
ABSTRACT
Purpose: Gene therapy brings opportunities to discover cures for diseases for which there are no adequate treatments. As most gene therapies target rare diseases, several challenges are associated with their clinical development, such as limited population size, lack of established clinical pathways for development, and sometimes the absence of validated endpoints. The objective of this study was to systematically review and evaluate the methodology and design of European clinical trials (CTs) utilising gene therapy medicinal products (GTMPs).
Methods: A systematic search of online CT databases was performed using keywords to identify CTs conducted with GTMPs in Europe, published from 1 January 1995 to 31 July 2019.
Results: The search identified 1571 CTs, of which 199 were identified as published articles. A total of 159 CTs remained following the elimination of duplicated CTs, non-gene therapy trials, and those conducted outside Europe. Of these, only nine CTs were randomised, double-blind, with or without parallel groups, and placebo-controlled.
Conclusions: The analysed randomised CTs were conducted in accordance with Good clinical practice with low risk of bias across domains. Only one CT was identified with some concerns of bias due to lack of information regarding the randomisation process and changes in protocol.
Introduction
Gene therapy (GT) is a relatively new and fast-growing field of medicine. Advances in GT have allowed the development of therapies for diseases that are not currently being treated successfully or for which no cure has been found. GTMPs are a type of advanced therapy medicinal product (ATMP) that are defined by the European Medicinal Agency (EMA) as biological medicinal products that contain a recombinant nucleic acid that lead to a therapeutic, prophylactic or diagnostic effect when administered to human beings with a view to regulate, repair, replace, add or delete a genetic sequence [Citation1]. The Committee for Advanced Therapies (CAT) was specifically established by the EMA to give recommendations on ATMP classification; assess their quality, safety and efficacy and provide scientific expertise for their development. In Europe, GTMPs are regulated by the same legal framework as conventional medicines. Since there are many serious unmet medical needs, the EMA has developed several mechanisms for early access and accelerated assessment to facilitate patient access to innovative therapies. However, there are significant additional regulatory challenges in the GTMPs approval process connected with the risks and concerns inherent with GT. This includes strict product definition and standardisation, toxicity and immune response studies to all GTMPs components, target tissue selectivity, and reproductive toxicology. Many GTMPs are developed for rare diseases for which there are not always established clinical pathways for development. Therefore, designing CTs with valid endpoints and strict requirements for statistical accuracy given the limited number of patients are some of the challenges in the development of these therapies [Citation2].
Randomised clinical trials (RCTs) have been accepted as the gold standard to provide evidence for the safety and efficacy of a new intervention and to seek subsequent marketing authorization. Despite the large number of CTs utilizing GTMPs, only seven are currently authorised in Europe.
Our aim was to systematically review and evaluate the methodology and design of CTs utilising GTMPs in Europe.
Methods
Search strategy
A systematic search of online databases using keywords was performed to identify CTs conducted with GTMPs in Europe published from 1 January 1995 to 31 July 2019 in English. Clinical trial registries such as the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT), World Health Organization International Clinical Trials Registry Platform (WHO ICTRP), the Cochrane Central Register of Controlled Trials (CCRCT) and Cochrane Database for systematic reviews (CDSR) were included in the search. The following key phrases were designed to maximize sensitivity for detecting clinical trials conducted with GTMPs: ‘gene therapy’ AND ‘clinical trial’ AND ‘vector’, ‘*’ AND ‘advanced therapy medicinal product’, ‘*’ AND ‘Melanoma’, ‘*’ AND ‘Talimogene laherparepvec’, ‘*’ AND ‘Lymphoma’, ‘Large B-Cell, Diffuse’, ‘*’ AND ‘Tisagenlecleucel’, ‘*’ AND ‘Leber Congenital Amaurosis’, ‘*’ AND ‘Retinitis Pigmentosa’, ‘*’ AND ‘Voretigene neparvovec’, ‘*’ AND ‘Autologous CD34+’, ‘*’ AND ‘Lymphoma’, ‘Follicular’, ‘*’ AND ‘Axicabtagene ciloleucel’, ‘CAR-T’, where ‘*’ = gene therapy.
