
ABSTRACT
Background: Cell and gene therapies are associated with uncertainty around their value claims at launch due to limitations of supporting clinical data; furthermore, their high costs present affordability issues for payers. Outcomes-based reimbursement can reduce payer decision uncertainty and improve patient access, however, requires data collection infrastructure and practice to be operational.
Objective: To identify indications most likely to see launch of cell or gene therapies in the UK over the next five years, and to perform a qualitative assessment of how conducive the existing data collection infrastructure and clinical practice is in facilitating adoption of outcomes-based reimbursement in the corresponding indications.
Methodology: Through secondary research, we identified target indications for cell or gene therapies at a mature clinical development stage (Phase III) with EU and/or US trial sites, and assessed availability of relevant data collection infrastructures in the UK. Secondary research findings were validated through primary research (expert interviews). Key parameters considered for the suitability of existing data collection infrastructure in supporting outcomes-based reimbursement include time horizon of data collection, whether data entry is mandatory and whether infrastructure is product or therapy area-specific.
Findings: We identified 58 cell or gene therapies, spanning 47 indications, 20 of which are in oncology. Oncology seems well placed for outcomes data collection (through the mandatory Systemic Anti-Cancer Treatment database), however data entry compliance can be an issue (due to resource limitations), and upgrading will be needed for the purpose of outcomes-based reimbursement. Among non-oncology indications ~two-thirds have data collection infrastructures in place, but only three come close to the requirements for outcomes-based reimbursement.
Conclusions: Existing data collection infrastructure in indications with potential cell or gene therapies launches in the next five years in the UK is overall not sufficient to facilitate outcomes-based reimbursement.
Introduction and background
As more advanced therapy medicinal products (ATMPs) obtain marketing authorisation, enabling patient access to these therapeutic innovations has become an increasingly debated topic. The advent of high-value cell and gene therapies launching has created a challenging balancing act for decision-makers in the National Health Service (NHS) in terms of ensuring patient access on one hand, and affordability for the NHS on the other, while rewarding manufacturers for improving patient health. Striking this balance is a particularly pertinent challenge in the case of ATMPs, firstly because the cost of developing, manufacturing, and delivering them is much higher than for the more conventional medicines (e.g., small molecules and biologics) and minimally manipulated cells; and secondly, because many cell and gene therapies have the potential to deliver life-long health improvements through a one-off treatment, rather than a long-term drug regimen of repeated administrations. Manufacturers therefore need to convincingly make the case for this long-term health benefit to regulators like the European Medicines Agency (EMA) and to health technology assessment (HTA) bodies and commissioners in the NHS [Citation1]. This is challenging as a new therapy typically will have one or two years of clinical trial data at launch, which is often not sufficient to provide conclusive proof of the long-term safety and/or benefit in the real world. Furthermore, many cell and gene therapies often target rare and/or severe indications, which makes it harder both to recruit patients and to design a trial according to the randomised controlled trial (RCT) ‘gold standard’. As a result, many pivotal trials in the cell and gene therapy space are conducted as e.g., small, single-arm trials with a short duration [Citation2]. This can reduce the statistical certainty in the results, making it more difficult both for regulators and payers to weigh the potential risks and benefits of adopting the new therapy against each other. Furthermore in the absence of an RCT design, making the comparative effectiveness value claims can be challenging.
From a regulatory perspective, the data uncertainty associated with cell and gene therapies has commonly been addressed in part by requiring that the manufacturer carries out post-marketing studies and submit results for review to the EMA [Citation3]. The duration of the post-marketing follow-up is established on a case-by-case basis, but for many gene therapies (e.g., those using integrating vectors or have the potential for latency followed by reactivation) it is usually an expectation to follow patients for 15 years [Citation4]. The marketing authorisation recently granted by the EMA for Yescarta® illustrates this further, where the requirement is to follow patients for 20 years [Citation5].
Data uncertainty is a challenge also for HTA bodies and payers, who want to be reasonably confident that the patient benefit seen in the trial will materialise in the real world and long-term, and thus that the NHS gets the value it pays for [Citation6]. Traditionally, payers have favoured simple discounts, paired with the potential for treatment discontinuation, to manage this uncertainty, in order to limit budget impact and the potential for overpayment (e.g., if the therapy does not perform as in the trial) [Citation7]. While this approach makes sense for managing conventional drugs (e.g., that are part of a regimen with repeated administrations), it is less useful for one-off cell and gene therapies. If the conventional model of paying for the treatment in full upfront is applied to one-off therapies, it will not be possible to terminate the treatment, and thereby limit the financial exposure in cases where the desired health outcome is not achieved. This creates a challenge both for payers, who face a substantial upfront payment and uncertain outcomes, and for manufacturers who struggle to get their therapies adopted under terms that are commercially viable.Footnote1Footnote2
The experience of the ATMPs that have launched to date illustrates this. No ATMP has yet achieved widespread reimbursement and access across the five biggest European countries (EU5: France, Germany, Italy, Spain and the UK), and as shows, among those that have been reimbursed, many have been restricted beyond the regulatory label.
Table 1. Reimbursement status in EU5 (France, Germany, Italy, Spain and the UK) of cell and gene therapies with EMA marketing authorisationFootnote1.
