
ABSTRACT
Background: Spinal muscular atrophy (SMA) is a rare genetic neuromuscular disease.
Objective: Characterize direct costs associated with SMA management.
Data source: Truven Health Analytics MarketScan claims data (2012–2016).
Patients: Eligible patients had ≥2 SMA-related medical claims ≥30 days apart. Patients were matched (1:1) to controls by birth year, gender, and geographic region. Patients were categorized as having infantile, child, or juvenile SMA based on diagnosis at age <1, 1–3, or 3–18 years, respectively.
Main outcome measures: Annual inpatient and outpatient insurance claims and costs (2019 USD) for cases versus controls.
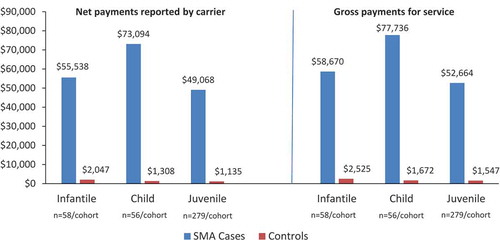
Results: Fifty-eight, 56, and 279 cases and controls comprised the infantile, child, and juvenile cohorts, respectively. Cases had more inpatient claims than controls (infantile: 60.3% vs 1.7%; child: 35.7% vs 3.6%; juvenile: 47.0% vs 4.3%; all P ≤ 0.002). Mean net payments for inpatient admissions were higher for cases versus controls (infantile: $118,609.00 vs $58.79; child: $26,940.01 vs $143.56; juvenile: $39,389.91 vs $701.21; all P ≤ 0.01), as were mean net payments for outpatient services (infantile: $55,537.83 vs $2,047.20; child: $73,093.66 vs $1,307.56; juvenile: $49,067.83 vs $1,134.69; all P ≤ 0.0002).
Conclusions: Direct costs of SMA are tremendous, often >50-fold higher compared with matched controls. Efforts are needed to reduce costs through improved standards of care.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder caused by homozygous deletion or mutation of the survival of motor neuron 1 (SMN1) gene [Citation1,Citation2]. An estimated 1 in 54 people in the USA are carriers of SMN1 genetic mutations or deletions, which corresponds to a disease frequency of about 1 in 11,000 live births [Citation3].
SMA is characterized by progressive dysfunction and loss of alpha motor neurons in the spinal cord, which results in muscle weakness and atrophy, loss of reflexes, and denervation. In severe cases, SMA can cause breathing and swallowing difficulties, paralysis, and death. Despite being a rare disease, SMA is one of the most common genetic causes of death in infants [Citation2–5].
The clinical severity of SMA is correlated with several factors such as age at disease onset, achieved milestones, and copy number of SMN2, a low-functioning paralogue of SMN1 [Citation2,Citation5]. In an estimated 50% to 60% of patients, SMA manifests in the first 6 months of life and the disease is severe; without treatment and supportive care, these patients cannot walk or sit without support, require intensive respiratory and feeding support as they quickly lose the ability to swallow, speak, and eat. Ultimately, they may quickly lose the ability to breathe and thus have a life expectancy of less than 2 years [Citation2]. Approximately 30% of patients develop SMA with intermediate severity, which is typically diagnosed between 6 and 18 months of age. While this group is defined as having the ability to sit, many may either lose this ability later in life or on the other end of the spectrum may achieve the ability to stand or walk with assistance [Citation6]. Additionally, these patients may develop difficulties involving chewing, swallowing, respiration, as well as progressive scoliosis [Citation7]. Patients typically have a life expectancy well into adulthood. However, due to the nature of the disease, without treatment, patients with symptoms of intermediate severity develop progressive muscle weakness in their limbs and trunk, followed by muscle atrophy across all body systems. Many people with this SMA phenotype develop difficulty swallowing and respiratory insufficiency [Citation2,Citation8–10]. When SMA onset occurs at ≥18 months of age (~12% of SMA cases), the disease is usually mild, causing minimal physical disability. Most patients with this mild SMA phenotype are able to walk without assistance and have a normal life expectancy [Citation3,Citation8,Citation9]. However, due to the progressive nature of SMA, some patients develop weakness in their hip and/or thigh muscles, which can cause an abnormal gait, and some patients lose ambulation during adolescence or adulthood [Citation2,Citation11].
