
ABSTRACT
Background and objective: To describe the clinical characteristics including the bronchoalveolar lavage fluid (BALF) characteristics of patients with antisynthetase syndrome (AS) associated interstitial lung disease (ILD) in a tertiary ILD outpatient clinic, their medical therapy and outcome.
Methods: Retrospective cohort study of patients with AS-ILD. All available data of clinical characteristics, pulmonary function tests, laboratory parameters, BALF analysis, histology, high-resolution computed tomography (HRCT) and treatment were collected from the patient files.
Results and conclusions: Twelve patients with AS-ILD were identified. Mean age at diagnosis was 55 years (range 45–69), 67% were female. Mean follow-up time was 7 years. The anti-aminoacyl tRNA-synthetase antibodies presented were anti-Jo1 (n = 6), anti-PL7 (n = 3), anti-PL12 (n = 2) and anti-EJ (n = 1). HRCT patterns were mainly non-specific interstitial pneumonia (75%). Four patients had BALF-eosinophilia (two of four anti-Jo1 patients) and two anti-PL12 positive patients had BALF-neutrophilia. Two AS-ILD patients improved on rituximab (RTX) as induction treatment and three out of four patients were stabilised on RTX as maintenance treatment. Two patient obtained remission by cyclophosphamide. Four patients were stabilised on azathioprine alone or in combination with oral corticosteroids.
Our cohort of AS-ILD patients showed clinical characteristics in accordance with previous reports at baseline and was comparable to other cohorts. Most patients can be stabilised with immunosuppressive treatment.
Introduction
Antisynthetase syndrome (AS) is defined by the occurrence of IgG anti-aminoacyl RNA-synthetase (anti-ARS) antibodies and associated types of autoimmune manifestations, mainly myositis. The autoimmunity accounts for important clinical manifestations, such as interstitial lung disease (ILD), arthritis, fever, Raynaud´s phenomenon and mechanic’s hand, which can be present at disease onset or appear later as the disease progresses [Citation1]. At present, eight anti-aminoacyl tRNA-synthetase antibodies (anti-ARS) have been identified: anti-Jo1 (histidyl), anti-PL7 (threonyl), anti-PL12 (alanyl), anti-EJ (glycyl), anti-OJ (isoleucyl), anti-KS (asparaginyl), anti-ZO (phenylalanyl) and anti-YRS/HA (tyrosyl). The most commonly encountered antibody is anti-Jo1 that accounts for up to 60–80% [Citation2–Citation4]. ARS are cytoplasmic enzymes that play a vital role in protein synthesis. Defect of these enzymes can theoretically cause adverse events from every organ. AS is a rare, but probably underdiagnosed disease with a reported prevalence for Caucasians of 87/100,000 in a Norwegian study [Citation5]. The reported incidence was 6–10 per million inhabitants per year diagnosed by presence of anti-ARS and polymyositis/dermatomyositis (PM/DM). Approximately, 25–30% of PM/DM patients had anti-ARS [Citation5]. In a US population, the incidence of AS is considered as low as one in 3–4 million with a diagnostic delay of more than 2 years [Citation6].
The typical triad of ILD, myositis and arthritis accounts for up to 90% of AS [Citation1]. However, more recent studies report increased number of cases with mono-organ involvement at the time of diagnosis, most likely due to earlier diagnosis caused by increasing awareness [Citation7]. In rare cases, cardiac and renal involvement have been reported [Citation8,Citation9].
ILD is a major contributor to morbidity and mortality [Citation3,Citation10], and recent data suggest that different AS-phenotypes, especially anti-PL7 and 12, could be related to incidence and severity of ILD. The pulmonary damage may be irreversible, which is consistent with early scarring of the interstitium [Citation7]. The most common radiological and histopathological pattern is non-specific interstitial pneumonia (NSIP)[Citation11].
The optimal treatment regimen has yet to be established. Due to a high relapse rate in patients treated with corticosteroid alone, a combination of corticosteroid and other immunosuppressants, such as methotrexate, azathioprine (AZA), cyclophosphamide and mycophenolate is now frequently used. In severe or rapidly progressive ILD, rituximab (RTX) has been used with some success [Citation12].