Against this backdrop, many stakeholders have sought to find a solution that enables patients to benefit from therapeutic innovation, while addressing HTA bodies’ and healthcare payers’ concerns around data uncertainty [Citation24]. Performance- or outcomes-based reimbursement schemes, whereby payments to manufacturers are made conditional on achieving and/or sustaining a certain patient benefit, is a means that has been discussed extensively in the literature [Citation1,Citation7,Citation24–Citation29]. In practice, however, payers and HTA bodies in Europe have historically conveyed a low appetite for reimbursement schemes that require the collection of outcomes data, due to concerns around their administrative complexity [Citation30]. Efforts to establish data management infrastructures that can link patient outcomes and reimbursement exist in countries such as Italy and Estonia, but are scarce elsewhere in Europe [Citation31,Citation32].
Despite the complexity in executing such schemes, the opportunity they present in terms of reducing data uncertainty and enabling patient access mean they remain an option that is often considered. A notable example is in the regenerative medicines study of England’s main HTA body, NICE, where strategies for managing decision uncertainty and enabling access to high-cost one-off therapies such as cell and gene therapies was exploredFootnote3 [Citation33]. One of the solutions considered in this exercise is where a one-off therapy is paid for as if it were a chronic treatment. NICE dubbed this ‘lifetime leasing’, and described it as paying for the one-off therapy ‘…for each period of time that the health [benefit] is delivered at the individual patient level. That is, if the observed effectiveness in clinical practice was equal to the expected effectiveness, the manufacturer would receive the full value of the technology over the agreed period. However, if observed effectiveness was less than expected, payment would stop and the risk to the health system of over-payment would be limited.’ [Citation33]
This means that payments for a one-off therapy would be staged over time, and that the NHS would only be liable to pay as long as the pre-agreed patient health outcome materialises and is sustained. There are several important and beneficial implications to this approach. Firstly, it reduces the uncertainty around the cost-effectiveness of the therapy, which is one of the central factors affecting NICE’s recommendation for reimbursement in the NHS. Secondly, it reduces the budget impact at launch, which improves (short-term) affordability. Thirdly, from the patients’ point of view, this provides an opportunity to speed up access to therapeutic innovation. Lastly, from the manufacturers’ point of view, this can provide an alternative to applying simple discounts on the upfront payment, which reduce the reward for the therapeutic innovation and diminishes the value of therapies with long-term benefits.Footnote4
Research objectives
Despite being a conceptually fairly simple idea, an outcome-based reimbursement scheme like the staged payment approach considered by NICE can only function properly if it is rooted in a data collection practice and infrastructure that provides the appropriate framework for such schemes.
In previous work, we built on the NICE regenerative medicines study [Citation33] to understand the cost and resource implications of implementing an outcomes-based, staged payment approach like ‘lifetime leasing’ in acute lymphoblastic leukaemia (ALL) [Citation34]. Our findings show that in ALL, there is a requirement to follow patients routinely and collect their data into the Systemic Anti-Cancer Treatment (SACT) database [Citation35], meaning that an infrastructure is already in place that could potentially support the adoption of an outcomes-based reimbursement scheme.
However, the question remains what the situation is in the other therapy areas that are likely to see launches of cell and gene therapies in the coming years. The objective of this research is therefore two-fold:
To provide a top-level scan of the indications most likely to see a launch of a cell or gene therapy (ATMP) in the UK over the next five years; and
To perform a qualitative assessment of what (if any) data collection infrastructure and practice is in place in the UK in the relevant indications, and to provide directional insights on how conducive the current environment is for facilitating the adoption of an outcomes-based reimbursement scheme
Methodology
Identifying the indications most likely to see launch of cell or gene therapies in the UK over the next five years
In order to identify the indications most likely to see a launch of cell or gene therapies in the UK over the next five years we:
Identified cell or gene therapies at a mature development stage, i.e., Phase III clinical trial (or in registrational trial stage where this is identifiableFootnote5 ) with EU and/or US trial sites, as these are more likely to enter the UK market in the time horizon of our analysis (unlike trials in geographies with distinctly different regulatory frameworks, e.g., China, Korea), and
Recorded the target indications for the products/trials identified
A number of sources were screened through secondary research during May 2018 to identify cell and gene therapies in clinical trials. The sources of information used include:
Data from the Alliance for Regenerative Medicine (ARM) detailing the global Phase III cell and gene therapy trials (as used in their Quarterly Data Reports [Citation36])
The European Clinical Trial Registry [Citation37]
The Advanced Therapies Investment Report 2017 from Phacilitate [Citation38]
The horizon scanning services provided by the UK Medicines Information (UKMi) [Citation39] and the UK Innovation Observatory [Citation40]
Review of company pipelines online
For each of the results we detailed:
Product names
Sponsors/manufacturers
Trial stage
Indications and therapeutic category
Whether the trial has sites in the European Union (EU) and/or the USA (US)
Identifying the existing UK data collection infrastructure and practice
We applied a combination of secondary and primary research to determine what data collection infrastructure exists in the UK, and how it is used in routine practice.
Secondary research: Internet searches to identify potentially relevant UK registries and databases in the indications identified
Primary research: Structured interviews with key opinion leader (KOLs) clinicians in the different target indications to test our secondary research findings, and to explore the extent to which the current environment can facilitate long-term data collection for the purposes of enabling performance-based reimbursement
KOLs were identified through Internet searches, and needed to have a minimum of 10 years’ experience, involvement in trials and/or a peer review publication experience in the relevant therapy areas.
