
ABSTRACT
Background
Standard of care (SoC) for transfusion-dependent β-thalassemia (TDT) requires lifelong, regular blood transfusions as well as chelation to reduce iron accumulation.
Objective
This study investigates the cost-effectiveness of betibeglogene autotemcel (‘beti-cel’; LentiGlobin for β-thalassemia) one-time, gene addition therapy compared to lifelong SoC for TDT.
Study design
Microsimulation model simulated the lifetime course of TDT based on a causal sequence in which transfusion requirements determine tissue iron levels, which in turn determine risk of iron overload complications that increase mortality. Clinical trial data informed beti-cel clinical parameters; effects of SoC on iron levels came from real-world studies; iron overload complication rates and mortality were based on published literature.
Setting
USA; commercial payer perspective
Participants
TDT patients age 2–50
Interventions
Beti-cel is compared to SoC.
Main outcome measure
Incremental cost-effectiveness ratio (ICER) utilizing quality-adjusted life-years (QALYs)
Results
The model predicts beti-cel adds 3.8 discounted life years (LYs) or 6.9 QALYs versus SoC. Discounted lifetime costs were $2.28 M for beti-cel ($572,107 if excluding beti-cel cost) and $2.04 M for SoC, with a resulting ICER of $34,833 per QALY gained.
Conclusion
Beti-cel is cost-effective for TDT patients compared to SoC. This is due to longer survival and cost offset of lifelong SoC.
Introduction
Transfusion-dependent β-thalassemia (TDT) is a rare, severe genetic disease characterized by reduced or absent production of the β-globin protein in adult hemoglobin due to mutations in the hemoglobin subunit beta (HBB) gene, leading to ineffective red blood cell production and profound anemia. While the overall prevalence of thalassemia in the USA (US) has increased by 7.5% over the last 50 years due to changes in immigration patterns [Citation1], β-thalassemia remains extremely rare, with the most severe form, TDT, affecting only about 1,000 people nationwide [Citation2]. Although allogeneic hematopoietic stem cell transplant (allo-HSCT) can be curative, it carries significant risk of transplant-related mortality that increases with age, graft versus host disease (GVHD), and graft rejection. Hence, allogeneic transplants are generally restricted to pediatric patients who have available human leukocyte antigen (HLA) matched sibling donors [Citation3–7]. An international (US, China, India) study examining characteristics and outcomes of 1110 patients undergoing allo-HSCT for beta-thalassemia between 2000 and 2016 found that 61% were performed using HLA-matched sibling donors, 16% developed GraedeII-IV acute GVHD and 11% experienced chronic GVHD [Citation7].
Standard of care (SoC) for TDT consists of lifelong, regular (typically every 2 to 5 weeks) blood transfusions to maintain a pre-transfusion hemoglobin level above 9 g/dL [Citation8,Citation9]. This only transiently addresses symptoms of TDT and inevitably leads to significant iron accumulation. Widespread organ damage from transfusion-induced iron overload – primarily due to cardiac, liver, and endocrine dysfunction – is the major cause of morbidity and mortality, with cardiac dysfunction the leading contributor to reduced life expectancy [Citation10–12]. For example, in the UK Hospital Episode Statistics database, 612 patients with TDT were identified: 40% had endocrine disorders (excluding diabetes), 34% diabetes, 40% osteoporosis, and 18% cardiac disease. The crude 10-year mortality was 6.2%, significantly greater than the 1.2% age and sex-adjusted mortality in the general population (p < 0.001) [Citation13].
Iron chelation is used to mitigate iron overload [Citation8] but can be associated with serious adverse effects in multiple organ systems [Citation14–16]. Thus, patients receiving iron chelation must be closely monitored to individualize their chelation as well as monitor for associated toxicities [Citation14,Citation15]. Moreover, iron chelation may be less efficacious in real-world clinical practice, in part because of poor compliance [Citation16]. A 2019 retrospective study using US insurance claims data estimated annual costs for β-thalassemia patients regularly transfused to be $128,062 with chelation accounting for 50% and outpatient care (including blood transfusions and iron monitoring) for 47% of these costs [Citation17].
Betibeglogene autotemcel (beti-cel; previously known as LentiGlobin for β-thalassemia) is a one-time gene addition therapy for TDT [Citation18]. The goal of treatment with beti-cel is to correct ineffective erythropoiesis and enable lifelong, stable production of functional adult hemoglobin at levels sufficient to result in transfusion independence. By addressing the underlying cause of TDT, a single administration of beti-cel offers the possibility of eliminating the need for ongoing transfusions for life [Citation19] and, thus, further long-term sequelae of the disease. Beti-cel treatment consists of transplantation of autologous CD34+ hematopoietic stem cells (HSC) transduced ex-vivo with the BB305 lentiviral vector containing functional copies of an engineered β-globin gene with a threonine to glutamine substitution at position 87. CD34+ HSC are mobilized and collected via apheresis of peripheral blood prior to genetic modification; the patient undergoes full myeloablative conditioning prior to intravenous (IV) infusion of the modified cells (e.g., in the clinical trials, this has included single-agent busulfan). Risks of myeloablative conditioning must be taken into consideration when deciding on treatment with beti-cel and include acute toxicities, infertility, secondary malignancy, etc. Also, theoretical gene therapy safety risks (e.g., insertional oncogenesis) need to be monitored.