Traditionally, SMA has been classified as type I (severe), type II (intermediate), or type III (mild) [Citation9]. However, as our understanding of the disease improves, clinical and genetic phenotypes are evolving, and diagnosis of SMA is often not type-specific [Citation6,Citation12,Citation13]. Therefore, many researchers and health professionals are now using categorizations of ‘infantile,’ ‘child,’ ‘juvenile,’ or ‘adult’ SMA based on age at diagnosis [Citation14–18].
Until recently, there were no effective treatments for SMA, with the standard of care including, but not limited to, requirements for neurology, respiratory, orthopedic, gastrointestinal, nutritional, physical and occupational therapy, and aquatic support [Citation19–22]. However, the SMA treatment paradigm is now shifting following the US Food and Drug Administration approval of two novel therapies: the antisense oligonucleotide nusinersen (approved in December 2016) and the gene-replacement therapy onasemnogene abeparvovec-xioi (approved in May 2019) [Citation14,Citation16,Citation19,Citation23,Citation24]. These therapies have been shown to significantly improve clinical outcomes for patients with SMA [Citation14,Citation16,Citation24]. Additionally, risdiplam, a small-molecule SMN2 splicing modifier designed to increase and sustain SMN protein levels, was recently approved by the US Food and Drug Administration for the treatment of SMA [Citation25,Citation26]. Results of recently completed clinical trials have shown that in patients with type I, II, or III SMA, treatment with risdiplam improved motor function [Citation27,Citation28].
As new treatments continue to become available, the economic impact of SMA on patients and healthcare systems has been debated [Citation29], despite limited availability of economic data [Citation19,Citation30,Citation31]. To understand and quantify this impact, it is important to have baseline data on the economic burden of SMA prior to the availability of pharmacotherapies. These data may be used to inform cost-effectiveness analyses and payer policy decisions related to the use of novel therapies.
The purpose of this study was to conduct a claims database analysis to characterize the direct costs associated with the management of SMA during the 4-year period before approval of the first pharmacotherapy for SMA in a commercially insured subset of patients. These results will provide insights into the economic burden of SMA and can provide baseline data to allow researchers to track cost-related changes over time as the use of new treatments increases.
Methods
Data source
This retrospective cohort study evaluated claims data from the Truven Health Analytics MarketScan Commercial Claims and Encounters and Medicare Supplemental databases between 1 October 2012 and 31 December 2016 to identify patients with SMA. The Truven MarketScan Commercial Claims and Encounters database contains de-identified patient details for over 30 million individuals covered by 1 of more than 300 employer-sponsored private health insurance plans, including birth year, gender, geographic region, insurance plan, inpatient and outpatient prescription claims, clinical resource utilization, and healthcare expenditures. Covered individuals included active employees and dependents, early (non-Medicare) retirees and dependents, and COBRA enrollees. The Truven MarketScan Medicare Supplemental database captures information on the subset of Medicare beneficiaries who have employer-paid supplemental insurance, including data on costs covered by employers and through Medicare, as well as beneficiary out-of-pocket costs [Citation32]. Information in these databases can be used to calculate overall healthcare costs, reimbursements, and out-of-pocket expenses paid by patients, providers, and payers [Citation32]. These databases are the Health Insurance Portability and Accountability Act (HIPAA)-compliant and have rigorous protections in place to ensure patient privacy [Citation32].
Patient cohorts
SMA cases were identified as patients having two or more medical claims at least 30 days apart based on the following International Classification of Diseases (ICD) diagnostic codes: ICD-9 335.19 (other SMA), ICD-9 335.10 (SMA, unspecified), ICD-10 G129 (SMA, unspecified), or ICD-10 G128 (other SMAs and related symptoms). Patients were required to have continuous enrollment for at least 1 month prior to their first SMA-related claim to increase the likelihood that the first observed SMA-related diagnosis was the first time they were diagnosed. Patients with gaps in their coverage were excluded. Patients were followed from the index date (defined as the first diagnosis of SMA) to the end of the study period, the end of continuous health plan enrollment, the end of health plan eligibility, or death. (Note: Death is not captured in this database and is therefore not reported here.)
Patients meeting SMA eligibility criteria were further classified as having infantile, child, or juvenile SMA. These classification cohorts were used instead of the more traditional SMA type I/II/III because of the limited nature of insurance claims data. Specifically, there are no ICD-9 or ICD-10 codes for SMA type I, II, or III; the age of disease onset is not known with certainty; SMN2 copy number is not known; and there are no details available on patients’ motor function status.