The key points explored in the research included:
Whether any data collection infrastructures such as registries or databases are in use for the target indication
Data collection practice, e.g., whether data collection and entry into databases/registries is mandatory
The time horizon that patients are followed-up in clinical practice (i.e., short- vs. longer-term)
The therapeutic remit of the registry (i.e., if it is specific to the indication or a specific therapy used for a subset of the target population)
Findings
The indications most likely to see launch of cell or gene therapies in the UK over the next five years
The ARM data provided the greatest number of Phase III trials among the sources used, with a total of product 71 entries, out of which 55 were included in the sample below. This was supplemented by three additional entries from the other sources.
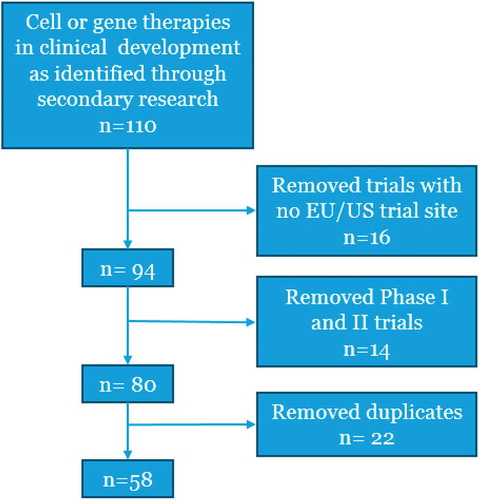
shows the results of our secondary research, detailing the number of results excluded due to not having a EU/US trial site or being Phase I/II, as well as duplicate results, leaving a total of 58 therapies/trials.
Figure 1. Flow diagram for the identification of the relevant therapies in late stage development in EU and/or US.
details the 58 cell or gene therapy products/trials that meet our criteria, and the associated target indications (47) and therapy areas (12). A minority of indications is targeted by more than one therapy in development (e.g., in heart failure, there are seven therapies in development).
Table 2. Cell or gene therapy products with clinical trial Phase III studies with sites in the EU and/or US with associated target indications and therapy areas.
shows the number and proportion of cell and gene therapies in Phase III trials with EU and/or US trial sites according to therapy area.
Figure 2. Number (and proportion) of cell and gene therapies in Phase III development (with US and/or EU trial site), according to therapy area.
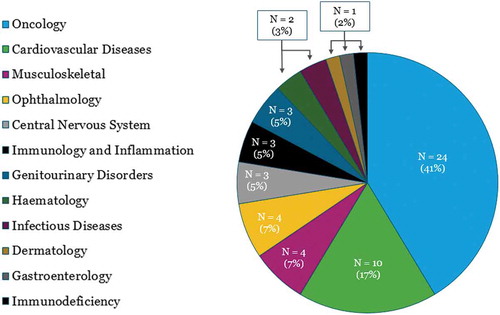
Our findings show that oncology is the therapy area has the highest likelihood to see UK launches of cell or gene therapies in the next five years, representing 40% (23/58) of the therapies identified, which is in line with other findings [Citation41–Citation43]. Cardiovascular disease comes second (also in line with other findings [Citation41–Citation43]) with 10 therapies in development, representing three indications, while the remaining therapy areas have four or fewer therapies in development.
The existing UK data collection infrastructure and practice in the indications of interest
Subsequently, we used secondary and primary research to identify the UK data collection infrastructures in place in the target indications identified,Footnote6 as well as the nature of patient follow-up and data collection in current clinical practice. Our previous work identified SACT database as mandatory to use for all oncology therapies [Citation34], and there was therefore no need to investigate each oncology indication individually. provides a graphical and numerical representation of our results (the grey part of the bar illustrates that the bar below is a subset of the bar above it), while summarises the key findings of our secondary research and KOL interviews per indication. The primary research component involved interviews with 20 KOLs across the 26 non-oncology target indications (some KOLs were able to speak to more than one target indication where indications were closely related, e.g., in ophthalmology).
Table 3. Data collection infrastructure and current practice in the target indications of interest.
Figure 3. Waterfall chart of the results according to the research criteria.
* Acute radiation syndrome (caused by e.g., nuclear meltdowns) was excluded from the analysis of infrastructure, due to the sporadic nature of the target indication; therefore the total number of indications for which data collection infrastructure and practice assessed is 46 (rather than the 47 identified in ).
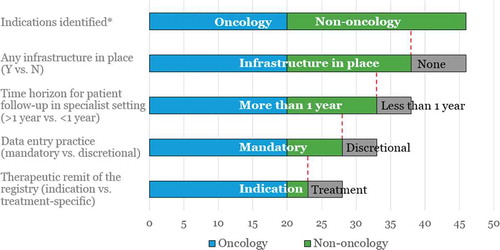
Our findings show that oncology (with its 20 indications), end-stage renal disease, haemophilia A and beta thalassaemia are the indications where a national data collection infrastructure is currently in place, and where patient follow-up and data collection is mandatory in the specialist setting beyond the one-year mark (in the latter three indications this is done indefinitely). These are the indications where the existing data collection infrastructure and clinical practice come close to the infrastructure needed to facilitate outcomes-based reimbursement schemes. But even in these indications upgrading is needed to enable the reimbursement mechanism and link all relevant stakeholders (clinicians, hospital pharmacists, finance, commissioners) while providing visibility for manufacturers and while maintaining patient confidentiality.