Some risks associated with gene therapy, such as insertional oncogenesis, remain theoretical. In a review of safety outcomes in clinical studies of hemopoietic stem cell gene therapy using lenti-viral vectors for non-oncologic diseases (N = 125), no cases of insertional oncogenesis occurred. Maximum follow-up time across these studies was 8 years [Citation20]. Nevertheless, patients should continue to be monitored for these adverse effects.
Clinical trials have evaluated beti-cel in patients with TDT regardless of genotype (HGB-204 [NCT01745120] and HGB-205 [NCT02151526]) and, in ongoing Phase III trials, specific subpopulations defined by genotype (patients with a non-β0/β0 genotype: HGB-207 [NCT02906202]; those with a β0 or β+IVS-I−110 mutation at both alleles of the HBBgene: HGB-212 [NCT03207009]). Beti-cel received conditional approval from the European Medicines Agency in May 2019 for the treatment of patients ≥12 years of age with TDT who do not have β0/β0 genotype and for whom allo-HSCT is appropriate, but an HLA-matched donor is not available. Beti-cel is not yet approved in the US.
Given that one-time gene therapy represents a new treatment paradigm, and that beti-cel targets a disease with a well-established SoC, its economic implications must be examined to understand its value and inform coverage decisions. To enable this assessment, a decision-analytic model was developed to estimate the incremental cost-effectiveness of beti-cel versus SoC for TDT, with analyses conducted from a US commercial payer’s perspective.
Methods
Model overview
The economic model simulates the course of TDT disease, treatment and outcomes. It was implemented in Microsoft Excel® using the discretely integrated condition event (DICE) platform [Citation21–23]. Design, implementation, and validation followed modeling good research practices recommendations from the International Society for Pharmacoeconomics and Outcomes Research and Society for Medical Decision Making [Citation24]. The DICE approach involves conceptualizing the decision problem in terms of the information handled by the model over time (‘conditions’) and the changes to those conditions that occur at specific points in time (‘events’). DICE is particularly suitable for time-to-event modeling, which was selected for this model due to the complexity of the clinical pathway, with the alternatives being compared having different trajectories and structures. Specifically, DICE enables implementation of event-driven models in Microsoft Excel without the need for specialized software. DICE also supports adding options to enable conducting scenario analyses and addressing structural uncertainty (more details in Supplemental Data).
Model design and assumptions
The model tracks individual patients over their lifetime, defining their disease state using several conditions (), primarily related to transfusion requirements and degree of iron deposition in the heart, liver, and serum. Based on these conditions, the patients may experience various events, including the development of iron-related complications and death. The time until each event occurs is randomly drawn for each individual from a distribution specified based on fits of the underlying data [Citation25].
Figure 1. Model overview
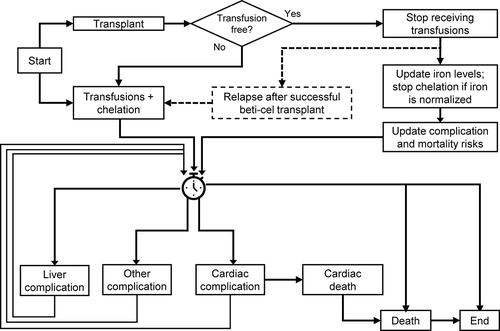
The clinical effects of treatment are primarily mediated through iron loading, with the risk of complications determined by the distribution of iron loading in each tissue. A key outcome in the model is transfusion independence, defined as no transfusions and a weighted average total hemoglobin of at least 9 g/dL for at least 12 continuous months after transplantation, in line with the primary endpoints in beti-cel trials. In patients who achieve transfusion independence (85.2% in the phase III trials), the need for chelation is eliminated after a normalization period, resulting in a minimal risk of further iron-related complications. This potentially curative scenario represents the base-case scenario modeled for beti-cel in the economic analysis.
Pediatric, adolescent, and adult patients aged 2 to 50 years (i.e., to match the clinical trial inclusion/exclusion criteria for age) with TDT and either β0/β0 or non-β0/β0 genotype (i.e., who carry a mutation of the β+ or βE type on at least one allele of their β-globin gene) enter the model at a Start event. Each patient is simulated twice, once with SoC consisting of continued transfusion plus chelation, and again with beti-cel. When simulating beti-cel, the model considers preparation for the gene therapy procedure, including hypertransfusion, apheresis for peripheral collection, and veno-occlusive liver disease prophylaxis in the inpatient hospital setting, followed by a myeloablative conditioning regimen (with single-agent busulfan) and beti-cel infusion. While engraftment failure has not been observed to date in the beti-cel clinical trials [Citation26], the possibility of failure cannot be excluded entirely. The model supports what-if scenario analyses for assessment of the possibility that some patients will experience engraftment failure within 90 days of transplantation [Citation18]. If engraftment failure were to occur, patients would receive a rescue infusion of unmodified autologous cells. The risk of death after failed engraftment is included as a parameter in the model. After engraftment success, the model allows for a proportion of patients to not achieve transfusion independence; these patients continue with SoC.