The aim of the present study was to describe the clinical characteristics, therapy and outcome of a cohort of patients with AS associated ILD (AS-ILD).
Methods
ICD-10 codes, J84 and M33, in the hospital registries were used to identify all patients with an AS-ILD diagnosis in our tertiary ILD referral centre in Aarhus between October 2006 and August 2017. The centre serves 1.9 million citizens from the central and north Denmark regions. All available data regarding clinical characteristics, pulmonary function (PF) tests, laboratory parameters, bronchoalveolar lavage fluid (BALF), high-resolution computed tomography (HRCT), treatment and disease course were collected from the patient files.
The diagnosis of AS was made by the presence of anti-ARS accompanied by key clinical features, including ILD and/or PM/DM and evaluated by an expert panel of rheumatologist, respiratory physicians and radiologist. Raynaud’s phenomenon, arthritis, fever and mechanic’s hands supported the diagnosis, but their presence was not mandatory. The presence of ILD was evaluated at a multidisciplinary team discussion with the presence of an expert ILD radiologist and expert ILD respiratory physicians.
Patients underwent clinical evaluation every 3–6 months. Follow-up included PF tests and laboratory parameters. A change in FVC of more than 10% points (absolute) and/or a change in DLCO of more than 15% points were considered significant whereas smaller changes in FVC and DLCO were regarded as stable disease.
Patient data were retrieved from the Danish National Registry for Interstitial Lung Diseases. The registry is approved by the Danish Health Authority. Patient confidentiality was maintained.
Results
Twelve patients with AS-ILD were identified between 2005 and 2017. Three patients were diagnosed with AS at our clinic whereas the remaining nine patients were referred from rheumatology outpatient clinics. The prevalence of AS-ILD was 6.3/1.000.000 population.
Median age at symptom onset was 55 years (range 44–68 years) and median age at the time of diagnosis was 57 years (range 45–69 years). Median diagnostic delay was 24 months (range 3–38 months) from the first visit at the rheumatology or respiratory outpatient clinic. Eight patients (66%) were female (). All were Caucasians except one who was African-American. Median follow-up time was 7 years (range 1–11 years).
Table 1. Demographics and clinical features of AS associated ILD.
Cough and/or dyspnoea were reported in 11 of 12 patients. Acute onset of dyspnoea was reported in three patients, gradual onset of dyspnoea in eight patients and one patient had no respiratory symptoms. The most common extrapulmonary symptoms were joint involvement (9/12), fever (6/12), mechanic’s hands (6/12), Raynaud’s phenomenon (5/12), Gottron’s sign (5/12) and sicca symptoms (2/12).
Five of six anti-Jo1 positive patients had PM/DM. Two anti-PL12-positive patients were amyopathic.
Antibody distribution, demographics and clinical features are shown in . Coexistence of anti-SSA/Ro5 was found in all 11 patients tested, and positive ANA in five patients. One patient with anti-SSA/Ro5 had sicca symptoms. Three patients were diagnosed with an overlap syndrome of systemic scleroderma and AS. Creatinine kinase was elevated in seven patients ().
Table 2. Demographics and clinical features according to serology.
Bronchoscopy data were available in nine patients. All cultures were negative, including culture for mycobacteria. BAL neutrophilia was seen in the two anti-PL12-positive patients, BAL lymphocytosis was seen in three patients and eosinophilia in four patients, two of those were anti-Jo1 positive, one anti-PL7 and one anti-EJ positive (). All four patients were non-smokers. There was no correlation between BAL differential count and HRCT pattern or disease progression.
Table 3. Bronchoalveolar lavage leukocyte differential count.
HRCT at onset revealed a NSIP-pattern in nine patients, a non-specific ILD-pattern in one patient and a usual interstitial pneumonia (UIP) pattern in two patients ().
Table 4. Treatment and outcome.