The SACT database in particular seems well poised to enable the collection of patient data longer-term, as it covers all cancer patients with solid and haematological malignancies in all of the NHS providers, and has been suggested as a means to support outcomes- and indication-based pricing [Citation34,Citation44]. The UK Renal Registry (UKRR) is another infrastructure that seems well suited for long-term outcomes collection. The UKRR links with the hospital episode statistics and the office for national statistics, and collects data on a quarterly basis from renal units across the country. Patients are seen by consultant physicians at least every three months, and have a variety of data collected, including haemoglobin, biochemistry data, types of treatments received, and adverse events. Similarly, in haemophilia, the National Haemophilia Database collects data quarterly from haemophilia centres regarding the treatment prescribed, factor level in the blood, adverse events, mortality, etc. In beta thalassaemia, safety, efficacy and adverse event data are collected long-term from patients’ annual visits and recorded in the National Haemoglobinopathy Registry. Data collection is mandatory, and compliance is thought to be good, meaning this is another area where the current clinical practice and infrastructure appears to be conducive to facilitating outcomes-based reimbursement.
Another infrastructure of particular interest is the British Bone Marrow Registry (BBMR), which is relevant for all patients undergoing bone marrow transplant (BMT), meaning it is relevant in several of the therapy areas identified (i.e., metachromatic leukodystrophy, adrenoleuko-dystrophy, Epstein-Barr virus infection, and cytomegalovirus infection, as well as certain thalassaemia patients). Data collection into the BBMR is mandatory and performed long-term. Although the BBMR is specific to the BMT treatment (in its current form), there are developments on the European level indicating that it may be used to facilitate data collection for Chimeric Antigen Receptor (CAR) T-cell therapies for regulatory purposes going forward [Citation45], however this is an area still in development. The BBMR is relevant also in adenosine deaminase severe combined immunodeficiency (ADA-SCID), where BMT is a common treatment option, however, this has now been complemented by the launch of Strimvelis®. Strimvelis® has an international, product-specific registry in place (for regulatory purposes), however, as with the BBMR, it is hard to say whether this can be leveraged also for the next gene therapy currently in development for ADA-SCID (OTL-101).Footnote7 Footnote8
Heart failure is the target indication with the highest number of therapies (seven) that could potentially launch over the next five years, and the National Heart Failure Audit (NHFA) is relevant for all patients that have been admitted to hospital with heart failure. It is mandatory to complete (for centres with a minimum of 20 admissions per month), and captures details such as 30-day readmission rate, mortality during stay and at one year, however, the majority of data is procedural rather than clinical outcomes (the NHFA is based on hospital episode data). Furthermore, there is a push to discharge heart failure patients from the specialist setting to primary care, and the vast majority (an estimated 90%) of patients are discharged from the specialist setting before the one-year mark (around 10% of patients, the most severe and unstable, are seen on a long-term basis by consultants or specialist nurses). Although this means that the more severe minority of patients are seen regularly and monitored longer-term, their data is not recorded in any national infrastructure (the NHFA is for hospital admissions only), but kept locally in respective hospitals.
Similarly, the majority of patients with burns, critical limb ischaemia and stress urinary incontinence are discharged to primary care typically within months, where data collection is currently thought to be very challenging. A clear message from many of the clinicians interviewed is that longer-term data collection in the primary care setting would require an increase in staff resources as well as ensuring access to the relevant data collection infrastructure.
Among the indications where no UK infrastructure was identified (relevant for the whole target population), there are examples where international infrastructures exist but where these are e.g., industry sponsored and/or focusing on patient-reported outcomes (e.g., REALITY LHON in leber hereditary optic neuropathy), or simply a registry of the patients with the disease in question, without capturing any outcomes data (e.g., the CHM registry in choroideremia). The case of biallelic RPE65-mediated inherited retinal disease is particularly pertinent as Luxturna® received a positive recommendation by the EMA’s Committee for Medicinal Products for Human Use (CHMP) for granting of marketing authorisation (pending European Commission confirmation at the time of writing) in October 2018 [Citation46]. There is no UK-wide data collection infrastructure in place that covers this target indication (the closest identified framework is the National Ophthalmology database, however, this is limited to cataract surgery patients only), so a new, possibly treatment-specific registry might need to be implemented if outcome-based reimbursement is going to be adopted for Luxturna®.
More broadly, our interviews with expert clinicians revealed that patients with chronic and severe conditions tend to be seen routinely and sometimes indefinitely in the specialist setting, where data collection and monitoring is engrained in routine clinical practice. Since longer-term data collection in the specialist setting is part of the current clinical practice for such indications, the barriers to implementing outcomes-based reimbursement seem quite modest if the appropriate data collection infrastructure were in place. Conversely, patients with conditions that are acute or require only short-term follow-up in the specialist setting (e.g., burns) are typically discharged into the primary care setting once they are stabilised, where long-term data collection is far more challenging.
Discussion
Managing data uncertainty in HTAs is a key challenge for NHS stakeholders when considering the adoption of a new therapy. In the above, we have detailed how this issue can be particularly pressing for cell and gene therapies that are administered as one-off treatments with the potential for providing long-term health benefits (but in the absence of long-term clinical data at launch). Furthermore, the issue of data uncertainty can be exacerbated for therapies that qualify for regulatory pathways such as the EMA’s priority medicines (PRIME) scheme or adaptive pathways, as these therapies can be granted a (conditional) marketing authorisation based on less mature trial data [Citation47,Citation48].