Following the achievement of transfusion independence, elevated levels of iron may persist, and iron chelation is continued for some time or patients may be phlebotomized to reduce iron. Based on the literature, beti-cel trial, and post-transplant iron data, the model incorporates a 1-year period of cardiac iron normalization, and a 3-year period of iron normalization for serum ferritin and liver iron, following successful transplantation; a longer iron normalization period of 6 years was considered in a scenario analysis [Citation27]. Although there is no scientific expectation of, nor data suggesting reversion to transfusion dependence, a time-dependent relapse possibility was considered in a scenario analysis. Following myeloablative conditioning, patients may be also at risk of infertility during a childbearing window. Clinicians mitigate this risk by offering a preservation strategy, so the associated costs are considered [Citation28,Citation29].
Iron levels at baseline are randomly assigned to patients using a categorical distribution (low, medium, or high overload) derived from data generated in the Thalassemia Longitudinal Cohort (TLC) study (refer to TLC Report in Supplemental Data). Patients receiving SoC at baseline and patients who receive beti-cel but do not achieve transfusion independence post-transplant are assumed to maintain constant iron levels over their lifetimes.
The risks for cardiac complications, liver complications, diabetes, or hypogonadism depend on iron levels. Rates of cardiac and liver complications are derived from the TLC report and MarketScan data, respectively (see Supplemental Data). Published risk equations were used to predict the incidence of diabetes and hypogonadism (). All complications are assumed to be permanent and incur ongoing management costs and QoL impairment.
Table 1. Model inputs
Validation
Face validity of the model was ascertained by a practicing hematologist with expertise in clinical aspects of TDT and an external panel of health economists with experience in TDT. The implementation was extensively verified by an independent modeler, following a documented and structured formal process. No independent predictive validation was undertaken because of unavailability of suitable data.
Model inputs
Main inputs are provided in (details in Supplemental Data). The modeled population is specified in terms of profiles of age, sex, weight, baseline comorbidity, iron level measured by serum ferritin, liver iron concentration (LIC), and myocardial magnetic resonance imaging (MRI) T2-star (T2*).
Based on pooled data from the Phase III HGB-207 and HGB-212 clinical trials as of March 2020 [Citation26], no engraftment failure for beti-cel was assumed and 85.2% achieve transfusion independence after engraftment. The remaining 14.8% continue to receive transfusions. Patients who achieve transfusion independence continue chelation therapy (75%) or begin phlebotomy (25%) to normalize iron levels for 3 years post-transplant, during which time associated annual costs and utility decrements are applied. Survival and quality of life (QoL) in patients who achieve transfusion independence without having developed iron-related complications are based on data for the age-matched general population, adjusted downward for potential long-term effects of undergoing myeloablative conditioning. Thus, an impairment of QoL (−0.02) and survival (1.25 standardized mortality ratio, SMR) are applied in the base case, with further decrements for any iron-related complications that were pre-existing or that developed prior to normalization. Continued transfusion dependence without cardiac complications carries an SMR of 3.9 [Citation35]. Cardiac complications also increase annual mortality to 13%.
Based on US MarketScan (insurance claims) data [Citation36], the breakdown of chronic chelation in TDT was 74% oral (assumed 73% deferasirox, 21% deferiprone, and 6% deferasirox+deferiprone); 7% subcutaneous (SC) deferoxamine mesylate; and 19% oral plus SC. Adverse events associated with chelation were based on published data (deferoxamine mesylate) [Citation37] and prescribing information (deferasirox [Citation14], deferiprone [Citation38]).
Utilities are summarized in (details in Supplemental Data). A vignette-based utility study of transfusion and oral chelation yielded a decrement of −0.20, and with subcutaneous chelation a decrement of −0.30 [Citation39]. A scenario analysis considered a 0.15 decrement, based on publications focusing on disutility of different chelation formulations [Citation40].
Table 2. Utility decrements
Cost inputs () in 2020 US dollars were based on MarketScan data [Citation36] or derived by micro-costing methods. Costs were inflated from prior years, if necessary, using the medical component of the US Consumer Price Index [Citation42]. At the time of these analyses, beti-cel had not yet been filed for approval with the US Food and Drug Administration and thus its price had not been set. Therefore, a base-case cost of $2,100,000 was assigned based on public comments made by the manufacturer (see https://www.wsj.com/articles/biotech-proposes-paying-for-pricey-drugs-by-installment-11546952520). Per the manufacturer’s proposed payment model, the acquisition cost of beti-cel is spread in equal annual payments over 5 years. The initial payment is incurred at the time of transplant, but the following payments are due only if a patient remains transfusion-independent at that time. A scenario analysis examines the impact of an alternative payment model where the full cost of beti-cel is incurred upfront, with an 80% rebate in subsequent years (maximum: 3) if the patient is not transfusion-independent. Other prices were considered in scenario analyses.
