
ABSTRACT
Background: Delandistrogene moxeparvovec (SRP-9001) is an investigational gene therapy that may delay progression of Duchenne muscular dystrophy (DMD), a severe, rare neuromuscular disease caused by DMD gene mutations. Early cost-effectiveness analyses are important to help contextualize the value of gene therapies for reimbursement decision making.
Objective: To determine the potential value of delandistrogene moxeparvovec using a cost-effectiveness analysis.
Study design: A simulation calculated lifetime costs and equal value of life years gained (evLYG). Inputs included extrapolated clinical trial results and published utilities/costs. As a market price for delandistrogene moxeparvovec has not been established, threshold analyses established maximum treatment costs as they align with value, including varying willingness-to-pay up to $500,000, accounting for severity/rarity.
Setting: USA, healthcare system perspective
Patients: Boys with DMD
Intervention: Delandistrogene moxeparvovec plus standard of care (SoC; corticosteroids) versus SoC alone
Main outcome measure: Maximum treatment costs at a given willingness-to-pay threshold
Results: Delandistrogene moxeparvovec added 10.30 discounted (26.40 undiscounted) evLYs. The maximum treatment cost was approximately $5 M, assuming $500,000/evLYG. Varying the benefit discount rate to account for the single administration increased the estimated value to >$5M, assuming $500,000/evLYG.
Conclusion: In this early economic model, delandistrogene moxeparvovec increases evLYs versus SoC and begins to inform its potential value from a healthcare perspective.
Introduction
Duchenne muscular dystrophy (DMD) is a rare, fatal, X-linked degenerative neuromuscular disease caused by DMD gene mutations, resulting in the absence of functional dystrophin protein [Citation1]. Dystrophin is a large protein that is integral to the dystrophin-associated protein complex (DAPC), which preserves muscle integrity by acting as a shock absorber and preventing damage during normal muscle contraction [Citation2–4]. Without dystrophin, the DAPC fails to assemble, leading to sarcolemmal membrane disruption, loss of muscle homeostasis, inflammation, segmental necrosis of fibers, impaired regeneration of fibers, replacement of myofibers with fat or fibrous connective tissue, progressive weakness, and irreversible loss of muscle function [Citation5]. DMD has an estimated prevalence of approximately 9,000–12,000 males in the United States (US) [Citation6,Citation7].
While there is evidence muscle damage begins before birth [Citation6,Citation8], DMD may not be visibly recognized until 2–3 years of age, with diagnosis around 4–5 years of age, as rapid motor development in the early stages of life often masks the ongoing muscle damage [Citation9,Citation10]. Initial signs and symptoms typically include delayed milestones, including sitting and walking, abnormal gait, and frequent falls, with subsequent progression following a predictable and inevitable course, including loss of ambulation (LoA) generally by the teenage years and ultimately premature death due to life-threatening complications (e.g., respiratory insufficiency and cardiomyopathy), with a median survival of 28.1 years for those born after 1990 with the introduction of corticosteroids for DMD [Citation9–12].
Currently, there is no cure for DMD. However, advancements have been made in the standard of care (SoC), starting with the introduction of systemic corticosteroids [Citation13,Citation14] and the establishment of comprehensive interdisciplinary care, including physical/occupational therapy and management for gastrointestinal, respiratory, and cardiac complications [Citation11]. Continuous corticosteroid use can initially delay LoA and preserve some function but is often associated with serious long-term adverse effects [Citation11,Citation15].
Dystrophin replacement therapies represent another therapeutic strategy for DMD [Citation16]. Recently, phosphorodiamidate morpholino oligomers (PMOs) capable of ‘skipping’ specific gene exons to allow for the production of truncated but functional dystrophin have been approved by the US Food and Drug Administration (FDA) for the treatment of a subset of patients with exon 51, 53, or 45 skip-amenable DMD mutations [Citation17–20]. Eteplirsen was the first PMO approved by the FDA via an accelerated approval pathway for patients with exon 51 skip-amenable mutations based on a significant increase from baseline in dystrophin over 48 weeks that was reasonably likely to predict clinical benefit [Citation21]. Subsequent studies of eteplirsen have confirmed dystrophin localization to the sarcolemma and accumulation over time [Citation21,Citation22], and functionally shown an attenuation in pulmonary decline and prolonged ambulation compared with mutation-matched controls, as well as increased survival in an indirect treatment comparison using real-world data [Citation23,Citation24]. Due to the specificity of the currently approved PMOs for select mutations, they are not a treatment option for ~70% of the DMD population [Citation25].
Gene therapies (GTs) represent another therapeutic strategy for DMD by utilizing a viral vector to deliver a functional gene to compensate for a mutated gene with the potential for a long-lasting treatment effect after a single administration. Delandistrogene moxeparvovec (SRP-9001) is an investigational GT that utilizes rAAVrh74 for targeted delivery of the SRP-9001 dystrophin transgene, expressed in skeletal and cardiac tissue under control of MHCK7 (a muscle-specific promoter) to produce shortened functional dystrophin [Citation9]. The clinical basis of the SRP-9001 dystrophin construct is based on the identification of a patient with Becker muscular dystrophy (BMD), who had an in-frame deletion of almost half of the DMD gene yet remained ambulant through 61 years of age [Citation26].
The goal of delandistrogene moxeparvovec treatment is to deliver the SRP-9001 transgene safely to enable production of shortened functional dystrophin protein in skeletal and cardiac muscles and ultimately maintain muscle function and delay disease progression, including LoA, need for assisted ventilation, and death. At the time of this analysis, delandistrogene moxeparvovec is being evaluated in ambulatory and non-ambulatory patients in both open-label trials and placebo-controlled trials with >140 patients dosed as of November 2022 [Citation27–32]. It has been granted FDA Fast Track designation for the treatment of DMD and is intended to be a single intravenous administration provided in an outpatient healthcare setting.
Since cost-effectiveness analyses (CEAs) are increasingly included in insurance coverage policies [Citation33–35], it is important to perform these assessments early and sometimes prior to FDA approval. However, previous research has described certain shortfalls with traditional CEA frameworks for single-administration treatments, including uncertainties in efficacy and durability as well as high upfront costs with potential lifetime benefits [Citation36–38]. These challenges may be especially profound for GTs targeting severe, progressive pediatric diseases, such as DMD, in which benefits have the potential to accrue over a longer period and may inadvertently result in assessments that place single-administration treatments at a disadvantage relative to chronic treatments [Citation36,Citation37,Citation39–41]. Many health technology assessments (HTAs) have also recognized the need for CEAs to accommodate treatment- and disease-specific attributes deemed important from a societal perspective (e.g., ultra-rare diseases and diseases with the greatest severity) [Citation42–47]. The present study sought to evaluate the potential value of delandistrogene moxeparvovec in patients with DMD from a US healthcare system perspective using a CEA.