Our findings identify the target indications and therapy areas most likely to see a launch of a cell or gene therapy in the UK over the next few years, and detail how conducive the existing clinical practice and data collection infrastructure are for collecting real-world data over time. Oncology stands out as the therapy area with the most cell and gene therapies in late stage development, and also seems well poised to enable the collection of patient data longer-term (through the SACT database). Despite the potential of the SACT database, the Association of the British Pharmaceutical Industry (ABPI) has voiced the concerns of their members around the quality and completeness of the SACT data, as well as around governance and access to the data. Given these shortcomings, ABPI expects most companies to rely on maturing data from clinical trials or post-marketing studies, to support reimbursement submissions rather than tying in with SACT [Citation49].
Among non-oncology target indications, we have shown that the majority have a data collection infrastructure in place, however, there are considerable differences between indications in terms of whether patients typically are followed up for more than one year, and whether data collection is mandatory. Although our findings show that in many indications, the current clinical practice would have to change to enable longer-term patient data collection, it is important to highlight that the introduction of a cell or gene therapy would likely require the clinical pathway to change considerably anyway. Recent marketing authorisations granted by the EMA demonstrate that many cell or gene therapies are required to follow patients for more than a decade to document the long-term safety and efficacy. This means that while the current clinical practice in therapy areas such as heart failure, burns and critical limb ischaemia is to discharge patients into the primary care setting where follow-up and data collection is highly challenging, the introduction of a cell or gene therapy would likely require patients to be followed regularly and to collect patient data routinely.
Aligning the post-launch evidence generation infrastructure to address requirements for regulatory and reimbursement purposes is increasingly recognised by industry as well as by regulators, payers, and HTA bodies as an attractive option for minimising the burden of post-launch data collection [Citation30,Citation45,Citation50,Citation51]. This opportunity was seized upon in Italy in 2005, when Italian health authorities faced an incoming pipeline of expensive oncology treatments, with high price tags and uncertainty in the clinical data [Citation32]. The Italian Medicines Agency (AIFA) established a registry that serves both regulatory and reimbursement purposes, by linking prescriptions and payments/rebates to clinical criteria and outcomes. This registry is flexible and has been expanded (from its original remit of oncology) to include many other therapy areas, and is adaptable to accommodate the collection of different patient outcomes as new indications are added [Citation32].
While there has been some debate around the operational success of the Italian system, (e.g., in terms of realising the amounts due for rebate by the manufacturers [Citation52]), the experience from Italy shows that it is possible to implement a universal data collection infrastructure across a country with 21 highly autonomous regions, each with their own healthcare authorities and information technology (IT) systems.
The need for such a universal data collection infrastructure was one of the key recommendations made in a recent review of current HTA principles and practices in Europe regarding ATMPs [Citation51], however the implementation of such an infrastructure is associated with considerable challenges. One key challenge identified during the EMA’s Patient Registries Workshop was that the plethora of different registries and registry holders (whether academic or manufacturers) means that there are not always the incentives in place to collaborate between them to collect data to meet needs that are not directly their own [Citation53]. From the UK perspective, a key challenge is that there is a plethora of registries and databases for different indications and/or types of treatments, some of which are used routinely with high compliance rates, and others that are driven by physician champions in a minority of centres. This means that system integration and interoperability will be a key issue where existing registries or databases are to be leveraged. Furthermore, ensuring that there is an appropriate resource of clinical staff to handle the data entry and management, to ensure high quality data entry is another central concern. The issue of staff resource was a recurring theme in our KOL interviews, where many of the interviewed clinicians said that access to research nurses or data managers was key to ensuring the quality and accuracy of data entry.
Clinicians we spoke to noted that the staff resource shortages are markedly worse in the primary care setting, and to the point where many speculated that long-term patient follow-up and data collection in this setting would be near impossible. This may not be an issue for cell and gene therapies at the first instance, as one would expect most of the patient follow-up to take place in the hospital setting. However, as this new therapeutic field matures, safety concerns are reduced, and cell and gene therapies deliver on the promise of curing patients, one could expect to see more patients being discharged to the primary care setting sooner; this begs the question of how to facilitate data collection in the ideal scenario, i.e., where patients are symptom-free and with good quality of life. Recent initiatives like the PROmics study at the University of Birmingham (UK), where patient-reported outcomes (PROs) are collected, illustrate the interest in collecting patient data after discharge from the hospital, however, it remains to be seen what role PROs will play for reimbursement purposes in the UK.
NHS England, has historically avoided complex pricing arrangements because of the potential burden on hospitals and clinicians in managing data collection and financial flows [Citation44]. While NHS England tends to favour simple discounts over outcomes-based reimbursement schemes, the need for such alternative pricing schemes that better address the opportunities and challenges that innovative therapies like ATMPs present for commissioners, is growing.
This need is more pressing especially in therapy areas where there are few good short-to-medium term indicators of long-term performance. E.g., in haematological cancers (the target of several CAR-T therapies) it is possible to establish curative status after patients have remained in remission for approximately three years. This means that the need for implementing long-term outcomes-based pricing schemes in such therapy areas is lower; this is illustrated by the reimbursement of Kymriah and Yescarta in England through the Cancer Drugs Fund where no sophisticated pricing scheme was applied (just a confidential discount on the list price). However, in other therapy areas like haemophilia, and thalassaemia, it is far more challenging to establish curative status at launch in the absence of long-term clinical data. We anticipate that in such therapy areas, longer-term outcomes-based pricing schemes will become increasingly important and relevant in avoiding erosion of product value through upfront discounts.
