a surrogate endpoint. Lancet Oncol. 2014;15(1):e42–e50.
- Liu LY, Yu H, Bai JL, et al. Verification of the correlation between progression-free survival and overall survival considering magnitudes of survival post- progression in the treatment of four types of cancer. Asian Pac J Cancer Prev. 2015;16(3):1001–1006.
- Pfeiffer BM, Kulakova M, Hashim M, et al. Can duration of response be used as a surrogate endpoint for overall survival in advanced non-small cell lung cancer? J Clin Oncol. [ Abstract] 2018;36(Suppl 15):9082.
- Brogan AP, DeMuro C, Barrett AM, et al. Payer perspectives on patient-reported outcomes in health care decision making: oncology examples. J Manag Care Spec Pharm. 2017;23(2):125–134.
- Hashem F, Merritt R. Supporting patients self-managing respiratory health: a qualitative study on the impact of the breathe easy voluntary group network. ERJ Open Res. 2018;4(1):pii:00076–2017.
- Moreira T. Understanding the role of patient organizations in health technology assessment. Health Expect. 2015;18(6):3349–3357.
- Dilla T, Lizan L, Paz S, et al. Do new cancer drugs offer good value for money? The perspectives of oncologists, health care policy makers, patients, and the general population. Patient Prefer Adherence. 2016;10:1–7.
- Siddiqi A, Given CW, Given B, et al. Quality of life among patients with primary, metastatic and recurrent cancer. Eur J Cancer Care (Engl). 2009;18(1):84–96.
- Bottomley A. The cancer patient and quality of life. Oncologist. 2002;7(2):120–125.
- Solem CT, Kwon Y, Shah RM, et al. Systematic review and benchmarking of quality-adjusted time without symptoms or toxicity (Q-TWiST) in oncology. Expert Rev Pharmacoecon Outcomes Res. 2018;188(3):245–253.
- Schnipper LE, Davidson NE, Wollins DS, et al. American society of clinical oncology statement: a conceptual framework to assess the value of cancer treatment options. J Clin Oncol. 2015;33(23):2563–2577.
- Miquel-Cases A, Schouten PC, Steuten LM, et al. (Very) Early technology assessment and translation of predictive biomarkers in breast cancer. Cancer Treat Rev. 2017;52:117–127.
- EMA (European Medicines Agency). Annual Report 2017, London UK, 2017
- Lage A, Crombet T. Control of advanced cancer: the road to chronicity. Int J Environ Res Public Health. 2011;8(3):683–697.
- Pauwels K, Huys I, Vogler S, et al. Managed entry agreements for oncology drugs: lessons from the European experience to inform the future. Front Pharmacol. 2017;8:171.