In this analysis, infantile SMA was defined as having two or more claims of either an SMA diagnosis or an SMA-related diagnosis before 1 year of age. SMA-related diagnoses were identified using keywords associated with the hallmark features of infantile SMA, including hypotonia, respiratory distress, dysphagia, poor feeding, weight loss, failure to thrive, poor weight gain, hypoxemia, respiratory intubation and mechanical ventilation, gastrostomy tube placement, and dependence on respirator [ventilator] (see Supplemental Table 1 for ICD codes for these SMA-related diagnoses). Child SMA was defined as having two or more claims of an SMA diagnosis, with the first claim occurring between 12 and 36 months of age, without an infantile SMA claim. Juvenile SMA was defined as having two or more claims of an SMA diagnosis, with the first claim occurring between 36 months and 18 years of age, without an infantile or a child SMA claim.
Patients with SMA were matched in a 1:1 ratio to controls without SMA based on birth year, gender, and geographic region (Northeast, North central, South, and West). Cases and controls were matched using the total enrollment population provided by the Truven MarketScan databases rather than inpatient or outpatient datasets. This provided a mix of patients with and without different types of claims, which is representative of healthcare utilization in the real-world US population [Citation32].
Economic analyses
In each subset of SMA (infantile, child, and juvenile), the number of claims was compared over the study period for cases versus controls based on the mean number of inpatient admissions per person per year; the number of patients having an inpatient claim; the mean, median, and range of days per inpatient stay among those with an inpatient stay; the mean number of outpatient services per person per year; and the number of patients having an outpatient claim. Inpatient and outpatient costs were annualized and adjusted to 2019 US dollars using the annual medical care component of the consumer price index and rounded up to the nearest whole dollar [Citation33]. Inpatient cost comparisons between cases and controls included the net and total gross payments to a hospital for covered services provided during an admission, total net payments for the admission, and total gross payments to all providers that submitted claims for covered services rendered during an admission. Any inpatient claim related to the birth of a child was excluded from the analyses. Outpatient cost comparisons between cases and controls included net payments reported by the insurance carrier and gross payments to a provider for a service (i.e., the amount eligible for payment under the medical plan terms after applying rules such as discounts but before applying coordination of benefits, copayments, and deductibles).
Results
Patient characteristics
A flowchart of the identification of eligible patients with SMA and matched controls is shown in . Based on outpatient claims and inpatient admissions from 2012 to 2016, there were 7,961 enrollees in the Truven Commercial Claims and Encounters database with at least 1 SMA claim. Of these patients, 58, 56, and 279 met eligibility criteria for infantile, child, and juvenile SMA, respectively.
Figure 1. Identification of SMA cases and controls included in the claims database analysis
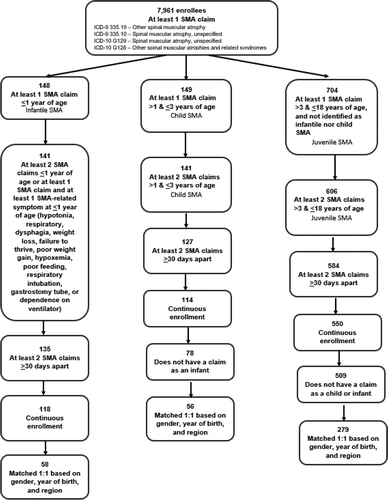
Demographic and insurance plan characteristics for cases and controls in the infantile, child, and juvenile SMA groups are shown in . Cases and controls were successfully matched by birth year, gender, and geographic region (P ≈ 1.0000 for cases vs controls for all matching characteristics). There were statistically significant differences between cases and controls for insurance plan type in the juvenile SMA analysis (p < 0.0001) and for employee classification in the child SMA and juvenile SMA analyses (p = 0.024 and p < 0.0001, respectively).