Seven patients presented with a restrictive PF pattern and two with an obstructive PF, one former smoker and one never smoker. The median FVC was 69% predicted (29–120%) and median DLCO 42% predicted (19–70%). One patient developed a restrictive pattern during follow-up.
Four different treatment regimens were used as initial therapy (). Each patient could receive more than one treatment regimen depending on treatment response. Seven patients were initially treated with steroid pulses and cyclophosphamide, which resulted in improved PF in two patients, unchanged in four and deterioration in one. Two patients were initially started on RTX with either AZA plus oral steroids (OS) or OS alone. Both patients had significant improvement of their PF. Two patients had OS as initial therapy and remained stable.
Maintenance therapy with AZA ± OS was given to five patients and stabilised four of them. Four patients received RTX in combination with other immunosuppressants, which resulted in improvement of one patient, stabilisation of two and one with deterioration. Only one patient (case 3) had no maintenance therapy due to mild symptoms and stable PF and HRCT, cases 4 and 10 were deemed terminally ill and it was decided to refrain from maintenance therapy. All other patients were on continuous maintenance therapy.
All three anti-PL7-positive patients had an initial low PF, but two patients improved on initial therapy. Two patients were stabilised on maintenance therapy.
One of the two anti-PL12-positive patients had a low PF and was stabilised with initial therapy but deteriorated on maintenance therapy (case 11). The other patient deteriorated in spite of treatment (case 10).
Changes in HRCT after treatment are presented in . Of the seven patients receiving steroid pulse courses and cyclophosphamide as initial therapy, three had HRCT improvement and three progressed despite treatment. Two patients received RTX, and one improved. During maintenance therapy, one patient was stabilised on RTX, one improved and one regressed from unspecific mild ILD to a NSIP pattern. Two patients were stabilised on AZA ± OS and three progressed in spite of treatment ( and ). Two patients (cases 2 and 6) progressed to a UIP pattern.
Figure 1. HRCT at debut. A NSIP pattern with consolidations, sub-pleural sparing and ground glass opacities in a perilobular localisation are seen in the basal lung zones.
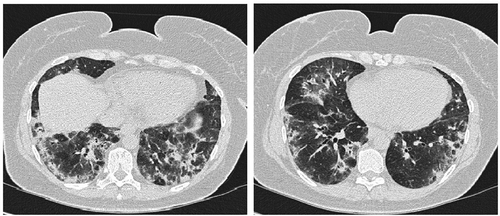
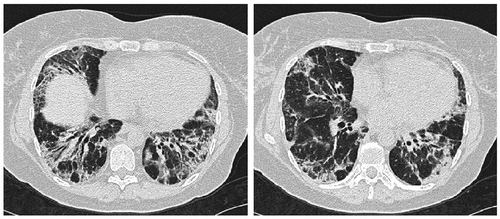
Figure 2. HRCT 3 years later. Distributions of the interstitial features are unchanged but the pattern has turned more fibrotic with the development of traction bronchiectasis and increased reticulation.
Four patients (cases 3, 5, 10 and 11) developed pulmonary hypertension diagnosed by echocardiography (verified by right heart catheterisation in two patients).
Four patients died during follow-up. Causes of death were colorectal cancer (case 4), pneumonia/sepsis (cases 5 and 11) and prolonged multi-organ failure (case 10).
Discussion
The present study reports the characteristics of a cohort of 12 patients with AS-ILD. The clinical characteristics are in accordance with previous reports with respect to gender and age distribution and the majority being anti-Jo1 positive. NSIP was the most common radiological pattern. We initially observed two definite UIP patterns, and two patients progressed to a UIP pattern during treatment. A large previous study did not report the presence of UIP in AS-ILD [Citation11].