Selection criteria
Type of therapy applied
Only CTs conducted with medicinal products confirmed in the trial protocol as GTMPs (section D.3.113.2.) or classified as such by the CAT (section D.3.11.3.5) were included in the systematic review.
Study design
Only CTs that were randomised, double-blind, and placebo-controlled or controlled against a standard/available therapy were included in the systematic review.
Data extraction
The review protocol used in this study was developed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement [Citation3]. Two reviewers (K.I. and B.B.) independently extracted data from the selected databases using the pre-defined inclusion criteria described above. Any differences between reviewers about compliance with the inclusion criteria were resolved by consensus before the start of data analysis.
Software and data processing
The following data were extracted and frequentist processing using MS Excel (2010) was performed: registration number, sponsor protocol number, CT name, official title of the CT, scientific title, type of the trial, phase, trial design, investigational medicinal product (IMP) identification as GTMP or not, trade name/international nonproprietary name (INN), principal sponsor, disease/indication (s) and URL. The search was conducted in August 2019.
Risk of bias assessment
Risk of bias was assessed using the Cochrane Risk of Bias tool 2.0 [Citation4].
Results
Study selection
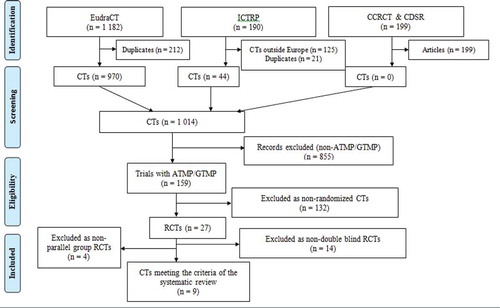
A total of 1571 CTs were discovered in the search, of which 199 were identified as published articles. Of the remaining 1372 CTs, 125 were conducted or ongoing outside Europe and 233 CTs were identified as duplicates. A total of 855 CTs were excluded as they utilized medicinal products that were not of GT origin. Of the 159 CTs using GTMPs, 132 trials were non-randomised. Of the 27 RCTs 14 were excluded as non-double blind CT and 4 were conducted without a parallel group. Only nine of the identified CTs were randomised, double-blind, and placebo-controlled or controlled versus a standard/available therapy (). Four of those were fully analysed. For the other four RCTs, officially published results were not available, which made a full review impossible. One of the nine studies was stopped before patient recruitment. The characteristics of the nine identified CTs are shown in .
Table 1. Characteristics of randomised, double-blind, parallel-group, placebo-controlled CTs included in the systematic review
Figure 1. PRISMA Study Flow Diagram. EudraCT: European Union Drug Regulating Authorities Clinical Trials; ICTRP: International Clinical Trials Registry Platform; CCRCT: Cochrane Central Register of Controlled Trials; CDSR: Cochrane Database of Systematic Reviews; CT: clinical trial; ATMP: advanced therapy medicinal product; GTMP: gene therapy medicinal product; RCT: randomized clinical trial; PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analyses
Analysis of the study results
CTs that were randomised, double-blind, and placebo controlled or controlled against standard/available therapies were analysed using the revised Cochrane Risk-of-Bias tool 2.0 (RoB2) for randomised trials [Citation4]. Six bias domains were evaluated based on signaling questions developed for randomised, parallel CTs; (1)bias arising from the randomisation process. (2) bias due to deviations from intended interventions. (3) bias due to missing outcome data. (4) bias in measurement of the outcome. (5) bias in the selection of the reported result, and (6) overall bias. Based on the assessment of each domain, the overall risk of bias was defined as low, moderate, or high, with the minimum overall risk being determined by the highest risk defined in each individual domain.