Table 3. Cost inputs
Analyses
The analytic perspective was that of a US commercial payer, focusing on direct lifetime medical costs, accumulated life years (LYs), and quality-adjusted life years (QALYs), all discounted at 3% per year [Citation47]. Given the stochastic nature of the model, the results vary each time the model is run. Based on analyses that showed when results stabilized (varied less than 1%), 150 replications were simulated for a run. Deterministic parameter uncertainty and scenario analyses assessed the impact of varying inputs over plausible ranges and of alternative structural assumptions. In probabilistic sensitivity analyses (PSA), 500 parameter sets were simulated with 150 replications each, varying clinical, cost, and disutility inputs with parameter uncertainty within their 95% confidence intervals.
Results
The model predicts an gain (undiscounted) of 13.8 LYs and 18.0 QALYs, 3.8 LYs and 6.9 QALYs (discounted) per patient treated with beti-cel as compared with SoC (). Lifetime (from mean entry age of 22.5 years) costs of treating the disease were $2,038,384 for SoC and $572,107 for beti-cel (excluding the cost of beti-cel itself). This large cost offset ($1,466,277) was due to lower costs associated with blood transfusions, iron chelation treatment, and iron monitoring. At the base case assumed price of beti-cel, the net cost is $238,709, resulting in an incremental cost-effectiveness ratio (ICER) of $34,833 per QALY gained for beti-cel.
Table 4. Base-case results
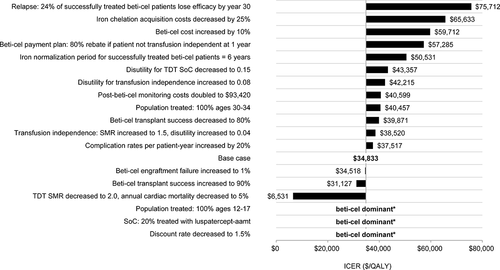
The impact of selected alternative scenarios on the ICER, always including the cost of beti-cel, is reported in . Beti-cel dominated (i.e., more effective, and less costly) SoC if the discount rate was 1.5% (costs and outcomes); and also in a subgroup of patients ages 12–17, and against a modified SoC that adds the lifetime use of luspatercept-aamt (approved for US use in November 2019) [Citation49]. This scenario assumed luspatercept-aamt acquisition costs from $119,000 to $239,000 annually, depending on body weight and dose; 50% improvement in the existing TDT SoC health state utility and 20% market uptake of luspatercept-aamt. In all scenarios, the ICER was below a common US cost-effectiveness threshold of $100,000/QALY (). The scenarios that decreased the probability of treatment success with beti-cel (i.e., transfusion independence) from 85.2% or increased the cost of beti-cel by 10% each yielded larger ICERs ($39,871 and $59,712, respectively), but beti-cel remained cost-effective. These results reflect the outcome-based payment plan in this analysis, where the majority of the beti-cel cost is not incurred if patients do not achieve transfusion independence.
Figure 2. Deterministic scenario analyses results tornado diagram
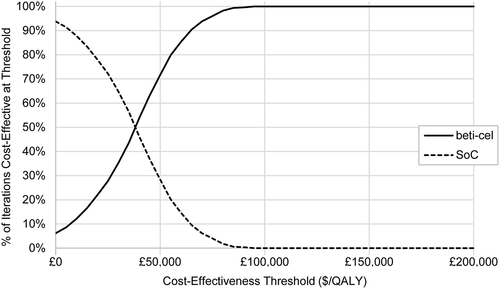
The scatterplot of PSA results () shows that beti-cel is more costly but more effective than SoC in 90% of the 500 parameter sets simulated, and dominant in the other 10%. Beti-cel is cost-effective versus SoC () in 100% of iterations at any willingness to pay above $89,000 per QALY gained.
Figure 3. Probabilistic sensitivity analyses results: scatter plot of incremental outcomes
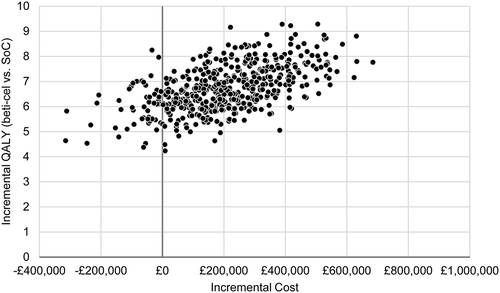
Figure 4. Cost-effectiveness acceptability curve
Discussion
The model results suggest that, for US payers, one-time gene therapy with beti-cel is a highly cost-effective treatment option for TDT as compared with current SoC. In the potentially curative base case scenario, beti-cel has the potential to extend life by decades and substantially improve QoL, as evidenced by the modeled discounted gain of 3.8 LYs and 6.9 QALYs. The higher up-front costs of beti-cel are offset in large part by the reduction in lifetime SoC costs for those patients achieving transfusion independence. Collectively, these benefits support an estimated ICER of $34,833 that is well below an even more stringent WTP threshold of $50,000 per QALY [Citation47].
Extensive scenario and sensitivity analyses confirmed the robust nature of the results. In some modeled scenarios, beti-cel is the dominant strategy. Since an outcomes-based agreement was modeled, any lower effectiveness of beti-cel in practice has limited impact on the ICER because the payer is compensated.