Our recommendation is for outcomes-based reimbursement schemes to be part of the solution to address uncertainty for HTA bodies and affordability for NHS decision-makers, so that patient access is accelerated. Furthermore, reimbursement schemes involving outcomes-based, staged payments (such as ‘lifetime leasing’ or annuities) have the added benefit of addressing payer concerns around realising the rebate payments, as they are essentially a rebate in reverse. While the concerns around the administrative burden of executing such schemes are legitimate, there is a case to be made for cell and gene therapies, that since there commonly is a long-term safety and efficacy data collection requirement already in place (for regulatory purposes), the incremental burden of executing outcomes-based reimbursement is lower.
Having the right data collection infrastructure in place is key for implementing such outcomes-based reimbursement schemes. A more detailed understanding of the ability of existing infrastructures to enable such schemes is needed, followed by a gap analysis to determine whether it is more useful to build on existing structures or to establish a universal registry like the one operated by AIFA in Italy. Regardless, an improved data collection infrastructure would be a key component in ensuring not only timely patient access, but also a better understanding and recognition of the value for the NHS, as well as an appropriate reward to manufacturers for innovation.
Acknowledgments
We acknowledge Lyndsey Scull at the Alliance for Regenerative Medicine (ARM) for kindly providing data on cell and gene therapies in Phase III trials at the time of this analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes
1 Luxturna® had at the time of writing this received a positive recommendation by the EMA’s Committee for Medicinal Products for Human Use (CHMP) for granting of marketing authorisation (pending European Commission confirmation).
2 Non-Italian patients will be required to travel to Italy for treatment (pending successfully completed reimbursement negotiations in their respective countries).
3 An HTA exercise for a hypothetical CAR-T cell therapy, exploring the impact of different reimbursement schemes on the potential reimbursement recommendation delivered by NICE.
4 It should be noted that this type of reimbursement scheme would also impact company cash flow as the payments are staged over time rather than in one upfront instalment.
5 Some cell and gene therapies aim for early or accelerated access using e.g., Phase II trial data where the unmet need and therapeutic potential is high.
6 Note that acute radiation syndrome (caused by e.g., nuclear meltdowns) was excluded from the analysis of infrastructure, due to the sporadic nature of the target indication; therefore the total number of indications assessed is 46 (rather than 47).
7 It should be noted that OTL-101 is developed by Orchard Therapeutics Limited, which also owns the license for Strimvelis®, so the competitive imperative for limiting the registry only to Strimvelis® is likely lower in this case than it would be if two competing companies owned the technologies.
8 The British Bone Marrow Transplant registry is also used for a subset of thalassaemia patients who undergo this type of treatment.
References
- Marsden G, Towse A, Pearson SD, et al. Gene therapy: understanding the science, assessing the evidence, and paying for value. 2017 [cited 2017 Apr 4]. Available from: https://www.ohe.org/publications/gene-therapy-understanding-science-assessing-evidence-and-paying-value
- Abou-El-Enein M, Grainger DW, Kili S. Registry contributions to strengthen cell and gene therapeutic evidence. Mol Ther. 2018;26(5):1172–13.
- The EuropeanParliament and the Council of the European Union. Regulation (EC) No 1394/2007 of the European parliament and of the council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. Brussels, Belgium: Official Journal of the European Union; 2007.
- European Medicines Agency (EMA). Guideline on safety and efficacy follow-up and risk management of Advanced Therapy Medicinal Products – Draft. 2018 [cited 2018 Sep 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2018/02/WC500242959.pdf
- European Medicines Agency (EMA). Assessment report - YESCARTA (EMA/481168/2018). 2018 [cited 2018 Sep 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004480/WC500254960.pdf
- Sabine Grimm MS, Brennan A, Wailoo A. framework for analysing risk in health technology assessments and its application to managed entry agreements. 2016. Available from: http://scharr.dept.shef.ac.uk/nicedsu/wp-content/uploads/sites/7/2016/03/DSU-Managed-Access-report-FINAL.pdf
- Ferrario A, Kanavos P. Managed entry agreements for pharmaceuticals: the European experience. 2013 [cited 2018 Sep 26]. Available from: http://eprints.lse.ac.uk/50513/1/__Libfile_repository_Content_Ferrario%2C%20A_Ferrario_Managed_%20entry_%20agreements_2013_Ferrario_Managed_%20entry_%20agreements_2013.pdf
- Garrison, L.P., Towse, A., Briggs, A., et al. Performance-based risk-sharing arrangements-good practices for design, implementation, and evaluation: report of the ISPOR good practices for performance-based risk-sharing arrangements task force. Value Health. 2013;16:703–719.
- Adamski, J., Godman, B., Ofierska-Sujkowska, G., et al. Risk sharing arrangements for pharmaceuticals: potential considerations and recommendations for European payers. BMC Health Serv Res. 2010;10:153.
- Carlson JJ, Chen S, Garrison LP. Performance-based risk-sharing arrangements: an updated international review. PharmacoEconomics. 2017;35(10):1063–1072.