RCTs evaluation of risk of bias (Cochrane risk of bias tool 2.0)
Risk of bias arising from the randomisation process
For this domain, RCTs were reviewed for allocation sequence generation and concealment. Proper accomplishment of these processes prevents selective enrolment of the patients based on prognostic factors and introduction of confounding. Baseline values of important prognostic factors were compared between intervention groups. Substantial differences in baseline values between groups suggest a problem with the randomisation process [Citation4]. Randomisation is the element that places RCTs in the highest level of evidence. An external party responsible for randomisation that is remote to the trial centres is the preferred method to achieve adequate concealment of the allocation sequence. This method was used in study nos. 2012–001700-37 and 2006–001246-13 [Citation5,Citation6]. Computer-generated random numbers used in study no. 2011–004761-33 indicate that the randomisation process was performed adequately [Citation7]. Study no. 2004–002508-13 did not meet the minimal criteria for a judgement of adequate concealment of the allocation sequence, as the randomisation process was not described in either the CT protocol or in the published results, which does not meet CONSORT 2010 requirements [Citation8]. In addition, there was no data on how allocation concealment was assured. In all RCTs, there was a balance between the intervention groups size, which leads to the conclusion that the randomisation process was performed properly. Baseline characteristics did not indicate substantial differences between the groups [Citation9,Citation10]. Only in study no. 2004–002508-13 was the risk of bias considered with ‘some concerns’ due to missing information regarding the randomisation process. In other RCTs, the risk of bias in this domain was estimated as low.
Risk of bias due to deviations from intended interventions
In this domain, reviewed RCTs were analysed for the presence of biases related to deviations from the intended interventions. All RCTs included in the analysis were double-blinded as predefined in the inclusion criteria. Proper blinding of the participants and trial personnel was discussed as it defines knowledge of the assigned intervention and possible deviations from it as well-statistical method used to estimate interventions effect.
Blinding of both participants and researches and the identical appearance of the investigated medicinal product (IMP) and placebo were specified in two of the RCTs, study nos. 2012–001700-37 and 2011–004761-33. In study nos. 2006–001246-13 and 2004–002508-13 only the sameness in treatment and placebo volumes and mode of administration were noted. This was considered adequate to avoid differences in health-related behaviors between the intervention groups. For these studies, there was no specific information in the protocol or published data on blinding of patients and researchers [Citation6,Citation9–11].
As outlined in RoB 2.0, when multiple analyses are reported for estimation of intervention effect, the effect of assignment to interventions should be estimated by intention-to-treat (ITT) analysis that includes all randomised participants [Citation4]. In two of the analysed RCTs (study nos. 2012–001700-37 and 2006–001246-13), ITT analyses were performed [Citation5,Citation11]. In study no. 2011–004761-33, analyses were performed in a per-protocol (PP) population defined as all randomised participants who received at least nine doses of assigned intervention. The reasons for discontinuation in the ITT population were similar between treatment and placebo groups and well described in the published paper [Citation7]. In study no. 2004–002508-13, efficacy analyses were performed in a modified ITT (mITT) population which included all randomised patients who had at least one post randomisation treadmill exercise test (273 (94.5%) vs 289) and additional PP patients data analyses were conducted [Citation12]. Safety endpoints on all randomised patients were analysed by the Wilcoxon-Mann-Whitney (WMW) test. Based on the above data, the risk of bias due to deviations from the intended interventions for all reviewed RCTs was evaluated as low. The published results did not mention non-adherence to the therapy, which could have negatively affected the results [Citation9,Citation10].
There was no data on patient adherence to the assigned interventions or discussion regarding the reasons for such deviations if they arose in any of the reviewed RCTs. In study no. 2006–001246-13, it was noted that some of the patients, depending on their clinical status and at the decision of the investigator, had more radiological examinations than planned in the protocol [Citation6]. However, this deviation reflects usual practices and cannot be considered as a basis for bias. The absence of such data made it very challenging for the review authors to estimate whether deviations from intended intervention arose because of the trial context or not. Based on the signaling questions for this domain in the RoB 2.0 tool, the risk of bias due to deviations from intended interventions was estimated as low for all RCTs reviewed.
Risk of bias due to missing outcome data
This domain addressed risk of bias due to missing outcome data, including biases introduced by procedures used to impute or otherwise account for missing outcome data [Citation4]. These biases rise not only because of missing data but most notably because of missing data mechanism. From the data given in the CTs synopses and published results, it was very challenging to identify whether the chance that the outcome was missing depended on its true value. For this reason, the assessment of the analysed RCTs was performed based on whether the outcome was measured in all participants. The size of the number of participants with missing outcome data and analyses was used to confirm that missing data id not significantly alter the estimated interventions effect.