Materials and methods
Model framework
A CEA was undertaken to compare relative costs and effects of delandistrogene moxeparvovec plus SoC to SoC alone from a US healthcare system perspective using a simulated cohort of 4-year-olds with DMD [Citation48]. SoC consisted of corticosteroids (prednisone/prednisolone), physical/occupational therapy, multidisciplinary assessments, and gastrointestinal/respiratory/cardiac management [Citation11]. PMOs were not selected as comparators because the majority of patients with DMD are ineligible for currently approved PMO treatments.
The model used a lifetime horizon to capture long-term costs and health benefits. The cohort was followed from treatment initiation to mortality. Outcomes included lifetime costs, quality-adjusted life years (QALYs), equal value of life years gained (evLYG), and maximum treatment cost at a given willingness-to-pay (WTP) threshold.
The model was developed based on clinical feedback and previously reported models () [Citation51,Citation52]. Patients started in the early ambulatory (EA) health state. Over time, patients progressed to the late ambulatory (LA) state, followed by the early non-ambulatory (ENA) state, and finally the late non-ambulatory (LNA) state [Citation52]. Patients progressed to health states in that specific order and could not return to an earlier health state. From LNA, patients could expire due to DMD causes; they could also expire at any point per general US male population mortality background rates. A patient-level simulation was developed to capture the heterogeneity of disease progression using Python programming language (Python Software Foundation).
Figure 1. Model structure. Loss of the ability to stand from supine in under 5 seconds signified the transition from the early ambulatory to late ambulatory state as that is associated with progressive mobility decline [Citation49,Citation50]. LoA was defined in this model as the inability to ambulate 10 meters [Citation49] and signified the transition from the late ambulatory to early non-ambulatory state. Progression to a Brooke score above 4 (loss of unweighted hand-to-mouth function) signified the transition from the early non-ambulatory to late non-ambulatory state. LoA, loss of ambulation.
![Figure 1. Model structure. Loss of the ability to stand from supine in under 5 seconds signified the transition from the early ambulatory to late ambulatory state as that is associated with progressive mobility decline [Citation49,Citation50]. LoA was defined in this model as the inability to ambulate 10 meters [Citation49] and signified the transition from the late ambulatory to early non-ambulatory state. Progression to a Brooke score above 4 (loss of unweighted hand-to-mouth function) signified the transition from the early non-ambulatory to late non-ambulatory state. LoA, loss of ambulation.](/cms/asset/646cf128-b8c5-4f9a-8339-783a86bf35a8/zjma_a_2216518_f0001_b.gif)
Risks with SoC
Risks for patients treated with SoC alone were age-specific and were obtained from McDonald et al. [Citation49] for non-fatal events. Loss of the ability to stand from supine in under 5 seconds signified the transition from EA to LA as that is associated with progressive mobility decline [Citation49,Citation50]. LoA was defined in this model as the inability to ambulate 10 meters [Citation49] and signified the transition from LA to ENA. Progression to a Brooke score above 4 (loss of unweighted hand-to-mouth function) signified the transition from ENA to LNA. Loss of unweighted hand-to-mouth function is important to QoL [Citation53] and is associated with FVC%p < 50% [Citation54], which is deemed to be a threshold whereby non-invasive nocturnal ventilation should be initiated [Citation55].
Risk of mortality due to DMD causes was age-specific and was estimated based on Broomfield et al. [Citation12] (patients born post 1970), Passamano et al. [Citation56], and Paramsothy et al. [Citation57]. US Centers for Disease Control and Prevention life tables were used for general male population mortality risk by age [Citation58].
Long-term treatment benefits
In both preclinical and clinical studies, delandistrogene moxeparvovec resulted in widespread SRP-9001 dystrophin expression in target muscle types and demonstrated evidence of DAPC reconstitution [Citation27–30,Citation59]. In ENDEAVOR, delandistrogene moxeparvovec-treated patients (ambulatory and ≥4 to <8 years of age at baseline) had a 54.2% increase in dystrophin expression as measured by western blot, a 48.3% increase in percent dystrophin-positive fibers, and a 66.5% increase in intensity of dystrophin expression from baseline at 12 weeks post dose [Citation60]. While biopsies were not taken at a later timepoint in ENDEAVOR, from Study 102, a Phase 2 placebo-controlled trial, patients treated with delandistrogene moxeparvovec had dystrophin expression at 12 weeks which continued through 60 weeks after treatment [Citation61]. In the longest running clinical trial, treatment with delandistrogene moxeparvovec has resulted in the maintenance of clinically meaningful improvements in motor function (as measured using the North Star Ambulatory Assessment [NSAA], a 17-item scale used to evaluate motor function in patients with DMD) from baseline over 4 years [Citation27], demonstrating a durable response and evidence of functional stabilization to date.
Based on clinical trial results thus far, the anticipated long-term treatment benefits (e.g., delaying key clinical events compared with SoC alone: losing the ability to stand in under 5 seconds, LoA, loss of unweighted hand-to-mouth function, and death) to patients treated with delandistrogene moxeparvovec were estimated by published literature and DMD clinical expert opinion. Specifically, the hazard ratios (HRs) from a published study [Citation62] that compared age at LoA and death in patients with DMD mutations with undetectable dystrophin vs those with >0% but <5% dystrophin were applied. In the absence of HRs for key clinical events from trial results, the published HRs described above (HR LoA, 0.16 applied to modeled non-fatal events; HR survival, 0.18 applied to modeled mortality) were determined by DMD clinical experts to be clinically representative of the anticipated trajectory of patients treated with delandistrogene moxeparvovec, including in the context of known PMO clinical results [Citation23,Citation24,Citation60]. The impact of delandistrogene moxeparvovec administration was modeled to be lifelong in the base case scenario.
Measurement of health utility and costs
As a rare disease, few data sources are available on the health state utilities and costs of DMD. Utilities were based on US DMD patient-reported data using the Health Utility Index-2 (HUI-2) () [Citation63]. Utility values decreased with disease progression (e.g., the median [interquartile range (IQR)] patient-reported HUI-2 value was 0.96 [0.86–0.99] for patients who were ambulatory with preserved upper limb function and was 0.35 [0.34–0.36] for patients who were non-ambulatory with loss of upper limb function and daytime ventilation) [Citation64].
Table 1. Key model inputs.
Health state costs were estimated based on Iff et al. [Citation65] and were inflated to 2021 USD (), showing average direct medical annual costs increase with disease progression, increasing from $17,688 in EA to $167,285 in LNA [Citation65]. Cost estimates did not include indirect medical or non-medical costs such as out-of-pocket expenditures or lost productivity. Additional costs associated with administration of delandistrogene moxeparvovec were included as were costs of prednisone/prednisolone treatment with increasing age/weight (). Costs were discounted using an annual rate of 3.0%.
Although a CEA for DMD is sensitive to granularity [Citation52], a five-state model was applied based on available data (). However, in contrast to previously published DMD CEAs [Citation51,Citation52], patient utility decreased linearly and costs increased linearly within a given health state (Supplemental Figure 1), reflecting the progressive nature of DMD. For example, the occurrence of LoA in DMD is not due to an acute, abrupt event (e.g., accident) but rather a progressive deterioration of skeletal muscle that results in increasing difficulty in walking over time until the eventual inability to walk 10 meters [Citation66]. The model assumes that at 2 years of age (median age of symptom onset [Citation67]), health begins to deteriorate from a starting utility of 1.0 (i.e., before the patient enters the model). Although it is not known if DMD progression of utilities and costs is linear over time, a continuous change in utility and costs within each of the high-level health states was deemed to be more reasonable than constant utility and costs within each health state.