While the impact of beti-cel is expected to be lifelong in patients who achieve transfusion independence [Citation50], a theoretical concern is that the effect of gene therapy may eventually diminish or be lost. For the effects of gene addition therapies such as beti-cel to be durable in the long term, a population of undifferentiated HSC that carry the therapeutic trans-gene of interest integrated into their genome must be established in the bone marrow. In the case of gene therapy with beti-cel, HSC that harbor the gene therapy-derived β-globin gene serve as a long-term reservoir for blood cells that express functional β-globin protein, thus ensuring durable expression [Citation51]. There is evidence from allo-HSCT that provides strong support for the long-term durability of transplanted stem cells [Citation52,Citation53] and improvement in patient QoL [Citation54,Citation55]. In the field of gene therapy specifically, there is also growing evidence in support of long-term durability [Citation56,Citation57]. Although in beti-cel clinical trials (through March 2020) no patient who has achieved transfusion independence has subsequently resumed transfusions, a scenario including such a risk was analyzed. Although it had the most impact on the cost-effectiveness of beti-cel, the ratio remained below 100,000. This reflects both the discounting of events that occur far in the future and avoidance of some permanent organ damage prior to relapse, yielding improved outcomes even if the direct effect is eventually lost.
Another strength of our study is the use of a disease modeling approach to allow for projection of clinical events over long periods despite an absence of observed data over that time. Consideration of the effect of beti-cel on intermediate outcomes (e.g., transfusion independence) that can be measured in the short term allows the link between the intermediate outcome (transfusion independence and iron normalization) and long-term outcomes (iron overload complications such as cardiac and liver toxicity and complication-related mortality) to be quantified based on the broader clinical literature. This approach introduces more components to the modeling, but their addition is facilitated by use of the DICE methodology.
Such an approach is likely to be appropriate for many one-time therapies in chronic conditions. Direct evidence of their impact on long-term outcomes will be lacking at the time they need to be evaluated. Using a modeling approach that connects short-term to long-term outcomes based on understanding of the disease process allows the use of the evidence where it is strongest: the direct clinical effects of treatment inform short-term efficacy, while the broader clinical literature provides decades of evidence on the chronic disease that can be used to translate that efficacy to long-term outcomes. This approach allows the impact of each step in the disease process to be assessed specifically and the impact of evidence gaps to be quantified. In this study, for example, several uncertainties were identified in the clinical evidence, including efficacy parameters and long-term mortality. These uncertainties were tested directly and found to have relatively limited impact on the estimated cost-effectiveness of beti-cel.
Although drug pricing in the US continues to receive much scrutiny, higher prices may be justifiable for innovative one-time therapies such as beti-cel that can provide lifelong benefits to individuals with serious conditions. These benefits can be difficult to value fully with QALYs because some desirable aspects do not translate into increased health-related QoL. Stakeholders may wish to value other important considerations, such as unmet need, the hope provided by the possibility of cure, or the avoidance of complex, cumbersome, and time-consuming alternatives. Although a WTP threshold up to $500,000/QALY has been used in ultra-rare conditions to account on a gross basis for these other factors [Citation47], it is unnecessary in the case of beti-cel given its cost-effectiveness. Moreover, broader societal impacts such as the effect on caregivers and changes in the productivity of patients that may also be delivered by successful treatment of TDT were not included in this analysis.
To address affordability concerns, reimbursement for recently approved gene therapies in the US is increasingly following an outcome-based payment approach. In this analysis, we incorporated an outcome-based payment over 5 years for beti-cel, and its impact on beti-cel cost-effectiveness was substantial. It should be noted that this specific payment model is not yet broadly implemented in the US. but risk-sharing agreements between payers and manufacturers via payment through an intermediary provide this option to payers and are executable in the current regulatory environment. An alternative payment model tested in our analyses (i.e., full up-front payment but 80% rebate if treatment success is not maintained) may be feasible under proposed rule changes being considered by the Centers for Medicare & Medicaid Services. Although payment mechanisms are also being developed to address the financial disconnect that can occur when high up-front costs accrue to one payer but downstream cost benefits accrue to another, these were outside the scope of this analysis. There may be statutory hurdles to such approaches, such as Medicaid’s best price policy [Citation58], which would need to be considered. Finally, our US payer perspective did not take into account accounting implications to payers of these types of payment models.
No model can predict how the treatment landscape for a condition might evolve years in the future. Recent changes in the treatment landscape for TDT were considered. One such important shift i is the introduction of effective oral chelators and the combination of oral and SC chelation. These agents and strategies can result in lower iron levels for patients and, thus, may improve outcomes in the decades to come. To address this in the model, it was assumed that complication rates are related directly only to iron concentrations. Thus, treatment only affects these rates through any reductions in iron levels. The effect of beti-cel is modeled as elimination of the need for transfusions, which in turn reduce iron overload, and thus, long-term organ damage. This enables use of a consistent set of data for each treatment – short-term transfusion requirements and iron loading – to predict intermediate and long-term outcomes. Variations in chelation practice across geographies or in different age groups can be accommodated in this approach if data on iron load is available.