In study no. 2012–001700-37, primary analysis of the primary and secondary endpoints was done in mITT population that comprised all randomised participants who received the intervention and excluded subjects who were AAV1 neutralizing antibody positive (243 patients, 97%). Secondary analyses were conducted with the ITT population, which consisted of all 250 randomised patients. Data obtained by both analyses were similar (ITT: n = 250, HR 95% CI, recurrent events: 0.92 (0.53–1.62); terminal events: 1.23 (0.71–2.14); mITT: n = 243, HR 95% CI, recurrent events: 0.93 (0.53–1.65); terminal events: 1.27 (0.72–2.24)). The joint frailty model, a semi-parametric analysis that takes into account recurrent clinical events, uneven follow-up periods between groups, and terminal events as a competing risk [Citation5], was used in the primary study analysis.
In study no. 2011–004761-33, the primary analysis was done in PP population predefined as participants who received at least nine doses of IMP or placebo. ANCOVA testing was conducted by the trial investigators to adjust the treatment effect in the IMP group versus placebo at 12 months follow up. The ANCOVA-adjusted treatment effect (3.7%, 95% CI 0.1–7.3; p = 0 · 046) was comparable with the treatment effect in patients in the ITT population (3.6%, 95% CI 0.2–7.0; p = 0.039) which demonstrated that probable values of the missing outcome data did not significantly alter the estimated intervention effect. The reasons for missing data between the two intervention groups were similar, well documented, and not related to the CT outcome itself [Citation7].
In study no. 2006–001246-13, the primary efficacy and safety analyses were done in the ITT population comprising all 732 patients enrolled in the CT. The main reasons for premature withdrawal were the sponsor’s decision to discontinue treatment with the CT drug, withdrawal initiated by the investigator, and IC withdrawn by the patient [Citation6].
In study no. 2004–002508-13, out of 289 randomised participants across the 4 patient groups, 273 patients (94.5%) that had at least one treadmill test were included in the efficacy analysis (mITT). The number of patients with missing test was similar across three treatment groups and placebo, and the reasons for not performing it were described in the published results. The WMW test, stratified by the presence or absence of diabetes mellitus at baseline was applied separately to three comparisons between each treatment group and the placebo group to compare the primary efficacy endpoint [Citation9]. Missing data from the 26-week peak walking time test (PWT), where the percentage change from baseline was used as primary efficacy endpoint, was input to the last-observation-carried-forward method (LOCF). LOCF is commonly used as a method of imputing missing data in longitudinal studies. The method has been criticized because, depending on last observed data, it assumes no disease progression or improvement after dropout, which leads to biased results [Citation13].
In two of the RCTs (study nos. 2012–001700-37 and 2006–001246-13) ITT analyses were performed and published. In study no. 2011–004761-33, the primary analysis was done in the PP population but used ANCOVA-adjusted treatment effects that demonstrated no significant difference in the results from ITT and PP analyses. In study no. 2004–002508-13, mITT analyses were performed. In all reviewed RCTs, in both intervention and placebo groups the size of the number of participants with missing outcome data and reasons for this were similar. Study nos. 2012–001700-37, 2011–004761-33 and 2006–001246-13, were assessed with low risk of bias in this domain. The proportion and reasons for missing outcome data in the experimental and placebo intervention groups in study no. 2004–002508-13 were similar, which suggest low risk of bias. Because of the missing clear justification for using LOCF [Citation4], the risk of bias due to missing outcome data was assessed as with ‘some concerns’.
Risk of bias in measurement of the outcome
This domain focuses mainly on differential errors related to intervention assignment. Such errors are less probable when outcome assessors are blinded to intervention assignment. Risk of bias in this domain depends on appropriateness of the method used for outcome measuring, the outcome assessor and whether the outcome assessor is blinded to intervention assignment [Citation4]. This data is important for assessing the risk of bias in outcome measurement, especially when knowledge of the intervention can influence the judgment of the outcome assessor.