Base case
The maximum treatment cost was estimated using the QALY (Scenario A) and evLYG (Scenario B), as is done in assessments by the Institute for Clinical and Economic Review when a treatment is assumed to increase survival [Citation68]. For the evLYG assessments, a utility of 0.851 was assumed during additional survival to align with the expected utility of the general population [Citation69–71].
The WTP threshold per evLYG was varied from $150,000 to $250,000 (Scenario C) and included a $500,000 threshold (Scenario D) based on WTP adjustments implemented by other global HTAs for severe diseases [Citation45,Citation46,Citation72] and suggested higher WTP thresholds for treatments for ultra-rare diseases (Supplemental Table 1) [Citation42,Citation73]. Scenario D represents a more equitable assessment for an ultra-rare treatment that is modeled to increase survival of a pediatric disease with severe unmet need. Furthermore, the impact of adjusting the annual benefit discount rate of evLYG from the standard 3.0% to 1.5% (Scenario E) or removing discounting of benefits altogether (Scenario F) was evaluated, encompassing discounting standards used in other countries [Citation74,Citation75]. Finally, the impact of simultaneous adjustments was assessed to understand the interactions between these variables (Scenarios G and H). These analyses will help inform whether a future price is above or below a given WTP threshold.
Model validation
The model was validated by experts in DMD and health economics. The trajectory of the SoC-only cohort was simulated against (input) Kaplan-Meier curves to validate disease progression and mortality with age. Extreme value testing was performed to ensure logical results, including setting HRs and utilities to 0 and 1 and setting costs to $0.
Scenario analyses
Probabilistic sensitivity analyses were conducted based on the maximum treatment cost calculated in the first two scenarios (A and B) to highlight potential differences in outcomes between a CEA using the QALY and one using the evLYG. In total, 1,000 model iterations were performed to assess the uncertainty of inputs (HRs, risks, direct medical costs, and utilities) and results given the fact that DMD is rare; therefore, 1) the treated population is likely to be small and 2) confidence intervals (CIs) around inputs are fairly wide.
For multivariate sensitivity analyses (Scenarios A and B), high and low values for each set of inputs (HRs, risks, direct medical costs, and utilities) were based on reasonable values from the literature (Supplemental Table 2) [Citation64,Citation65]. Each category of input was varied to the high and then low values to ensure reasonable scenarios (e.g., all utilities set to their low value and then high value to maintain the consistent decrease in utility as the patient progressed). In scenario analyses, non-lifetime durability was assessed and modeled at 10, 20, and 30 years. In the absence of clinical data describing how a ‘waning’ effect may occur, treatment efficacy was assumed to be ‘all or nothing’ in these scenarios to provide a range of potential results if durability is not lifelong. After the assessed time frame, the patient’s incremental risk resumed as per the SoC-only cohort, which would be less than an untreated patient with DMD of the same age. Additional scenario analyses considered the impact of constant costs and utilities within each health state and applied a utility value of 1.0 during extended survival when assessing evLYG.
Results
Base case
Median ages for key clinical events for patients treated with delandistrogene moxeparvovec plus SoC and SoC alone were estimated. In this model, compared with SoC alone, treatment with delandistrogene moxeparvovec delayed the median age of all key clinical events: losing the ability to stand in under 5 seconds, 10.5 vs 18.1 years; LoA, 13.5 vs 26.4 years; loss of unweighted hand-to-mouth function, 19.7 vs 38.5 years; and death, 25.2 vs 48.5 years.
In the base case analysis, the total discounted lifetime direct medical costs (non-treatment costs) was $1,164,783 for the delandistrogene moxeparvovec-treated cohort and $1,105,932 for the SoC-only cohort. The increase of $58,851 over the patient’s lifetime for the treated patient is due to additional costs associated with increased survival. The QALYs for the delandistrogene moxeparvovec cohort were 30.55 without discounting and 17.70 with a 3.0% discount rate (42% reduction). The equal value of life years (evLYs) for the delandistrogene moxeparvovec cohort were 39.79 without discounting and 20.68 with a 3.0% discount rate, representing a 48% reduction due to discounting alone. In comparison, the QALYs for the SoC-only cohort (and the evLYs, which are equivalent to the QALYs for the SoC-only cohort by definition) were 13.39 without discounting and 10.39 with a 3.0% discount rate (22% reduction).
The value of delandistrogene moxeparvovec via CEA approaches is shown in . Using a QALY-based CEA and a WTP threshold of $150,000, the maximum treatment cost of delandistrogene moxeparvovec was approximately $1.0 M (Scenario A). Using the evLYG (holding all other inputs the same), the maximum treatment cost was $1.5 M (Scenario B). Increasing the WTP per evLYG threshold from $150,000 to $250,000 or $500,000 (Scenarios C and D, respectively) resulted in a maximum treatment cost of $2.5 M and $5.1 M, respectively. For scenarios with a WTP of $150,000/evLYG and reducing the benefit discount rate to 1.5% or 0.0% (Scenarios E and F, respectively), the maximum treatment cost was $2.3 M and $3.9 M, respectively. For scenarios in which simultaneous adjustments were assessed to understand the interactions between assumptions (Scenarios G and H), the maximum treatment cost reached $13.1 M.
Table 2. Value assessment of delandistrogene moxeparvovec.
Model validation
Extreme value testing yielded expected results, providing model validation. Additionally, the simulated trajectory of SoC-only treated patients matched the (input) Kaplan-Meier curves. Median ages of the delandistrogene moxeparvovec plus SoC and SoC-only cohorts of key disease milestones were validated by DMD clinical experts.
Sensitivity and scenario analyses
For the sensitivity analysis, the relative impact of uncertainty from the varied inputs was consistent across all scenarios described in . Cost-effectiveness acceptability curves generated from probabilistic sensitivity analyses of Scenarios A and B (using their respective calculated maximum treatment costs) indicated similar variations in the incremental cost-effectiveness ratio (ICER) when using the QALY or evLYG (Scenario A IQR: $128,844, $180,574; Scenario B IQR: $129,990, $169,642; ). Multivariate sensitivity analysis indicated that the results were most sensitive to changes in the HRs (Supplemental Figure 2). Utilities had a greater relative impact on the ICER with QALYs than the ICER with evLYG.
Figure 2. Cost-effectiveness acceptability curve for scenarios A and B. Scenario A includes a CEA with QALYs and a placeholder treatment cost of $1,038,093. Scenario B includes a CEA with evLYG and a placeholder treatment cost of $1,485,635. Both scenarios assume 3.0% discounting of costs and benefits.