Table 1. Demographic and insurance plan characteristics for cases and controls
Infantile SMA analyses
The infantile SMA analysis showed that inpatient claims were significantly more common for cases compared with controls; 60.3% (35/58) of cases and 1.7% (1/58) of controls had an inpatient claim during the study period (P < 0.0001). The mean (standard deviation [SD]) number of inpatient admissions per person per year was 1.3 (0.33) for cases and 0.01 (0.06) for controls (P = 0.0001). Among SMA cases with an inpatient admission, the mean (SD) length of each hospital stay was 10.0 (14.3) days and the median length of stay was 4 days (range 1–49.5 days). One control had a 1-day inpatient stay. Among SMA cases, respiratory illness was the most common (24/35) reason for inpatient admission. For controls, the only reason (1/1) for inpatient admission was related to a respiratory illness ().
Table 2. Most common diagnoses during an inpatient service for SMA cases versus controls for infantile, child, and juvenile SMA
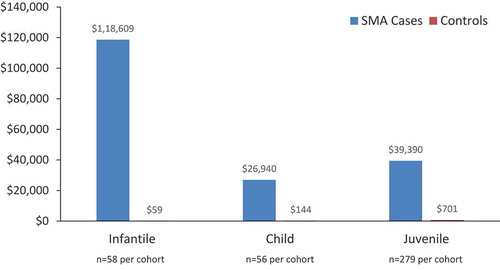
Inpatient costs for cases compared with controls in all cohorts are shown in . Notably, in the infantile analysis, the mean (SD) annualized total net payments for inpatient admission were 118,609 USD (338,118) for SMA cases compared with 59 USD (448) based on one inpatient admission in the control group; P = 0.01 ().
Table 3. Mean (SD) inpatient costs for SMA cases versus controls for infantile, child, and juvenile SMA (2019 USD)
Figure 2. Mean total net payments for inpatient admissions for SMA cases versus controls for infantile, child, and juvenile SMA (2019 USD).*
The mean (SD) number of outpatient claims per person per year was 234.4 (226.2) for infantile SMA cases and 25.8 (18.4) for controls (P < 0.0001). Outpatient costs for cases compared with controls in all cohorts are shown in . In the infantile analysis, mean (SD) net payments for outpatient services were 55,538 USD (73,375) for SMA cases compared with 2,047 USD (2,011) for controls (P < 0.0001). Among infantile SMA cases, a home health service (e.g., in-home skilled nursing or physical therapy) was the most common (n = 2219) type of outpatient claim. For controls, a non-specialty office visit (e.g., a routine infant or child health examination) was the most common (n = 647) type of outpatient claim ().
Table 4. Five most common outpatient services for SMA cases versus controls for infantile, child, and juvenile SMA
Figure 3. Mean outpatient costs for all years for SMA cases versus controls for infantile, child, and juvenile SMA (2019 USD)
Child SMA analyses
Similar to findings from the infantile SMA analysis, the child SMA analysis showed that inpatient claims were significantly more common for cases compared with controls; 35.7% (20/56) of cases and 3.6% (2/56) of controls had an inpatient claim during the study period (P = 0.002). The mean (SD) number of inpatient admissions per person per year was 0.62 (1.3) for child SMA cases and 0.04 (0.19) for controls (P = 0.0014). Among patients with an inpatient admission, the mean (SD) length of each hospital stay was 6.7 (6.9) days (median: 4.3 days; range: 1–32 days) for child SMA cases and 1 day for the two controls (P = 0.0015) that had an inpatient claim. The mean (SD) annualized total net payments for inpatient admission were more than 180-fold higher for child SMA cases compared with controls ($26,940 [64,909] for cases vs 144 USD [758] for controls; P = 0.0031) ( and ). Among cases, respiratory illness was the most common (17/20) principal diagnosis resulting in an inpatient admission. For controls, the two reasons for an inpatient admission were related to a respiratory illness and a skin burn, respectively ().
The mean (SD) number of outpatient claims per person per year was 248.7 (259.5) for child SMA cases compared with 16.9 (12.9) for controls (P < 0.0001). Mean (SD) net payments for outpatient services were more than 50-fold higher for child SMA cases compared with controls ($73,094 [136,889] for child SMA cases compared with 1,308 USD [2,018] for controls; P = 0.0002) (). Among child SMA cases, a home health service (e.g., in-home skilled nursing or physical therapy) was the most common (n = 2446) type of outpatient claim. For controls, a non-specialty office visit (e.g., a routine infant or child health examination) was the most common (n = 535) type of outpatient claim ().