Our analysis has several limitations. First, because of TDT’s rarity and the complexity of conducting gene therapy studies, the clinical evidence for beti-cel was obtained from open-label, single-arm trials with small sample sizes and limited duration of follow-up. Given this, comparison of beti-cel to SoC is associated with substantial uncertainty; however, sensitivity analyses varying clinical effectiveness parameters did not meaningfully alter the estimated cost-effectiveness of beti-cel. In addition, our analysis did not capture the clinical, QoL, or economic benefits for patients in beti-cel trials who obtained substantial reductions (>60%) in transfusion requirements but did not achieve transfusion independence. This simplification is conservative (i.e., it underestimates the economic efficiency of beti-cel).
Second, some uncertainties inherent to a novel therapy cannot be quantified until additional clinical experience accumulates. These include unexpected adverse effects appearing over the long-term and future advances in the management of TDT and iron chelation therapy. Nevertheless, scenario analyses evaluating the impact of variation in the SMR of transfusion dependence found that, due to the impact of discounting, uncertainties around long-term survival had a limited impact on the estimated cost-effectiveness of beti-cel.
Finally, our model considered only the direct costs of care and the main complications of TDT. Additional complications of TDT associated with anemia, transfusions, or chelation have not been valued, but as the main complications have been incorporated in the model, these effects on cost-effectiveness are anticipated to be minor. Costs of care considered in the model are limited to direct costs borne by the payer because of the perspective taken. A broader societal perspective would include indirect costs, such as caregiver burden and productivity. Because patients with TDT are young, valuing these additional outcomes would further improve the cost-effectiveness of beti-cel. The US Second Panel on Cost-effectiveness recommends a broader healthcare sector perspective that incorporates costs borne by other stakeholders, particularly out-of-pocket costs to the patient [Citation59]. Considering additional perspectives can highlight the multiple benefits that can be realized by different stakeholders – payers benefit from the reduced need for expensive chelation, patients from the improved clinical outcomes and reduced out-of-pocket costs, and ultimately society benefits from increased productivity.
Treatment with beti-cel is predicted to yield a large clinical and patient benefit compared to SoC, while reducing the substantial lifetime costs of the latter. These benefits are driven by extended survival, improved QoL, avoidance of cardiac complications, and avoidance of lifelong transfusions and chelation therapy.
Despite advances in conventional management of TDT, there remains significant unmet need for therapies that can address the genetic cause of the disease, eliminate the need for transfusion and chelation, and significantly improve morbidity and mortality. Beti-cel has the potential to transform patients’ lives and its benefits are shown to be cost-effective or cost-saving in the one-time treatment of TDT.
Supplemental Material
Download MS Word (2.1 MB)Supplementary material
Supplemental data for this article can be accessed here.
Disclosure statement
Reifsnider, Brand, Coughlan, Li, Caro are employed by Evidera, a consultancy which provides services to bluebird bio and the pharmaceutical industry. Kansal and Cragin were employed by Evidera at the time the work was completed. Hawkins is employed by Visible Analytics, a consultancy that provides services to bluebird bio and the pharmaceutical industry. Paramore and Dietz are employed by bluebird bio, the manufacturer of beti-cel. This work was funded by bluebird bio.
References
- Sayani FA, Kwiatkowski JL. Increasing prevalence of thalassemia in America: implications for primary care. Ann Med. 2015;47(7):592–13.
- Centers for Disease Control and Prevention. Thalassemia awareness. 2019 cited 2019 Jun 24]; Available from: https://www.cdc.gov/features/international-thalassemia/index.html.
- Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99(5):811–820.
- Baronciani D, Angelucci E, Potschger U, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European society for blood and bone marrow transplantation hemoglobinopathy registry, 2000-2010. Bone Marrow Transplant. 2016;51(4):536–541.
- Caocci G, Efficace F, Ciotti F, et al. Prospective assessment of health-related quality of life in pediatric patients with beta-thalassemia following hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2011;17(6):861–866.
- Lucarelli G, Isgro A, Sodani P, et al. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med. 2012;2(5):a011825.
- Li C, Mathews V, Kim S, et al. Related and unrelated donor transplantation for beta-thalassemia major: results of an international survey. Blood Adv. 2019;3(17):2562–2570.
- Capellini MD, Cohen A, Porter J, Taher A, Viprakasit V. Thalassemia international federation: guidelines for the management of transfusion dependent thalassaemia (TDT). Third ed. Nicosia, Cyprus: Thalassaemia International Federation; 2014.
- Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. N Engl J Med. 2021;384(8):727–743.
- Ladis V, Chouliaras G, Berdoukas V, et al. Survival in a large cohort of Greek patients with transfusion-dependent beta thalassaemia and mortality ratios compared to the general population. Eur J Haematol. 2011;86(4):332–338.
- World Health Organization (WHO). European detailed mortality database. 2014 cited 2019 Jun 24]; Available from: http://data.euro.who.int/dmdb/Help/help.htm.
- Kwiatkowski JL, Kim H-Y, Thompson AA, et al. Chelation use and iron burden in North American and British thalassemia patients: a report from the thalassemia longitudinal cohort. Blood. 2012;119(12):2746–2753.