In Study no. 2012–001700-37, all patients, physicians and outcome assessors were blinded to treatment assignment. The study was an event-driven trial and all clinical events were reviewed by both a non-blinded Data Monitoring Committee and an independent blinded Clinical Endpoints Committee. Any clinical events served as an automatic trigger for safety evaluation by the blinded Clinical Endpoints Committee [Citation12].
In study no. 2011–004761-33, the CT was monitored at intervals by an independent Data Monitoring and Ethics Committee (DMEC), which verified the blinded group data and provided written confirmation that the trial could continue without modification. Clinical examination results, including pulmonary function, gas transfer markers, and systemic inflammation indicators were examined in a masked manner by the DMEC [Citation14].
The available data for study no. 2006–001246-13 did not provide specific information on the masking of participants. However, the outcomes reported, including overall survival (OS) and the laboratory variables (complete blood count and chemistry panel), did not involve judgement that would potentially lead to bias. The data from secondary efficacy endpoints, and progression-free survival were adjudicated by blinded peer review [Citation6].
In study no. 2004–002508-13, the methods of outcome measurement were very specific and the same measurement methods and thresholds in both the intervention and placebo groups were used at comparable time points. There was no specific data provided on trial participants masking. However, the primary efficacy parameter was assessed by PWT, which did not involve outcome assessor judgement. After the first interim assessment of unblinded patient safety data, assessment by the independent Data Monitoring Committee (DMC) was blinded [Citation9].
Only in one of the reviewed RCTs (study no. 2012–001700-37) was detailed information for blinding of all participants in the trial provided. All RCTs included in the analyses were predefined as double-blind. The definition of the CT as double-blind was likely considered by the trialists as sufficient, with no need for additional data regarding of outcome assessors masking. The risk of bias in the evaluation of results for all RCTs was assessed as low.
Risk of bias in the selection of the reported result
In this domain, RCTs were reviewed for risk of bias that arose because of selection of the reported results [Citation4]. To ensure better consistency in reported outcomes in trials in the same clinical area, core outcome sets (COS) have been developed. These sets serve to advise on a number of essential outcomes that should be measured and reported in all clinical trials for a specific condition [Citation15]. A review of available COS for the clinical field of analysed RCTs during the period they were conducted was performed as well as a comparison between trial reports and their protocols concerning consistency in planned and published outcome measures and analyses.
There was no consensus reached on optimal phase II endpoints in acute or chronic heart failure trials [Citation16] and study no. 2012–001700-37 was analysed based on the trial protocol and published data. The efficacy and safety endpoints analyses were performed as planned in the CT protocol. There were amendments in the protocol, which were presented in detail in the published data (https://www.clinicaltrialsregister.eu/ctr-search/trial/2012-001700-37/results). Post-hoc analyses for the primary and secondary endpoints, stratified by randomisation in the study before and after the protocol amendments, were performed to ensure that no meaningful differences in the treatment effect were observed (primary outcome: HR 0.86 [95% CI 0.32–2.27] before amendment vs 1.05 [0.53–2.08] after amendment; secondary outcome: 1.14 [0.53–2.44] vs 1.38 [0.59–3.25]) [Citation5].
Standardised outcome measures for the cystic fibrosis (CF) CT were extensively discussed [Citation17], but there was no consensus (http://www.comet-initiative.org/studies/details/882). Study no. 2011–004761-33, was designed after extensive study of the published CF RCTs [Citation14]. A detailed plan for statistical analysis was approved and finalised before the database was locked and the study unblinded. The relevant data were extracted once the database was locked and the researchers were unblinded [Citation14]. No deviations were observed between planned and published outcome measurements and analyses.
Currently, a program that is still ongoing started developing COS for CTs in renal cancer (http://www.comet-initiative.org/studies/details/1406). The clinical outcomes in study no. 2006–001246-13 were common for cancer therapy (patient survival and progression-free survival). There was no deviation from the researchers’ pre-specified intentions for planned outcome measurements and analyses [Citation11].