Juvenile SMA analyses
Consistent with findings from the infantile and child SMA analyses, the juvenile SMA analysis showed that inpatient claims were significantly more common for SMA cases compared with controls; 47.0% (131/279) of cases and 4.3% (12/279) of controls had an inpatient claim during the study period (P < 0.0001). The mean (SD) number of inpatient admissions per person per year was 0.66 (1.3) for juvenile SMA cases and 0.02 (0.11) for controls (P < 0.0001). Among patients with an inpatient admission, the mean (SD) length of each hospital stay was 5.8 (5.3) days for juvenile SMA cases and 4.4 (5.2) days for controls (P = 0.41). The mean (SD) annualized payments for inpatient admissions were more than 50-fold higher for juvenile SMA cases compared with controls ($39,390 [143,229] for cases vs 701 USD [8,484] for controls; P < 0.0001) ( and ). Among juvenile SMA cases, respiratory illness was the most common (61/131) reason for an inpatient admission; for controls, appendicitis and depression were the most common (3/12 for each) reasons for inpatient admission ().
The mean (SD) number of outpatient claims per person per year was 170.3 (217.1) for juvenile SMA cases compared with 12.2 (13.4) for controls (P < 0.0001). Mean (SD) net payments for outpatient services were more than 40-fold higher for juvenile SMA cases compared with controls ($49,068 [95,613] for SMA cases compared with 1,135 USD [2,750] for controls; P < 0.0001) (). Among juvenile SMA cases, a home health service (e.g., in-home skilled nursing or physical therapy) was the most common (n = 7541) type of outpatient claim. For controls, a non-specialty office visit (e.g., a routine infant or child health examination) was the most common (n = 1122) service for an outpatient claim.
Discussion
Results of these analyses show that the economic burden of direct inpatient and outpatient costs associated with untreated SMA is tremendous and significantly higher than for age-, gender-, and region-matched controls. As expected, this analysis showed that costs are highest for patients with infantile SMA, the most severe form of the disease. Annualized inpatient costs for identified cases of infantile SMA exceeded 100,000 USD per patient, and median inpatient hospital stays were 4 times longer than for controls. Outpatient costs in the infantile SMA cohort for all years evaluated were more than 50,000 USD per patient. Although costs were lower for child and juvenile SMA than for infantile SMA, costs in these SMA cohorts remained close to 50-fold higher than for controls.
Results of the current study support other recent analyses of the economic burden of SMA. For example, an analysis of hospitalization costs using data from the 2012 Kids’ Inpatient Database representing hospitals in 44 US states showed that the mean cost per admission for children with type I SMA (n = 237 admissions) was 150,921 USD compared with 19,261 USD per admission (n = 632,467 admissions) for children without complex chronic conditions (P < 0.0001) [Citation34]. In another study, hospitalization data obtained from the Healthcare Cost and Utilization Project and the Center for Health Information and Analysis indicated that the mean annualized cost per patient with severe SMA (n = 229) was 104,197 USD [Citation35]. Additionally, an analysis of claims data from the Symphony Health Integrated Dataverse opens claims database (n = 349 patients with type I SMA and 45 patients with type I SMA receiving treatment with nusinersen) showed that total mean healthcare costs excluding the cost of treatment with nusinersen were between 92,618 USD and 137,627 USD per person per year [Citation19]. Furthermore, an analysis of Department of Defense Military Healthcare System data showed that among 239 patients with a mean age at SMA diagnosis of 7.5 years, median (range) outpatient costs were 53,152 USD ($23,902-$136,150) and inpatient costs were 11,258 USD ($0-$51,987) over a mean of 4.8 years after the initial SMA diagnosis [Citation31].
In the current study, the primary cost driver for infantile SMA was inpatient expenses, most commonly related to respiratory infection and/or respiratory failure. For the less severe disease manifestations of child and juvenile SMA, outpatient costs were the primary cost drivers, especially costs associated with in-home health services. These findings are consistent with results from another recent analysis of Truven MarketScan claims data from 1,120 patients with SMA, which found that outpatient spending drove costs, with 73.5% of SMA patients utilizing home health services and 60.6% of patients requiring neurology and/or pulmonology outpatient services [Citation36].