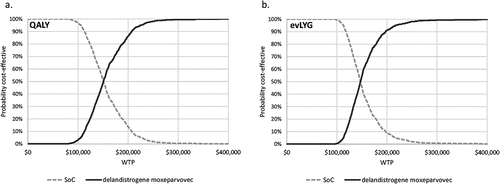
Additional scenario analyses indicated that the results are sensitive to assumptions around the modeled durability with an ICER with evLYG approximately 2.5 times higher with a 10-year durability compared with lifetime (). The impact of varying durability is not linear on the ICERs with QALYs or evLYG, reflecting the fact that the results are more sensitive to survival gains versus QoL improvements. Applying constant costs and utilities per health state had a small impact on the overall value assessment (4% improvement). In addition, increasing the utility value during extended survival from 0.851 to 1.0 improved the ICER with evLYG by 10%, with no impact on the ICER with QALYs (by definition).
Table 3. Scenario analysis using scenarios A and B.
Discussion
The results of this early CEA show that the potential of delandistrogene moxeparvovec to alter the course of DMD is evident, with 7.31 discounted projected QALYs gained and 10.30 discounted evLYG with this treatment compared with SoC alone. These QALY gains are considerably greater than those reported in analyses of other novel treatments. A recent assessment determined a median QALY gain of 3.39 for cell therapies and GTs, 0.05 for drugs, and 0.07 for biologics [Citation76]. Similar results were observed for QALYs gained for cancer treatments (0.380 [Citation77]) and treatments included in at least one FDA expedited review program (0.182 [Citation78]). Moreover, the modeled results of this study suggest that treatment with delandistrogene moxeparvovec has the potential to delay key clinical markers of DMD substantially (including prolonging ambulation by ~13 years and survival by at least two decades vs SoC alone), which is more in line with the observed natural history of patients with BMD (a milder form of muscular dystrophy) than those with DMD [Citation79]. These substantial health benefits are driven by both the high unmet need for this population (as demonstrated by the approximate 50 life years lost compared with the general male population [Citation58]) as well as the projected transformative nature of the treatment.
In addition to examining the potential health benefits obtained from delandistrogene moxeparvovec, the present study also sought to analyze its potential value from a US healthcare system perspective. Although the specific market price for delandistrogene moxeparvovec has not been established, it is critical to perform CEAs early, even before a regulatory approval, to align resource allocation with value. Indeed, given the growing importance of CEAs and their inclusion in insurance policies, value assessments of other GTs have been increasingly reported ahead of an FDA approval [Citation80,Citation81]. The present study suggests that delandistrogene moxeparvovec would be determined to be cost-effective compared with SoC alone if it were priced up to $5.1 M using a CEA appropriate for ultra-rare treatments with projected survival gains (Scenario D). After the benefit discount rate was reduced to reflect the approach used in other countries that future benefits should not be discounted at the same rate as costs, the estimated value was >$5.1 M at a WTP threshold of $500,000/evLYG [Citation82,Citation83]. Although this analysis does not conclude the potential price of delandistrogene moxeparvovec, it directly informs its value, which is an important and necessary consideration for pricing and reimbursement. Other attributes of DMD treatments, such as budget impact, are also important factors when determining the value of a new treatment within the context of affordability.
Although the QALY is the most commonly used benefit metric for HTAs worldwide, its use in CEAs of treatments for diseases that are rare or result in disability has been criticized [Citation34,Citation84,Citation85]. As described in recent literature [Citation86–88], it is more equitable in this instance to use the evLYG metric given the expected survival gains obtained with delandistrogene moxeparvovec and the bias of the QALY approach for diseases with significant disability [Citation87–89]. Furthermore, a WTP threshold of $500,000 may be reasonable for this assessment given the ultra-rarity of the disease [Citation42] and severity of DMD [Citation44–46,Citation90,Citation91]. This is also reflective of society’s preference to prioritize treatments for ultra-rare diseases as well as those diseases with the highest severity, both criteria that describe DMD [Citation42–47].
The treatment costs for GTs are incurred upfront, but GTs may have substantial health benefits that accrue over many years. As a result, GTs are very sensitive to discounting and the selected discount rate, which was highlighted in these analyses [Citation37,Citation92]. Traditional CEAs would therefore favor a chronic lifetime treatment over an equivalent single-administration treatment (despite the convenience and lack of adherence issues) even if the two treatments theoretically provided the same benefit to patients and had the same lifetime costs [Citation41]. This impact is further amplified in treatments for severe diseases in pediatric populations, in which benefits could potentially span many decades of life. The concerns of benefit discounting for evaluations of pediatric treatments have been previously described, particularly as many incur high upfront costs with deferred benefits, potentially and unfairly lowering the perceived health benefit [Citation93]. This impact is likely even greater in those pediatric diseases with severe unmet need. In the present study, decreasing the health benefit discount rate also increased the value assessment of delandistrogene moxeparvovec nearly threefold, consistent with previous studies evaluating the cost-effectiveness of other GTs [Citation37,Citation80,Citation81,Citation92]. To address this, some prominent HTAs have adopted rate ranges that reduce the benefit discount rate such that it is up to 1.5% lower than the cost discount rate [Citation75]. In other cases, ad hoc adjustments have been made to the benefit discount rate for a short-term treatment with substantial long-term benefits [Citation94]. Thus, it is important to consider the appropriateness of the benefit discount rate given the impact it has on the value assessment of all GTs, including delandistrogene moxeparvovec.
Value assessments are increasingly growing in importance, especially in the US. It is necessary to understand the potential shortcomings of traditional CEAs for evaluating innovative treatments, particularly for rare and severe diseases with a high unmet need [Citation33]. In contrast to some other GTs, the overall value assessment for delandistrogene moxeparvovec is negatively affected by the low background costs associated with SoC, arguably a reflection of the lack of effective treatments for the majority of patients with DMD. The presence of an alternative treatment would have increased the potential for cost offsets, such as those seen in hemophilia or other rare neuromuscular diseases [Citation95], resulting in a higher cost-effective value for delandistrogene moxeparvovec. For example, the value assessment for onasemnogene abeparvovec GT was substantially higher when compared with nusinersen, a chronic treatment, than it was when compared with best supportive care, although the modeled incremental treatment benefit of onasemnogene abeparvovec was lower when compared with nusinersen [Citation95]. While the opportunity for cost offsets is an important consideration in a value assessment, it is also important to recognize when low-cost offsets are driven by a high unmet need to ensure that an underserved patient population is not prohibited from accessing a novel treatment simply because there are no other treatments available.