The COS for peripheral arterial disease was developed in 2018 [Citation18]. In study no. 2004–002508-13, all eligible reported results for the outcome corresponded to all intended outcome measurements [Citation9,Citation10]. The CT data were analysed using the predetermined methods of analysis. There were changes made in the protocol, but they were not specified, raising concerns about bias in selection of the reported results.
Although no COSs in respective therapeutic areas were available during the RCTs design, the most common clinical field outcome measures were planned and performed. There were no observed inconsistencies between outcome measures and analyses intentions and publications in the reviewed RCTs. The risk of bias due to the choice of reported results for all RCTs was considered low.
Overall risk of bias
Based on the reviewed data, the overall risk of bias in study nos. 2012–001700-37, 2011–004761-33, and 2006–001246-13 was estimated as low. Concerning study no. 2004–002508-13, the mechanism of randomisation was not described and changes in the protocol from the original version were not specified. Given that the minimum overall risk is usually determined by the highest risk identified in each domain, the overall risk of bias for study no. 2004–002508-13 was estimated with ‘some concerns’.
Discussion
This review indicates that most CTs conducted with GTMPs have a very small number of patients, are single-arm trials, and are conducted without comparative therapy or placebo. This is due to the fact that a large number of GTs are developed for the treatment of rare diseases (113 [71%]) of the 159 clinical trials identified in this search. Thus, there are few or no therapeutic alternatives for patients, not always established clinical pathways for development, and the limited number of patients does not generate the data required for treatment approval. Оf the 9 RCTs, 5 (56%) were for studying GTs for rare diseases (cystic fibrosis, locally advanced or metastatic renal clear cell adenocarcinoma and LHON). All three diseases are more common than other rare diseases, which allowed the recruitment of a sufficient number of patients [Citation19–21]. The analysed CTs were performed according to RCT requirements, following standards for their planning and results reporting. Statistical analysis based on the disease studied and the size of the study population adequately reflected the obtained outcomes with low risk of bias across the domains. In three CTs, the risk in all bias domains was rated as low and only one CT identified a moderate risk of bias due to lack of information regarding the randomization process.
Identified pitfalls were missing information in reported and published data regarding (a) sequence generation and allocation concealment; (b) blinding mechanism for trial participants; (c) adherence to assigned intervention.
In one of the RCTs the mechanism used to apply a random patient distribution sequence was not described. In three of the reviewed RCTs, blinding was described only with the broad term ‘double blind’. Explicit reporting on the blinding mechanism for patients and trial personnel was given only in one of the RCTs. In addition, only limited data were provided in the reviewed RCTs regarding deviations from intended intervention that were inconsistent with the trial protocol. In study no. 2011–004761-33, initiation of a treatment non-compliant with the protocol was mentioned as a reason for dropout of patients. In EudraCT only synopses for RCTs results that contain modest information are available. The principles of the CONSORT statement should be widely followed for improving the quality of reporting of RCTs. Challenges in conducting CT with ATMP are also posed by their biological nature and specific manufacturing process. In study no. 2006–001246-13, some of the batches of IMP were contaminated with the wild-type virus used for gene delivery which affected the clinical potential of the IMP. The need for better understanding of the pathophysiology of some of diseases and the mechanism of action of the therapies, along with limitations in recruitment of sufficient number of patients, makes generating adequate and robust data on the therapy safety and efficacy difficult. It also impedes the conduct of more RCTs with GTs. Bias in planning CT design could be minimised by cooperation between academia worldwide similar to the CF Therapeutics Development Network (CF-TDN) introduced in 1998 by the Cystic Fibrosis Foundation with the aim to speed the delivery of new therapies through efficient study design, optimized clinical trial execution, and high-quality data [Citation22].
Selection of reliable and well-defined objective endpoints which demonstrate clinical benefits and design plans for managing missing outcomes will minimize the bias in outcome analysis. In clinical areas where potential GTs may offer treatment options, academia should consider the development of COS to evaluate efficacy and safety similar to the coreHEM project (http://www.comet-initiative.org/studies/details/997).