In addition to high direct inpatient and outpatient costs, rare diseases such as SMA are associated with tremendous burdens for caregivers. Statistics included in a 2018 report issued by the National Alliance for Caregiving indicate that caregivers for children with rare diseases spend an average of 53 hours per week on caregiving responsibilities, which requires many people (52%) to cut back their work hours. As a result of lost wages and the costs of care, 86% of caregivers for patients with rare diseases experience financial hardship. Concerns about finances, as well as the everyday stresses and strains of caregiving, can have a profound negative impact on caregivers’ emotional and physical health [Citation37]. Studies specific to SMA have found that there are substantial productivity losses as well as supportive care, opportunity, and psychosocial costs for both patients and their caregivers, all of which are associated with reduced health-related quality of life [Citation30,Citation38]. In a recent survey of parents of children with SMA (n = 93) and adults living with SMA (n = 29), 46% of respondents reported work productivity losses and 56% reported activity impairment over the last 7 days [Citation39]. Supportive care costs of SMA can include wheelchairs, walkers, orthotics, enteral feeding products, speech and occupational therapy, equipment to improve respiration (e.g., mechanically assisted cough devices, invasive ventilation, continuous positive airway pressure [CPAP], and bilevel positive airway pressure [BiPAP]), and home care services [Citation36,Citation40]. A qualitative study of the costs incurred by parents of children with type II or type III SMA in Australia reported that there are several often-overlooked economic consequences of disease, including struggles to afford specialized wheelchair-accessible housing, seating and mobility equipment, and specialized vehicles and bedding. Caregivers for patients with SMA reported having to work more hours, sacrifice retirement savings and disposable income, and/or rely on community or charity donations to afford necessary care and equipment. These increased financial hardships were often magnified, because in many cases, parents were forced to give up paid employment, limit career advancement, or take lower-paying hourly work to care for their child with SMA [Citation41]. Quantifying the indirect costs associated with SMA, including caregiver burden and lost productivity, is an important area for future research.
Until recently, receiving a diagnosis of SMA for a child was associated with devastating psychological effects on parents, who were forced to confront the likelihood of their child’s premature death or loss of functional ability, as well as the loss of the parents’ independence, increased stress, fear of stigma, and uncertainty about their financial future [Citation42]. With the recent approvals of novel therapies to treat or potentially cure SMA, patients and their caregivers can now have hope for better long-term survival, physical functioning, and quality of life [Citation26,Citation29,Citation43].
Novel therapies, including nusinersen, onasemnogene abeparvovec, and risdiplam, have the potential to transform the SMA treatment landscape and substantially reduce the burden of disease for patients and caregivers [Citation23,Citation26]. However, the economic value of these new therapies has been debated [Citation23].
Although it is widely acknowledged that higher cost-effectiveness thresholds may be appropriate for ultra-rare diseases like SMA [Citation29,Citation43], modeling the costs associated with SMA has been challenging, given the lack of available data on the economic burden of disease and the effects of treatment on this burden [Citation19]. The current study provides important data on the economic burden of SMA, which are needed to adequately inform assessments of the cost-effectiveness of new therapies. Results of this study also provide baseline data for the economic burden of untreated SMA, which may change over time with the broad use of new pharmacologic therapies. Additionally, this study provides insights into the real-world rates of hospitalization among patients with SMA, which are notably lower than rates reported in controlled clinical trial settings [Citation44].
This study has some notable limitations. Importantly, claims database analyses are always limited by the potential for coding errors and other inaccuracies. In the case of SMA, diagnostic codes do not distinguish between type I, II, or III SMA so age at diagnosis is only an approximate surrogate for disease severity and it may be possible that some patients were misclassified to an older cohort. This is a shared limitation of similar insurance claim studies on SMA patients using age at diagnosis as a surrogate for disease severity [Citation31,Citation40]. This dataset is limited to providing SMA prevalence, not incidence rates, and age at diagnosis is an assumption based on the date of the first claim in the database. Therefore, caution should be used when generalizing the results in SMA prevalence estimates. Use of diagnostic claims data for SMA is also limited because if genetic testing for SMN1 mutations is not performed at an early age, delayed diagnosis or misdiagnosis are likely given the rare nature of the disease and the number of other diseases with clinical features that are similar to SMA [Citation45,Citation46]. Thus, there is no way to confirm if the first reported diagnostic claim for SMA represents an accurate diagnosis or if earlier claims related to SMA were not captured because of previous misdiagnoses. Another limitation is that this study population represents a predominantly commercially insured segment of the US population, so results may not be generalizable to all patients with SMA, specifically those covered by state Medicaid programs and/or Medicaid Managed Care programs. Another limitation of this study is mortality data is not captured in this dataset. As treatments become available for the SMA community, and thus increasing survival rates, the overall and long-term costs reflected in here in each cohort may be underestimated. Finally, it is important to recognize that the direct costs reported herein represent only a portion of the overall economic burden of SMA incurred by patients and their families throughout their lifetime. Equipment costs for products that improve mobility and accessibility can be substantial (e.g., costs for motorized wheelchairs, accessible vans, lifts, communication devices, and home renovations to improve accessibility) [Citation38].