While having to extrapolate the long-term treatment benefits beyond clinical trial results without accounting for heterogeneity of response is a common limitation with CEAs performed before a regulatory approval, it was critical to make these assumptions to assess the potential value of delandistrogene moxeparvovec given the high unmet need of DMD with SoC. Nonetheless, these assumptions were mitigated in part by contextualizing treatment benefits relative to long-term eteplirsen outcomes, and potentially even underestimating the treatment benefit expected clinically, given that delandistrogene moxeparvovec targets muscle cells that either do not replicate or replicate slowly, allowing for persistent transgene expression. This differs from other CEAs for DMD that were based on hypothetical benefits rather than clinical trial data [Citation51,Citation52]. Furthermore, although current clinical trial data focus primarily on functional outcomes involving ambulation, delandistrogene moxeparvovec has the potential to benefit upper limb function and respiratory and cardiac outcomes long term, which will be validated with ongoing clinical trials. As with benefits, durability was also assumed beyond that currently known (up to 4 years in clinical trial data [Citation27]). Nonetheless, the impact of delandistrogene moxeparvovec administration was modeled to be lifelong in the base case scenario, which is in line with DMD clinical expert opinion and other CEAs of GTs [Citation69,Citation80,Citation81,Citation95]. Due to a lack of data, non-lifetime durability scenarios were simplified and may not reflect the actual decline in function if treatment durability were to wane. Furthermore, the general population mortality did not exclude DMD mortality; however, given the rarity of DMD, this limitation would have had a negligible effect on the analysis. In addition, the various data sources may have had different definitions for the modeled health states; therefore, the risks do not perfectly align with the data for costs and utilities. However, the model was not especially sensitive to those inputs. Additionally, a utility of 0.851 was assumed for the evLYG assessments to align with the expected utility of the general population [Citation69–71]; however, this may not be an appropriate constant for pediatric conditions. Future research may also benefit from the inclusion of indirect and non-medical costs of the disease, which substantially increase over time, due to the progressive nature of the disease [Citation96–99]. Finally, value assessments should reflect treatment attributes that matter to society. A societal perspective that includes potential consequences extending beyond the healthcare system, such as productivity, scientific spillover, or caregiver impact, would be informative in understanding the full value of a treatment. For example, the potential impact delaying DMD progression has on increased working years and presenteeism for caregivers [Citation97,Citation100], or the increased aspirations and opportunities for patients with DMD to work [Citation101] were not considered. Similarly high vector processing and manufacturing costs associated with GTs [Citation82,Citation83] are not included in this value assessment. It is also conceivable that delandistrogene moxeparvovec may be steroid sparing supported by the low steroid use by patients with BMD [Citation102–104], which would reduce the morbidities associated with SoC (e.g., weight gain and vertebral compression fractures). These benefits were not considered in the current model. Thus, this analysis may have underestimated the potential value of delandistrogene moxeparvovec whereby other elements of value should be independently considered [Citation105].
Conclusion
In summary, this model suggests delandistrogene moxeparvovec has the potential to be a transformative treatment that substantially delays life-limiting disease milestones and prolongs survival for boys with DMD. It is the first GT for DMD under FDA review to restore functional dystrophin with the potential to impact the management of DMD. The results of this value assessment suggest that delandistrogene moxeparvovec has the potential to be cost-effective up to $5.1 M compared with SoC alone at a WTP threshold of $500,000/evLYG. Addressing the analytical biases of benefit discounting in CEAs for single-administration treatments resulted in the estimated value being >$5.1 M at a WTP threshold of $500,000/evLYG. As with most CEAs, other components of value that extend beyond the healthcare system were not captured in these scenarios [Citation105]. Nonetheless, in this model, delandistrogene moxeparvovec substantially increases overall QALYs and evLYs as compared with SoC alone, resulting in a potential treatment option for DMD with significant value for a severe, underserved population. These results will help to inform the potential value of delandistrogene moxeparvovec.
Supplemental Material
Download MS Word (568.3 KB)Acknowledgments
The authors would like to thank Basia Rogula from Broadstreet Health Economics for statistical analysis support. They are also grateful to Joel Iff for providing additional data on direct medical cost by health state. Medical writing and editorial support were provided by Katherine M. Alfond, PharmD, RPh, of Sarepta Therapeutics, Inc., and Marjet Heitzer, PhD, of 360 Medical Writing and was funded by Sarepta Therapeutics, Inc.
Disclosure statement
Alexa C. Klimchak, Lauren E. Sedita, Louise R. Rodino-Klapac, and Katherine L. Gooch are employees of Sarepta Therapeutics, Inc., and may own stock/options in the company. Jerry R. Mendell has reported receiving grants from Parent Project Muscular Dystrophy; receiving personal fees from Sarepta Therapeutics, Inc., and Nationwide Children’s Hospital outside the submitted work; and holding a pending patent to micro-dystrophin cassette for gene therapy and an issued patent to rAAV.SGCA delivery isolated limb infusion. Craig M. McDonald served as a consultant on clinical trials of DMD for Astellas, Avidity Biosciences, Capricor Therapeutics, Catabasis, Edgewise Therapeutics, Entrada Therapeutics, Epirium Bio (formerly Cardero Therapeutics), FibroGen, Italfarmaco, Pfizer, PTC Therapeutics, Roche, Santhera Pharmaceuticals, and Sarepta Therapeutics, Inc. He reports honoraria for presentations from PTC Therapeutics, Sarepta Therapeutics, Inc., Solid Biosciences, Santhera Pharmaceuticals, Capricor Therapeutics, and Catabasis. He has received compensation for participation in advisory boards from PTC Therapeutics, Sarepta Therapeutics, Inc., Avidity Biosciences, Edgewise Therapeutics, and Santhera Pharmaceuticals. He has received research support for clinical trials from Capricor Therapeutics, Catabasis, Italfarmaco, Pfizer, PTC Therapeutics, Santhera Pharmaceuticals, and Sarepta Therapeutics, Inc., and reports grants from the U.S. Department of Defense, U.S. National Institutes of Health (NINDS), Parent Project Muscular Dystrophy, and U.S. National Institute on Disability, Independent Living, and Rehabilitation Research (NIDILRR). Daniel C. Malone has served as a consultant to Sarepta Therapeutics, Inc.
Data availability statement
Qualified researchers may request access to the data that support the findings of this study from Sarepta Therapeutics, Inc., by contacting [email protected].
SUPPLEMENTARY MATERIAL
Supplemental data for this article can be accessed online at https://doi.org/10.1080/20016689.2023.2216518
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010 Jan;9(1):77–14.
- Aartsma-Rus A, Van Deutekom JC, Fokkema IF, et al. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006 Aug;34(2):135–144.
- Johnson EK, Zhang L, Adams ME, et al. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE. 2012;7(8):e43515. DOI:10.1371/journal.pone.0043515
- Verhaart IE, Heemskerk H, Karnaoukh TG, et al. Prednisolone treatment does not interfere with 2’-O-methyl phosphorothioate antisense-mediated exon skipping in Duchenne muscular dystrophy. Hum Gene Ther. 2012;23(3):262–273. DOI:10.1089/hum.2011.127
- Duan D, Goemans N, Takeda S, et al. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021 Feb 18;7(1):13.
- Mendell JR, Khan N, Sha N, et al. Comparison of long-term ambulatory function in patients with Duchenne muscular dystrophy treated with eteplirsen and matched natural history controls. J Neuromuscul Dis. 2021;8(4):469–479. DOI:10.3233/JND-200548
- Crisafulli S, Sultana J, Fontana A, et al. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis. 2020 Jun 5;15(1):141.