One of the limitations of the current review is that only a small number of RCTs were identified in our search. According to the Global Gene Therapy Clinical Trials Registry [Citation23], 2686 CTs were conducted in a shorter time period than the current systematic review (1 January 1995–31 December 2018), which is a far greater number than that found in the current search. This may be due to lower keyword sensitivity of the databases employed in this review or the possibility that not all CTs with GTMPs conducted in Europe are registered in EudraCT and ICTRP. Another limitation of this review is that only RCTs were considered for analysis. This significantly limits the conclusions that can be drawn about the methodology and design of CTs with GTMPs due to the small number of identified RCTs. Only qualitative methods were used for assessing the RCTs. The use of a statistical tool would be beneficial for drawing empirical evidence for how the presence of bias in different domains influences the estimation of the intervention effect.
Disclosure of interests
The authors report no conflict of interest.
References
- https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
- Schneider CK, Salmikangas, P., Jilma, B. et al. Committee for Advanced Therapies (CAT), CAT Scientific Secretariat. Challenges with advanced therapy medicinal products and how to meet them. Nat Rev Drug Discov. 2010;9:195–8.
- Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Available from: http://www.prisma-statement.org/
- Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898.
- Greenberg B, Butler J, Felker GM, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387(10024):1178–1186.
- TroVax® Protocol TV3/001/06 Synopsis, www.clinicaltrialsregister.eu/ctr-search/trial/2006-001246-13/results
- Alton EWFW, Armstrong DK, Ashby D, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3(9):684–691.
- Schulz KF, Altman DG, Moher D, et al. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. PLoS Med. 2010;7(3):e1000251.
- Ad2/HIF-1α/VP16, Abbreviated Clinical Study Report: PADHIF00704. Available from: https://www.clinicaltrialsregister.eu/ctr-search/trial/2004-002508-13/GB
- Creager МА, Olin JW, Belch JJ, et al. Effect of hypoxia-inducible factor-1α gene therapy on walking performance in patients with intermittent claudication. Circulation. 2011;124(16):1765–1773.
- Amato RJ, Hawkins RE, Kaufman HL, et al. Vaccination of metastatic renal cancer patients with MVA-5T4: a randomized, double-blind, placebo-controlled phase III study. Clin Cancer Res. 2010;16(22):5539–5547.
- Greenberg B, Yaroshinsky A, Zsebo KM, et al. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail. 2014;2(1):84–92.
- Mavridis D, Salanti G, Furukawa TA, et al. Allowing for uncertainty due to missing and LOCF imputed outcomes in meta-analysis. Stat Med. 2019;38(5):720–737.
- Alton EWFW, Armstrong DK, Ashby D, et al. A randomised, double-blind, placebo-controlled trial of repeated nebulisation of non-viral cystic fibrosis transmembrane conductance regulator (CFTR) gene therapy in patients with cystic fibrosis. Efficacy Mech Eval. 2016;3(5):1–210. Available from: https://www.ncbi.nlm.nih.gov/books/NBK373650/.
- Clarke M. Standardising outcomes for clinical trials and systematic reviews. Trials. 2007;8(1):39.
- Zannad F, Garcia AA, Anker SD, et al. Clinical outcome endpoints in heart failure trials: a European society of cardiology heart failure association consensus document. Eur J Heart Fail. 2013;15(10):1082–1094.
- Döring G, Elborn JS, Johannesson M, et al. Clinical trials in cystic fibrosis. J Cyst Fibros. 2007;6(2):85–99.
- Rieß HC, Debus ES, Schwaneberg T, et al. Indicators of outcome quality in peripheral arterial disease revascularizations – a Delphi expert consensus. Vasa. 2018;47(6):491–497.
- Rowe SM, Borowitz DS, Burns JL, et al. Progress in cystic fibrosis and the CF therapeutics development network. Thorax. 2012;67(10):882–890.
- EU/3/06/429. Available from: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu306429
- EU/3/11/860. Available from: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu311860
- Cystic fibrosis foundation. Available from: https://www.cff.org/Research/Researcher-Resources/Therapeutics-Development-Network/Working-With-the-TDN/
- Gene therapy clinical trials worldwide. Available from: http://www.abedia.com/wiley/years.php