In summary, results of the current study indicate that the direct inpatient and outpatient costs associated with infantile, child, and juvenile SMA are tremendous averaging more than 100,000 per patient per year from 2012 to 2016. In many cases, these costs are more than 50-fold higher than those for matched controls. Uniform adoption and implementation of the current standards of care guidelines could result in fewer complications of SMA. In addition, the availability of new treatments and widespread SMA newborn screening has the potential to not only reduce the economic burden of SMA but also to significantly improve long-term clinical outcomes and patient and caregiver quality of life [Citation47].
Supplemental Material
Download MS Word (12.2 KB)Acknowledgments
Editorial and writing support were provided by Cherie Koch, PhD (Synchrogenix). This study was financially supported by the SMA Industry Collaboration with funding to Cure SMA. At the time financial support was provided, members of the SMA Industry Collaboration included Novartis Gene Therapies, Biogen, Genentech/Roche Pharmaceuticals, Novartis, Astellas, Cytokinetics, and Scholar Rock.
Disclosure statement
Lisa Belter, Rosángel Cruz, and Jill Jarecki are employees of Cure SMA. Sierra Kulas and Omar Dabbous are employees of Novartis Gene Therapies, a Novartis Company, and own stock in Novartis. Emily McGinnis is a former employee of Novartis and owns stock in Novartis.
Supplementary material
Supplemental data for this article can be accessed here.
References
- Finkel RS, McDermott MP, Kaufmann P, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014;83(9):810–13.
- Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51(2):157–167.
- Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20(1):27–32.
- Lefebvre S, B¸rglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165.
- Feldkotter M, Schwarzer V, Wirth R, et al. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–368.
- Tizzano EF, Finkel RS. Spinal muscular atrophy: a changing phenotype beyond the clinical trials. Neuromuscul Disord. 2017;27(10):883–889.
- Zerres K, Rudnik-Schöneborn S, Forrest E, et al. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146(1):67–72.
- Bharucha-Goebel D, Kaufmann P. Treatment advances in spinal muscular atrophy. Curr Neurol Neurosci Rep. 2017;17(11):91.
- Jha NN, Kim JK, Monani UR. Motor neuron biology and disease: A current perspective on infantile-onset spinal muscular atrophy. Future Neurol. 2018;13(3):161–172.
- Wirth B, Karakaya M, Kye MJ, et al. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu Rev Genomics Hum Genet. 2020;21:231–261.
- Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33(4):831–846.
- Darras BT, De Vivo DC. Precious SMA natural history data: A benchmark to measure future treatment successes. Neurology. 2018 Aug 21;91(8):337–339.
- Rossor AM, Oates EC, Salter HK, et al. Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2. Brain. 2015;138(Pt 2):293–310.
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–1732.
- Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–3026.
- Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–635.
- Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86(10):890–897.
- Lipnick SL, Agniel DM, Aggarwal R, et al. Systemic nature of spinal muscular atrophy revealed by studying insurance claims. PLoS One. 2019;14(3):e0213680.
- Droege M, Sproule D, Arjunji R, et al. Economic burden of spinal muscular atrophy in the USA: a contemporary assessment. J Med Econ. 2020;23(1):70–79.
- Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027–1049.
- Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103–115.
- Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy: part 2: pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018;28(3):197–207.
- Pearson SD, Thokala P, Stevenson M, et al. The effectiveness and value of treatments for spinal muscular atrophy. J Manag Care Spec Pharm. 2019;25(12):1300–1306.
- Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–1722.
- Sturm S, Günther A, Jaber B, et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br J Clin Pharmacol. 2019;85(1):181–193.
- FDA approves oral treatment for spinal muscular atrophy [press release]. 2020 Aug 7. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy
- Mercuri E, Barisic N, Boespflug-Tanguy O, et al. SUNFISH Part 2: efficacy and safety of risdiplam (RG7916) in patients with type 2 or non-ambulant type 3 spinal muscular atrophy. At the 2nd International Scientific Congress on Spinal Muscular Atrophy; 2020 Feb 5-7; Évry, France.