- Jobanputra M, Paramore C, Laird SG, et al. Co-morbidities and mortality associated with transfusion-dependent beta-thalassemia in patients in England: a 10-year retrospective cohort analysis. Br J Haematol. 2020;191(5):897–905.
- Novartis. Prescribing information: JADENU® (deferasirox) tablets for oral suspension. 2005 cited 2019 Oct 2]; Available from: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/jadenu.pdf.
- Bonifazi F, Conte R, Baiardi P, et al. Pattern of complications and burden of disease in patients affected by beta thalassemia major. Curr Med Res Opin. 2017;33(8):1525–1533.
- Vekeman F, Sasane M, Cheng WY, et al. Adherence to iron chelation therapy and associated healthcare resource utilization and costs in Medicaid patients with sickle cell disease and thalassemia. J Med Econ. 2016;19(3):292–303.
- Weiss M, Sheth JM, Sheth S. S, Clinical and economic burden of regularly transfused adult patients with β-thalassemia in the USA: a retrospective cohort study using payer claims. Am J Hematol. 2019;94(5):E129–132.
- Thompson AA, Walters MC, Kwiatkowski J, et al. Gene Therapy in Patients with Transfusion-Dependent beta-Thalassemia. N Engl J Med. 2018;378(16):1479–1493.
- Sadelain M. Recent advances in globin gene transfer for the treatment of beta-thalassemia and sickle cell anemia. Curr Opin Hematol. 2006;13(3):142–148.
- Seneca N, Negre O, Smith F, Lewis K. A retrospective literature review of the safety outcomes of clinical studies of haemotopoietic stem cell gene therapy using lentiviral vectors in non-oncologic diseases, in 27th Annual Congress of the European Society of Gene and Cell Therapy British Society for Gene Therapy. 2019, Human Gene Therapy: Barcelona, Spain. p. A116.
- Caro JJ. Discretely integrated condition event (DICE) simulation for pharmacoeconomics. Pharmacoeconomics. 2016;34(7):665–672.
- Moller J, Davis S, Stevenson M, et al. Validation of a DICE simulation against a discrete event simulation implemented entirely in code. Pharmacoeconomics. 2017;35(10):1103–1109.
- Pennington B, Filby A, Owen L, et al. Smoking cessation: a comparison of two model structures. Pharmacoeconomics. 2018;36(9):1101–1112.
- Caro JJ, Briggs AH, Siebert U, et al. Modeling good research practices–overview: a report of the ISPOR-SMDM Modeling Good Research Practices Task Force–1. Value Health. 2012;15(6):796–803.
- Caro JJ, Moller J, Karnon J, Stahl J, Ishak J. Discrete event simulation for health technology assessment. Boca Raton; London: CRC Press, Taylor & Francis Group. xx; 2016. p. 354.
- Hongeng S, Thompson AA, Kwiatkowski JL, et al. 1 - efficacy and safety of betibeglogene autotemcel (beti-cel; LentiGlobin for β-thalassemia) gene therapy in 60 patients with transfusion-dependent β-thalassemia (TDT) followed for up to 6 years post-infusion. Transplant Cell Ther. 2021;27(3):S1.
- Busca A, Falda M, Manzini P, et al. Iron overload in patients receiving allogeneic hematopoietic stem cell transplantation: quantification of iron burden by a superconducting quantum interference device (SQUID) and therapeutic effectiveness of phlebotomy. Biol Blood Marrow Transplant. 2010;16(1):115–122.
- Kyweluk MA, Diane Chen JR, Chen D. Fertility preservation legislation in the USA: potential implications for transgender individuals. LGBT Health. 2019;6(7):331–334.
- Lyttle Schumacher B, Grover. N, Mesen T, et al. Modeling of live-birth rates and cost-effectiveness of oocyte cryopreservation for cancer patients prior to high- and low-risk gonadotoxic chemotherapy. Hum Reprod. 2017;32(10):2049–2055.
- Fryar CD, Gu Q, Ogden CL, Flegal KM. Anthropometric reference data for children and adults: USA, 2011-2014. Vital Health Stat. 2016;3(39):1–46.
- New England Research Institutes (NERI). A secondary analysis of the thalassemia longitudinal cohort data to evaluate outcomes of standard medical care for the treatment of beta-thalassemia (THAL-1698), version 1.0. February, 2018 (data on file).
- Galambrun C, Pondarré C, Bertrand Y, et al. French multicenter 22-year experience in stem cell transplantation for beta-thalassemia major: lessons and future directions. Biol Blood Marrow Transplant. 2013;19(1):62–68.
- Walters MC, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16(2):263–272.
- Ang AL, Tzoulis P, Prescott E, et al. History of myocardial iron loading is a strong risk factor for diabetes mellitus and hypogonadism in adults with beta thalassemia major. Eur J Haematol. 2014;92(3):229–236.
- Delea TE, Sofrygin O, Thomas SK, et al. Cost effectiveness of once-daily oral chelation therapy with deferasirox versus infusional deferoxamine in transfusion-dependent thalassaemia patients: US healthcare system perspective. Pharmacoeconomics. 2007;25(4):329–342.