- Timonen A, Lloyd-Puryear M, Hougaard DM, et al. Duchenne muscular dystrophy newborn screening: evaluation of a new GSP®neonatal creatine kinase-MM kit in a US and Danish population. Int J Neonatal Screen. 2019;5(3):27. DOI:10.3390/ijns5030027
- Asher DR, Thapa K, Dharia SD, et al. Clinical development on the frontier: gene therapy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2020 Mar;20(3):263–274.
- Nascimento Osorio A, Medina Cantillo J, Camacho Salas A, et al. Consensus on the diagnosis, treatment and follow-up of patients with Duchenne muscular dystrophy. Neurologia (Engl Ed). 2019;34(7):469–481.
- Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018 Mar;17(3):251–267.
- Broomfield J, Hill M, Guglieri M, et al. Life expectancy in Duchenne muscular dystrophy: reproduced individual patient data meta-analysis. Neurology. 2021 Dec 7;97(23):e2304–2314.
- Scully MA, Pandya S, Moxley RT. 3rd. Review of phase II and phase III clinical trials for Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2013;1(1):33–46.
- Mendell JR, Moxley RT, Griggs RC, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320(24):1592–1597. DOI:10.1056/NEJM198906153202405
- Liu D, Ahmet A, Ward L, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy, Asthma Clin Immunol. 2013 Aug 15;9(1):30.
- Fortunato F, Farne M, Ferlini A. The DMD gene and therapeutic approaches to restore dystrophin. Neuromuscular Disorders. 2021 Oct;31(10):1013–1020.
- US Food and Drug Administration. AMONDYS 45 prescribing information. 2021. Available from: https://www.amondys45.com/Amondys45casimersenPrescribingInformation.pdf
- US Food and Drug Administration. VYONDYS 53 (golodirsen) prescribing information. 2019. Available from: https://www.accessdata.fda.gov/drugsatfdadocs/label/2019/211970s000lbl.pdf
- US Food and Drug Administration. VILTEPSO (viltolarsen) injection, for intravenous use. Prescribing information. 2020. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf
- US Food and Drug Administration. EXONDYS 51 (eteplirsen) prescribing information. 2019. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf
- McDonald CM, Shieh PB, Abdel-Hamid HZ, et al. Open-label evaluation of eteplirsen in patients with Duchenne muscular dystrophy amenable to exon 51 skipping: PROMOVI trial. J Neuromuscul Dis. 2021;8(6):989–1001. DOI:10.3233/JND-210643
- Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–647. DOI:10.1002/ana.23982
- Iff J, Done N, Tuttle E, et al. Survival in eteplirsen-treated vs Duchenne muscular dystrophy (DMD) natural history controls: an indirect treatment comparison using real-world data. Presented at the 27th International Hybrid Annual Congress of the World Muscle Society; 2022 October 11–15; Halifax, Nova Scotia, Canada.
- Mitelman O, Abdel-Hamid HZ, Byrne BJ, et al. A combined prospective and retrospective comparison of long-term functional outcomes suggests delayed loss of ambulation and pulmonary decline with long-term eteplirsen treatment. J Neuromuscul Dis. 2022;9(1):39–52. DOI:10.3233/JND-210665
- Aartsma-Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30(3):293–299. DOI:10.1002/humu.20918
- England SB, Nicholson LV, Johnson MA, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343(6254):180–182. DOI:10.1038/343180a0
- Mendell JR, Sahenk Z, Lehman K, et al. Phase 1/2a trial of delandistrogene moxeparvovec in patients with DMD: 4-year update. Paper presented at: 17th International Congress on Neuromuscular Diseases. 2022 July 5-9; Brussels, Belgium.
- Mendell JR, Sahenk Z, Lehman K, et al. Assessment of systemic delivery of rAavrh74.Mhck7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020 Sep 1;77(9):1122–1131.
- Mendell JR, Sahenk Z, Lehman K, et al. Phase 1/2a trial of SRP-9001 in patients with Duchenne muscular dystrophy: 3-year safety and functional outcomes. Paper presented at: World Muscle Society Virtual Congress. 2021 Sep 20-24.
- Mendell JR, Shieh PB, Sahenk Z, et al. A randomized, double-blind, placebo-controlled, gene-delivery clinical trial of rAavrh74.Mhck7.micro-dystrophin for Duchenne muscular dystrophy. Paper presented at: 2021 MDA Virtual Clinical & Scientific Conference; 2021 Mar 15–18.
- Sarepta Therapeutics. Community Bulletin: eMBARK study (SRP-9001-301) enrollment update. 2022. Available from: https://www.sarepta.com/community-bulletin-embark-study-srp-9001-301-enrollment-update
- Ingram D, Rodino-Klapac LR SRP-9001: new clinical data and integrated analysis. 2022 Jul 6. Available from: https://investorrelations.sarepta.com/static-files/b58a68ba-d97a-4505-9929-a097a12006e8
- Margaretos NM, Bawa K, Engmann NJ, et al. Patients’ access to rare neuromuscular disease therapies varies across US private insurers. Orphanet J Rare Dis. 2022 Feb 5;17(1):36.
- Enright D, Panzer A, Chambers J. PNS48 do US Commercial health plans cite ICER reports in their specialty drug coverage policies? An empirical analysis. Value Health. 2021;24:S181.
- Kim DD, Silver MC, Kunst N, et al. Perspective and costing in cost-effectiveness analysis, 1974-2018. Pharmacoeconomics. 2020 Oct;38(10):1135–1145.
- Institute for Clinical and Economic Review. Adapted value assessment methods for high-impact “single and short-term therapies” (SSTs). 2019 Nov 12. Available from: https://icer.org/wp-content/uploads/2020/10/ICER_SST_FinalAdaptations_111219.pdf
- Henderson N ISPOR issue panel roundup: are our HTA methods fit for purpose for gene therapies? Available from: https://www.ohe.org/news/ispor-issue-panel-roundup-are-our-hta-methods-fit-purpose-gene-therapies
- Pearson SD, Ollendorf DA, Chapman RH. New cost-effectiveness methods to determine value-based prices for potential cures: what are the options? Value Health. 2019 Jun;22(6):656–660.
- National Institute for Health and Care Excellence. Interim process and methods of the highly specialised technologies programme updated to reflect 2017 changes. 2017. Available from: https://www.nice.org.uk/media/default/about/what-we-do/nice-guidance/nice-highly-specialised-technologies-guidance/hst-interim-methods-process-guide-may-17.pdf
- Taylor C, Jan S, Thompson K. Funding therapies for rare diseases: an ethical dilemma with a potential solution. Aust Health Rev. 2018 Feb;42(1):117–119.
- Klimchak AC, Sedita LE, Gooch KL, et al. Assessing the impact of single or short-term administration on a therapy’s cost-effectiveness: a hypothetical disease-agnostic model. J Med Econ. 2023 Jan-Dec;26(1):594–602.
- Institute for Clinical and Economic Review. Modifications to the ICER value assessment framework for treatments for ultra-rare diseases. 2020 Jan 31. Available from: https://icer.org/wp-content/uploads/2020/10/ICER_URD_Framework_Adapt_013120.pdf
- National Institute for Health and Care Excellence. Clarifying meanings of absolute and proportional shortfall with examples. 2013 Sep 17. Available from: https://www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-technology-appraisals/OHE-Note-on-proportional-versus-absolute-shortfall.pdf
- Angelis A, Kanavos P, Montibeller G. Resource allocation and priority setting in health care: a multi-criteria decision analysis problem of value? Global Policy. 2017;8(S2):76–83.