- Carlson, J.J., Gries, K.S., Yeung, K., et al. Current status and trends in performance-based risk-sharing arrangements between healthcare payers and medical product manufacturers. Appl Health Econ Health Policy. 2014;12(3):231–238.
- Klemp M., Fronsdal KB., Facey K. What principles should govern the use of managed entry agreements? Int J Technol Assess Health Care. 2011;27(1):77–83.
- Morel, T., Arickx F., Befrits G., et al. Reconciling uncertainty of costs and outcomes with the need for access to orphan medicinal products: a comparative study of managed entry agreements across seven European countries. Orphanet J Rare Dis. 2013;8(1):198.
- Bouvy JC, Sapede C, Garner S. Managed entry agreements for pharmaceuticals in the context of adaptive pathways in Europe. Front Pharmacol. 2018;9:280.
- Estonian Health Insurance Fund. Overview of Estonian experience on inclusion of medicines in benefits package and pricing. 2017 [cited 2019 Jan 8]. Available from: https://www.haigekassa.ee/sites/default/files/pressile/20170421_washingtondc_estonia.pdf
- Montilla, S., Xoxi, E., Russo, P., et al. Monitoring registries at italian medicines agency: fostering access, guaranteeing sustainability. Int J Technol Assess Health Care. 2015;31(4):210–213.
- Hettle, R., Corbett, M., Hinde, S., et al. The assessment and appraisal of regenerative medicines and cell therapy products: an exploration of methods for review, economic evaluation and appraisal. Health Technol Assess. 2017;21(7):1–204.
- Kefalas, P., Jørgensen, J., Merryfield, N., et al. Establishing the cost of implementing a performance-based, managed entry agreement for a hypothetical CAR T-cell therapy. J Mark Access Health Policy. 2018;6(1):1511679.
- National Cancer Registry and Analysis Service (NCRAS) at Public Health England. Systemic Anti-Cancer Therapy (SACT) dataset. 2018. Available from: http://www.chemodataset.nhs.uk/
- Alliance for Regenerative Medicine (ARM). Quarterly data reports. 2018 [cited 2018 Sep 21]. Available from: http://alliancerm.wpengine.com/publications-presentations/
- EU Clinical Trials Register. 2018 [cited 2018 Sep 21]. Available from: https://www.clinicaltrialsregister.eu
- Phacilitate & Biotech and Money. Advanced therapies investment report 2017. 2017 [cited 2018 Sep 6]. Available from: https://www.phacilitate.co.uk/sites/default/files/clarion_phacilitate/pdfs/advanced_therapies_investment_report_phacilitate.pdf
- The UKMI Horizon Scanning and Medicines Evaluation (HSME) Service. New medicines. 2018 [cited 2018 Sep 21]. Available from: https://www.sps.nhs.uk/category/new-medicines/
- National Institute for Health Research (NIHR). Innovation observatory. 2018 [cited 2018 Sep 21]. Available from: http://www.io.nihr.ac.uk/
- de Wilde, S., Guchelaar, H.J., Zandvliet, M.L., et al. Clinical development of gene- and cell-based therapies: overview of the European landscape. Mol Ther Methods Clin Dev. 2016;3:16073.
- Hanna, E., Rémuzat, C., Auquier, P., et al. Advanced therapy medicinal products: current and future perspectives. J Mark Access Health Policy. 2016;4. DOI:10.3402/jmahp.v3404.31036.
- Hanna, E. Gene therapies development: slow progress and promising prospect. J Mark Access Health Policy. 2017;5(1):1265293.
- Towse A, Cole A, Zamora B The debate on indication-based pricing in the U.S. and five major european countries. 2018 [cited 2018 Sep 26]. Available from: https://www.ohe.org/publications/debate-indication-based-pricing-us-and-five-major-european-countries
- Euroean Society for Bone and Marrow Transplantation (EBMT). Cell therapy registry and pharma collaboration. 2018 [cited 2018 Oct 3]. Available from: https://www.ebmt.org/ebmt/news/cell-therapy-registry-and-pharma-collaboration
- European Medicines Agency (EMA). Summary of opinion (initial authorisation) - Luxturna (voretigene neparvovec). 2018 [cited 2018 Oct 2]. Available from: https://www.ema.europa.eu/documents/smop-initial/chmp-summary-positive-opinion-luxturna_en.pdf
- European Medicines Agency (EMA). Adaptive pathways. 2018 [cited 2018 Sep 26]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000601.jsp&mid=WC0b01ac05807d58ce
- European Medicines Agency (EMA). PRIME: priority medicines. 2018 [cited 2018 Sep 26]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000660.jsp&mid=WC0b01ac05809f8439
- McConaghie A England’s ‘world-first’ cancer database – can it deliver on its promise? 2016 [cited 2018 Sep 24]. Available from: https://pharmaphorum.com/views-and-analysis/englands-world-first-cancer-database-can-deliver-promise/
- European Medicines Agency (EMA). Report on CAR T-cell therapy Registries - Workshop 9 February 2018 - Patient Registries Initiative. 2018 [cited 2018 Oct 3]. Available from: https://www.ema.europa.eu/documents/report/report-car-t-cell-therapy-registries-workshop_en.pdf
- Jönsson, B., Hampson, G., Michaels, J., et al. Advanced therapy medicinal products and health technology assessment principles and practices for value-based and sustainable healthcare. Eur J Health Econ. 2018. DOI:10.1007/s10198-018-1007-x. Epub ahead of print.