- Truven Health Analytics, Characteristics and costs of transfusion-dependent thalassemia patients. Data from the marketscan commercial, medicaid and medicare databases. Time Period: October 1, 2011 to September 30, 2016. December 2017.
- Fisher SA, Brunskill SJ, Doree C, Gooding S, Chowdhury O, Roberts DJ. Desferrioxamine mesylate for managing transfusional iron overload in people with transfusion-dependent thalassaemia. Cochrane Database Syst Rev. 2013 Aug 21;(8):CD004450. doi: https://doi.org/10.1002/14651858.CD004450.pub3. PMID: 23963793.
- FERRIPROX® (deferiprone) oral solution for oral use. Available from: https://ferriprox.com/healthcare-providers/healthcare-provider-home/#
- Matza LS, Paramore LC, Stewart KD, et al. Health state utilities associated with treatment for transfusion-dependent beta-thalassemia. Eur J Health Econ. 2020;21(3):397–407.
- National Institute for Health and Care Excellence (NICE). Highly specialised technologies guidance [HST7]: strimvelis for treating adenosine deaminase deficiency–severe combined immunodeficiency. 2018 cited 2019 Oct 2]; Available from: https://www.nice.org.uk/guidance/hst7.
- Fryback DG, Dasbach EJ, Klein R, et al. The Beaver Dam Health Outcomes Study: initial catalog of health-state quality factors. Med Decis Making. 1993;13(2):89–102.
- USA Department of Labor and Bureau of Labor Statistics. Medical Care CPI 2018. cited 2019 Sept 24]; Available from: http://data.bls.gov/timeseries/CUUR0000SAM?output_view=pct_12mths
- InHealth. Physicians’ Fee and Coding Guide. 2020. Atlanta, GA: InHealth Professional Services; 2020.
- IBM. Micromedex® RED BOOK® 2020 24 September]; Available from: https://www.micromedexsolutions.com/home/dispatch.
- Agency for Healthcare Research and Quality (AHRQ), HCUPnet: statistics by principal procedure. Weighted national estimates from HCUP National Inpatient Sample (NIS). 2017.
- Angelucci E, Muretto P, Lucarelli G, et al. Phlebotomy to reduce iron overload in patients cured of thalassemia by bone marrow transplantation. Italian cooperative group for phlebotomy treatment of transplanted thalassemia patients. Blood. 1997;90(3):994–998.
- Institute for Clinical and Economic Review (ICER). Modifications to the ICER value assessment framework for treatments for ultra‐rare diseases. 2017 January 2020 15 Apr 2021]; Available from: https://icerorg.wpengine.com/wp-content/uploads/2020/10/ICER_URD_Framework_Adapt_013120.pdf.
- The Wall Street Journal. Biotech Proposes Paying for Pricey Drugs by Installment. 8 January 2019]; Available from: https://www.wsj.com/articles/biotech-proposes-paying-for-pricey-drugs-by-installment-11546952520.
- AccessData.FDA.gov. REBLOZYL® (luspatercept-aamt) for injection, for subcutaneous use. Prescribing information. Initial U.S. Approval: 2019. Revised: 04/2020. 2020 [cited 2020 Oct 26]; Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761136orig2lbl.pdf.
- European Medicines Agency (EMA). Zynteglo Summary of Product Characteristics. 2 October 2019]; Available from: https://www.ema.europa.eu/en/documents/product-information/zynteglo-epar-product-information_en.pdf.
- Doulatov S, Notta F, Laurenti E, et al. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–136.
- Angelucci E. Hematopoietic stem cell transplantation in thalassemia. Hematology Am Soc Hematol Educ Program. 2010;2010(1):456–462.
- Baerlocher GM, Rovó A, Müller A, et al. Cellular senescence of white blood cells in very long-term survivors after allogeneic hematopoietic stem cell transplantation: the role of chronic graft-versus-host disease and female donor sex. Blood. 2009;114(1):219–222.
- Caocci G, Vacca A, Piras E, et al. Return to normal life after hematopoietic stem cell transplantation for thalassemia: a study of patients transplanted from matched sibling donors. Bone Marrow Transplant. 2016;51(12):1640–1641.
- Longest living transplant survivor: Nancy’s story. [cited 2021 Mar 31]; Available from: https://www.bmtinfonet.org/story/longest-living-transplant-survivor-nancys-story.
- Cavazzana M, Six E, Lagresle-Peyrou C, et al. Gene therapy for x-linked severe combined immunodeficiency: where do we stand? Hum Gene Ther. 2016;27(2):108–116.
- Ferrua F, Aiuti A. Twenty-five years of gene therapy for ADA-SCID: from bubble babies to an approved drug. Hum Gene Ther. 2017;28(11):972–981.
- Sachs R, Bagley N, Lakdawalla DN. innovative contracting for pharmaceuticals and medicaid’s best-price rule. J Health Polit Policy Law. 2018;43(1):5–18.
- Sanders GD, Neumann PJ, Basu A, et al. Recommendations for conduct, methodological practices, and reporting of cost-effectiveness analyses: second panel on cost-effectiveness in health and medicine. JAMA. 2016;316(10):1093–1103.