- Ottersen T, Forde R, Kakad M, et al. A new proposal for priority setting in Norway: open and fair. Health Policy. 2016;120(3):246–251. DOI:10.1016/j.healthpol.2016.01.012
- Reckers-Droog VT, van Exel NJA, Brouwer WBF. Looking back and moving forward: on the application of proportional shortfall in healthcare priority setting in the Netherlands. Health Policy. 2018 Jun;122(6):621–629.
- Szabo SM, Audhya IF, Feeny D, et al. Societal perspectives on disease and treatment attributes characterizing rare diseases: a qualitative study from the United States. J Patient Rep Outcomes. 2022 Jan 24;6(1):9.
- Soim A, Wallace B, Whitehead N, et al. Health profile of preterm males with Duchenne muscular dystrophy. J Child Neurol. 2021 Oct;36(12):1095–1102.
- McDonald CM, Henricson EK, Abresch RT, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018 Feb 3;391(10119):451–461.
- Muntoni F, Domingos J, Manzur AY, et al. Categorising trajectories and individual item changes of the North Star ambulatory assessment in patients with Duchenne muscular dystrophy. PLoS ONE. 2019;14(9):e0221097. DOI:10.1371/journal.pone.0221097
- Institute for Clinical and Economic Review. Deflazacort, eteplirsen, and golodirsen for Duchenne muscular dystrophy: effectiveness and value. 2022 Apr 22. Available from: https://icer.org/wp-content/uploads/2020/10/Corrected_ICER_DMD-Final-Report_042222.pdf
- Landfeldt E, Alfredsson L, Straub V, et al. Economic evaluation in Duchenne muscular dystrophy: model frameworks for cost-effectiveness analysis. Pharmacoeconomics. 2017 Feb;35(2):249–258.
- Peay H, Kennedy A, Fischer R, et al. Promoting meaningful clinical trial outcome measures for Duchenne muscular dystrophy. Neuromuscular Disorders. 2016;26:S187.
- McDonald CM, Gordish-Dressman H, Henricson EK, et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: long-term natural history with and without glucocorticoids. Neuromuscular Disorders. 2018 Nov;28(11):897–909.
- Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018 Apr;17(4):347–361.
- Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012;31(2):121–125.
- Paramsothy P, Wang Y, Cai B, et al. Selected clinical and demographic factors and all-cause mortality among individuals with Duchenne muscular dystrophy in the muscular dystrophy surveillance, tracking, and research network. Neuromuscular Disorders. 2022 Jun;32(6):468–476.
- Arias E, Xu JQ. United States life tables, 2018. National Vital Stat Rep. 2020;69(12):1–44.
- Potter RA, Griffin DA, Heller KN, et al. Dose-escalation study of systemically delivered rAavrh74.Mhck7.micro-dystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7–8):375–389.
- Zaidman C, Proud C, McDonald C, et al. One-year ENDEAVOR data (ambulatory, ≥4-to <8-year-olds): phase 1b trial of delandistrogenemoxeparvovecin Duchenne muscular dystrophy (DMD). Paper presented at: 17th International Congress on Neuromuscular Diseases (ICNMD). 2022 July 5-9; Brussels, Belgium.
- Mendell JR, Shieh PB, Sahenk Z, et al. A Phase 2 clinical trial evaluating the safety and efficacy of delandistrogene moxeparvovec in patients with DMD. Presented at the 27th International Annual Congress on the World Muscle Society (WMS) 2022. 2022 October 11-15. Halifax, Canada.
- De Feraudy Y, Ben Yaou R, Wahbi K, et al. Very low residual dystrophin quantity is associated with milder dystrophinopathy. Ann Neurol. 2021 Feb;89(2):280–292.
- Horsman J, Furlong W, Feeny D, et al. The Health Utilities Index (HUI): concepts, measurement properties and applications. Health Qual Life Outcomes. 2003 Oct 16;1(1):54.
- Audhya IF, Szabo SM, Bever A, et al. Estimating health state utilities in Duchenne muscular dystrophy (DMD) using the EQ5D and Health Utilities Index (HUI). Paper presented at: World Muscle Society. 2022 Oct 11-15; Halifax, Canada.
- Iff J, Zhong Y, Gupta D, et al. Disease progression stages and burden in patients with Duchenne muscular dystrophy using administrative claims supplemented by electronic medical records. Adv Ther. 2022 Jun;39(6):2906–2919.
- Bello L, Morgenroth LP, Gordish-Dressman H, et al. DMD genotypes and loss of ambulation in the CINRG Duchenne natural history study. Neurology. 2016;87(4):401–409. DOI:10.1212/WNL.0000000000002891
- Soim A, Smith MG, Kwon JM, et al. Is there a delay in diagnosis of Duchenne muscular dystrophy among preterm-born males? J Child Neurol. 2018 Jul;33(8):537–545.
- Institute for Clinical and Economic Review. Cost-effectiveness, the QALY, and the evLYG. 2023 Apr 7. Available from: https://icer.org/our-approach/methods-process/cost-effectiveness-the-qaly-and-the-evlyg/
- Institute for Clinical and Economic Review. Betibeglogene autotemcel for beta thalassemia: effectiveness and value. Final evidence report. 2022 Jul 19. Available from: https://icer.org/wp-content/uploads/2021/11/ICER_Beta-Thalassemia_Final-Report_071922.pdf
- Institute for Clinical and Economic Review. Anti B-cell maturation antigen CAR T-cell and antibody drug conjugate therapy for heavily pre-treated relapsed and refractory multiple myeloma final report. 2021 Jul 9. Available from: https://icer.org/wp-content/uploads/2020/10/ICER_Multiple-Myeloma_Final-Report_Update_070921.pdf
- Pickard AS, Law EH, Jiang R, et al. United States valuation of EQ-5D-5L health states using an international protocol. Value Health. 2019 Aug;22(8):931–941.
- National Institute for Health and Care Excellence. NICE health technology evaluations: the manual. 2022 Jan 31. Available from: https://www.nice.org.uk/process/pmg36/resources/nice-health-technology-evaluations-the-manual-pdf-72286779244741
- Garrison LP, Jackson T, Paul D, et al. Value-based pricing for emerging gene therapies: the economic case for a higher cost-effectiveness threshold. J Manag Care Spec Pharm. 2019 Jul;25(7):793–799.
- Zorginstituut Nederland. Guideline for economic evaluations in healthcare. 2016 [cited 2022 Jul 14]. Available from: https://english.zorginstituutnederland.nl/binaries/zinl-eng/documenten/reports/2016/06/16/guideline-for-economic-evaluations-in-healthcare/Guideline+for+economic+evaluations+in+healthcare.pdf
- John J, Koerber F, Schad M. Differential discounting in the economic evaluation of healthcare programs. Cost Eff Resour Alloc. 2019;17(1):29.