- Seabrook TJ, Baranello G, Servais L, et al. P.064 FIREFISH Part 1: 1-year results on motor function in infants with Type 1 spinal muscular atrophy (SMA) receiving risdiplam (RG7916). Can J Neurol Sci. 2019;46(s1):S31–S.
- Garrison LP, Jackson T, Paul D, et al. Value-based pricing for emerging gene therapies: the economic case for a higher cost-effectiveness threshold. J Manag Care Spec Pharm. 2019;25(7):793–799.
- López-Bastida J, Peña-Longobardo LM, Aranda-Reneo I, et al. Social/economic costs and health-related quality of life in patients with spinal muscular atrophy (SMA) in Spain. Orphanet J Rare Dis. 2017;12(1):141.
- Armstrong EP, Malone DC, Yeh WS, et al. The economic burden of spinal muscular atrophy. J Med Econ. 2016;19(8):822–826.
- Kulaylat AS, Schaefer EW, Messaris E, et al. Truven health analytics MarketScan databases for clinical research in colon and rectal surgery. Clin Colon Rectal Surg. 2019;32(1):054–60.
- US Bureau of Labor Statistics: Consumer Price Index. Measuring price change in the CPI: medical care. [cited 2020 May 6] Available from: https://www.bls.gov/cpi/factsheets/medical-care.htm.
- Cardenas J, Menier M, Heitzer MD, et al. High healthcare resource use in hospitalized patients with a diagnosis of spinal muscular atrophy type 1 (SMA1): retrospective analysis of the kids’ inpatient database (KID). Pharmacoecon Open. 2019;3(2):205–213.
- Lee M Jr., França UL, Graham RJ, et al. Pre-nusinersen hospitalization costs of children with spinal muscular atrophy. Pediatr Neurol. 2019;92:3–5.
- Goble K, Dai D, Boulos F, et al. The economic burden of spinal muscular atrophy patients in a commercially-insured population in the USA. Neurology. 2019;92(15Suppl):P1.6–054.
- National Alliance for Caregiving. Rare disease caregiving in America 2018. [cited 6 May 2020] Available from: https://www.caregiving.org/wp-content/uploads/2018/02/NAC-RareDiseaseReport_February-2018_WEB.pdf.
- Graham RJ, Rodday AM, Weidner RA, et al. The impact on family of pediatric chronic respiratory failure in the home. J Pediatr. 2016;175:40–46.
- Belter L, Cruz R, Jarecki J, editors. HUI, WPAI, and Neuromuscular PedsQL scores in patients with spinal muscular atrophy: findings from the 2019 Cure SMA Community Update Survey. Presented at the 2019 Annual Child Neurology Society Meeting; 2019 Oct 23-26; Charlotte, NC, USA.
- Tan H, Gu T, Chen E, et al. Healthcare utilization, costs of care, and mortality among patients with spinal muscular atrophy. J Health Econ Outcomes Res. 2019;6(3):185–195.
- Farrar MA, Carey KA, Paguinto S-G, et al. Financial, opportunity and psychosocial costs of spinal muscular atrophy: an exploratory qualitative analysis of Australian carer perspectives. BMJ Open. 2018;8(5):e020907.
- Qian Y, McGraw S, Henne J, et al. Understanding the experiences and needs of individuals with spinal muscular atrophy and their parents: a qualitative study. BMC Neurol. 2015;15:217.
- Malone DC, Dean R, Arjunji R, et al. Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J Mark Access Health Policy. 2019;7(1):1601484.
- Al-Zaidy S, Pickard AS, Kotha K, et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr Pulmonol. 2019;54(2):179–185.
- Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 2017;12(1):124.
- Lin CW, Kalb SJ, Yeh WS. Delay in diagnosis of spinal muscular atrophy: a systematic literature review. Pediatr Neurol. 2015;53(4):293–300.
- Ellis A. Spinraza and Zolgensma for Spinal Muscular Atrophy: Effectiveness and Value. Institute for Clinical and Economic Review; 2019. Available at: https://icer-review.org/wp-content/uploads/2018/07/ICER_SMA_Final_Evidence_Report_040319.pdf.