- Garattini L, Curto A, van de Vooren K. Italian risk-sharing agreements on drugs: are they worthwhile? Eur J Health Econ. 2015;16(1):1–3.
- European Medicines Agency (EMA). Patient registries workshop, 28 October 2016 - observations and recommendations arising from the workshop. 2017 [cited 2018 Sep 12]. Available from: https://www.ema.europa.eu/documents/report/report-patient-registries-workshop_en.pdf
- Abou-El-Enein M, Elsanhoury A, Reinke P. Overcoming challenges facing advanced therapies in the EU market. Cell Stem Cell. 2016;19(3):293–297.
- European Biotechnology Life Science and Industry Magazine. Uniqure withdraws €1m drug Glybera from market. 2017 [cited 2017 Apr 28]. Available from: http://european-biotechnology.com/up-to-date/latest-news/news/uniqure-withdraws-eur1m-drug-glybera-from-market.html
- National Institute for Health and Care Excellence (NICE). Talimogene laherparepvec for treating unresectable metastatic melanoma - technology appraisal guidance [TA410]. 2016 [cited 2017 Apr 25]. Available from: https://www.nice.org.uk/guidance/ta410/chapter/1-Recommendations
- Touchot N, Flume M. Early insights from commercialization of gene therapies in Europe. Genes (Basel). 2017;8(2):78.
- National Institute for Health and Care Excellence (NICE). Talimogene laherparepvec for treating unresectable metastatic melanoma - technology appraisal guidance. 2016 [cited 2018 Sep 24]. Available from: https://www.nice.org.uk/guidance/ta410/resources/talimogene-laherparepvec-for-treating-unresectable-metastatic-melanoma-pdf-82604596507333
- Haute Autorité de Santé (HAS). Commission de la transparence Avis 20 juillet 2016 - cellules épithéliales cornéennes humaines autologues amplifiées ex vivo contenant des cellules souches. 2016. Available from: http://www.has-sante.fr/portail/upload/docs/evamed/CT-15190_HOLOCLAR_PIC_INS_Avis2_CT15190.pdf
- Agenzia Italiana del Farmaco (AIFA). Pubblicazione schede di monitoraggio Registro HOLOCLAR. [cited 2017 Mar 1]. 2017 Available from: http://www.aifa.gov.it/content/pubblicazione-schede-di-monitoraggio-registro-holoclar-01032017
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Informe de Posicionamiento Terapéutico de células epiteliales corneales humanas autólogas, expandidas ex vivo, entre las que se encuentran células madre (Holoclar®) en el tratamiento de pacientes adultos con deficiencia de células madre limbares debida a quemaduras oculares por agentes físicos o químicos. 2016 [cited 2018 Sep 24]. Available from: https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-celulas-epiteliales-Holoclar-deficiencia-celulas-limbares.pdf
- National Institute for Health and Care Excellence (NICE). Holoclar for treating limbal stem cell deficiency after eye burns - technology appraisal guidance. 2017 [cited 2018 Sep 24]. Available from: https://www.nice.org.uk/guidance/ta467/resources/holoclar-for-treating-limbal-stem-cell-deficiency-after-eye-burns-pdf-82604910595525
- National Institute for Health and Care Excellence (NICE). Strimvelis for treating adenosine deaminase deficiency–severe combined immunodeficiency - highly specialised technologies guidance. 2018 [cited 2018 Sep 24]. Available from: https://www.nice.org.uk/guidance/hst7/resources/strimvelis-for-treating-adenosine-deaminase-deficiencysevere-combined-immunodeficiency-pdf-1394905926085
- Molmed. MolMed: Board of Directors approved Full Year 2017 Results. 2018 [cited 2018 Sep 17]. Available from: https://www.molmed.com/sites/default/files/uploads/press-releases/3265/3265_1520621626.pdf
- CO.DON AG. CO.DON AG: spherox - largest product launch in the company’s history. 2017 [cited 2018 Sep 26]. Available from: http://www.codon.de/news-overview/press-release/read-news/article/codon_ag_spherox_groesster_produktlaunch_der_unternehmensgeschichte.html?L=1&cHash=57889048b5ad69a67bd9b22280a7cddb
- National Institute for Health and Care Excellence (NICE). Autologous chondrocyte implantation using chondrosphere for treating symptomatic articular cartilage defects of the knee. 2018 [cited 2018 Sep 26]. Available from: https://www.nice.org.uk/guidance/ta508
- NHS England. NHS England strikes deal for ground breaking cancer treatment in a new European first. 2018 [cited 2018 Oct 12]. Available from: https://www.england.nhs.uk/2018/10/nhs-england-strikes-deal-for-ground-breaking-cancer-treatment-in-a-new-european-first/
- NHS England. NHS England announces groundbreaking new personalised therapy for children with cancer. 2018 [cited 2018 Sep 24]. Available from: https://www.england.nhs.uk/2018/09/nhs-england-announces-groundbreaking-new-personalised-therapy-for-children-with-cancer/
- Pena A NICE rejects kymriah for England’s NHS to treat aggressive DLBCL. 2018 [cited 2018 Oct 12]. Available from: https://lymphomanewstoday.com/2018/09/25/nice-rejects-kymriah-treating-aggressive-dlbcl-england-national-health-service/