- Chambers J, Silver MC, Lin PJ, et al. Pmu77 cell and gene therapies are associated with substantially larger quality-adjusted life year gains than conventional drugs and biologics. Value Health. 2019;22:S236.
- Sandberg EA, Thorat T, Neumann PJ, et al. Qaly gains in cancer comPared with cHronic diseases. Value Health. 2016;19(3):A1–318. DOI:10.1016/j.jval.2016.03.065
- Chambers JD, Thorat T, Wilkinson CL, et al. Drugs cleared through the FDA’s expedited review offer greater gains than drugs approved by conventional process. Health Aff (Millwood). 2017 Aug 1;36(8):1408–1415.
- Darras BT, Urion DK, Ghosh PS Dystrophinopathies. In: Adam MP, Everman DB, Mirzaa GM, et al, editors. Dystrophinopathies. Seattle (WA): University of Washington, Seattle; 1993-2022. [Updated 2022 Jan 20;cited 2000 Jan 20]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1119/
- Kansal AR, Reifsnider OS, Brand SB, et al. Economic evaluation of betibeglogene autotemcel (Beti-cel) gene addition therapy in transfusion-dependent beta-thalassemia. J Mark Access Health Policy. 2021 Jun 7;9(1):1922028.
- Malone DC, Dean R, Arjunji R, et al. Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J Mark Access Health Policy. 2019;7(1):1601484. DOI:10.1080/20016689.2019.1601484
- Harris E Breaking down pricing of cell & gene therapies. 2019 Jun 18. Available from: https://www.cellandgene.com/doc/breaking-down-pricing-of-cell-gene-therapies-0001#:~:text=Cost%20of%20goods%2Fmanufacturing%20alone,to%20provide%20access%20to%20patients.
- Salva MZ, Himeda CL, Tai PW, et al. Design of tissue-specific regulatory cassettes for high-level Raav-mediated expression in skeletal and cardiac muscle. Mol Ther. 2007;15(2):320–329. DOI:10.1038/sj.mt.6300027
- Institute for Clinical and Economic Review. 2020-2023 Value assessment framework. Accessed October 16, 2022. Available from: https://34eyj51jerf417itp82ufdoe-wpengine.netdna-ssl.com/wp-content/uploads/2020/10/ICER_2020_2023_VAF_102220.pdf
- National Institute of Health and Care Excellence. The guidelines manual. Process and methods [PMG6]. 2012. Accessed October 16, 2022. Available from: https://www.nice.org.uk/process/pmg6/chapter/assessing-cost-effectiveness
- Beresniak A, Medina-Lara A, Auray JP, et al. Validation of the underlying assumptions of the quality-adjusted life-years outcome: results from the ECHOUTCOME European project. Pharmacoeconomics. 2015 Jan;33(1):61–69.
- Prieto L, Sacristan JA. Problems and solutions in calculating quality-adjusted life years (QALYs). Health Qual Life Outcomes. 2003 Dec 19;1(1):80.
- Rand LZ, Kesselheim AS. Controversy over using quality-adjusted life-years in cost-effectiveness analyses: a systematic literature review. Health Aff. 2021 Sep;40(9):1402–1410.
- Shafrin J, Lakdawalla DN, Doshi JA, et al. A strategy for value-based drug pricing under the inflation reduction act. Health Aff Forefront. 2023; DOI:10.1377/forefront.20230503.153705
- National Institute for Health and Care Excellence. NICE health technology evaluations: the manual. Process and methods [PMG36]. 2022 [cited 2022 Oct 17]. Available from: https://www.nice.org.uk/process/pmg36/chapter/introduction-to-health-technology-evaluation
- Lakdawalla DN, Phelps CE. Health technology assessment with diminishing returns to health: the generalized risk-adjusted cost-effectiveness (GRACE) approach. Value Health. 2021;24(2):244–249.
- Whittington MD, McQueen RB, Ollendorf DA, et al. Long-term survival and value of chimeric antigen receptor t-cell therapy for pediatric patients with relapsed or refractory leukemia. JAMA Pediatr. 2018 Dec 1;172(12):1161–1168.
- Ungar WJ, Prosser LA, Burnett HF. Values and evidence colliding: health technology assessment in child health. Expert Rev Pharmacoecon Outcomes Res. 2013;13(4):417–419.
- National Institute for Health and Care Excellence. Mifamurtide for the treatment of osteosarcoma. 2011 Oct 26. Available from: https://www.nice.org.uk/guidance/ta235/resources/mifamurtide-for-the-treatment-of-osteosarcoma-pdf-82600372273093
- Institute for Clinical and Economic Review. Spinraza® and zolgensma® for spinal muscular atrophy: effectiveness and value. 2019 May 24. Available from: https://icer.org/wp-content/uploads/2020/10/ICER_SMA_Final_Evidence_Report_110220.pdf
- Cavazza M, Kodra Y, Armeni P, et al. Social/Economic costs and health-related quality of life in patients with Duchenne muscular dystrophy in Europe. Eur J Health Econ. 2016 Apr;17 Suppl 1(S1):19–29.
- Landfeldt E, Lindgren P, Bell CF, et al. The burden of Duchenne muscular dystrophy: an international, cross-sectional study. Neurology. 2014 Aug 5;83(6):529–536.
- Schreiber-Katz O, Klug C, Thiele S, et al. Comparative cost of illness analysis and assessment of health care burden of Duchenne and Becker muscular dystrophies in Germany. Orphanet J Rare Dis. 2014 Dec 18;9(1):210.
- Thayer S, Bell C, McDonald CM. The direct cost of managing a rare disease: assessing medical and pharmacy costs associated with Duchenne muscular dystrophy in the United States. J Manag Care Spec Pharm. 2017 Jun;23(6):633–641.
- Soelaeman RH, Smith MG, Sahay K, et al. Labor market participation and productivity costs for female caregivers of minor male children with Duchenne and Becker muscular dystrophies. Muscle Nerve. 2021 Dec;64(6):717–725.
- Schwartz CE, Biletch E, Stuart RBB, et al. Patient life aspirations in the context of Duchenne muscular dystrophy: a mixed-methods case-control study. J Patient Rep Outcomes. 2022 Sep 14;6(1):97.
- Clemens PR, Niizawa G, Feng J, et al. The CINRG Becker natural history study: baseline characteristics. Muscle Nerve. 2020 Sep;62(3):369–376.
- Cowen L, Mancini M, Martin A, et al. Variability and trends in corticosteroid use by male United States participants with Duchenne muscular dystrophy in the Duchenne Registry. BMC neurol. 2019 May 2;19(1):84.
- Johnston KM, Salhany RM, Ciafaloni E, et al. The natural history of Becker muscular dystrophy (BMD) - a systematic literature review. Paper presented at: 25th International Annual Congress of the World Muscle Society- Virtual; 2020 Sep 30-Oct 2.
- Lakdawalla DN, Doshi JA, Garrison LP Jr., et al. Defining elements of value in health care-a health economics approach: an ISPOR special task force report [3]. Value Health. 2018 Feb;21(2):131–139.