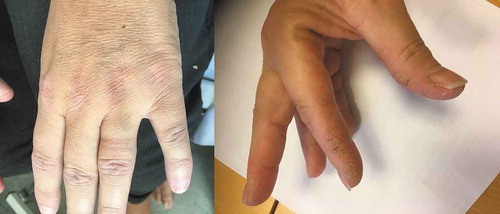
The clinical characteristics at the time of diagnosis are consistent with the classic triad of arthritis, myositis and ILD. However, the presence of the triad at diagnosis could be caused by diagnostic delay and cumulative, progressive symptoms [Citation7,Citation13]. Pinal-Fernandez et al. [Citation7] found that >60% of AS patients experienced pulmonary and muscle involvement but not necessarily simultaneously. This was also reported by Cavagna et al. [Citation1,Citation13] who recommend an earlier screening for anti-ARS in patients presenting with arthritis alone and even in those fulfilling the criteria for rheumatoid arthritis given the similarities of the diseases. Screening for anti-ARS in patients with mono-symptomatic arthritis would be costly and with a low positive hit rate and even risk of false positive or negative results. ILD may also be the only initial presenting manifestation of the disease, typically for anti-PL7 and anti-PL12, although we did not find this correlation in our cohort [Citation14]. Pinal-Fernandez et al. [Citation7] speculated that the pulmonary lesions in AS-ILD – especially for anti-PL12 patients – were irreversible, which is supported by our findings. Diagnostic delay is not uncommon in rare diseases, however early diagnosis and initiation of therapy is essential in order to slow down or hopefully prevent irreversible lung damage in AS-ILD [Citation15]. We therefore suggest to screen for anti-ARS in patients with clinical suspicion of connective tissue disease, females, middle age, symptoms or clinical signs of connective tissue disease and a HRCT pattern of NSIP. Especially a thorough investigation for skin manifestations, such as the red, scaly, elevated Gottron’s papules on the metacarpophalangeal and interphalangeal finger joints or mechanic’s hands with hyperkeratotic, scaly and fissured papules on the first web space () could lead to an earlier diagnosis.
Figure 3. Left: Gottron’s papules; right: mechanic’s hands.
The definition of ILS is non-coherent when different studies of AS-ILD are compared. We defined ILS by criteria for both PF and HRCT patterns, while other studies only used HRCT patterns. This compromises the comparability of the few published studies as the definition of AS varies among studies. In our study, an expert panel of radiologists, respiratory physicians and rheumatologists assessed every patient, but the diagnosis still rely on the expert opinions of the panel rather than on well-defined internationally accepted criteria and is opt to vary depending on, e.g. the level of experience and culture. The need for diagnostic criteria for AS is obvious.
Our study is the first to report BAL findings in AS-ILD. No specific pattern or relationship with HRCT patterns was found. All BALF was collected at diagnosis when AS was active. Passadore I et al. [Citation16] analysed BALF of four AS patients for protein content but did not present differential counts. Interestingly, both our patients with anti-PL12 had BALF neutrophilia revealing a possible trend. Four patients had eosinophilia. None were smokers in the months up to and at the time of the bronchoscopy. Thus, BALF differential count may have a role as a possible step towards a personalised treatment in AS-ILD in the future, but larger studies are needed.
Case 4 died of malignancy (colorectal cancer). It has previously been suggested that AS patients have increased risk of cancer. However, in recent studies, the incidence of malignancy seemed similar to the general population [Citation7].
Treatment of AS is challenging and based upon small case series rather than randomised, clinical trials. It is generally agreed that corticosteroids are the first line therapy with the addition of other immunosuppressants, such as mycophenolate, methotrexate, AZA or cyclophosphamide, but there is no consensus on which immunosuppressive agents to prefer. The choice depends primarily on the severity of the disease [Citation3,Citation17]. Treatment in our cohort followed these recommendations. Seven of our patients (case 4–6, 9–12) received high doses of corticosteroid and cyclophosphamide pulse courses as induction therapy. This resulted in improvement in PF of only two patients, but stabilised four. Schnabel et al. [Citation18] reported a favourable response to intravenous pulse cyclophosphamide in 10 patients with progressive ILD. We found disease progression in spite of cyclophosphamide therapy in three of seven patients, however, two of these patients were anti-PL12 positive, which correlates to other studies also finding a high risk of treatment failure in patients with anti-PL12 antibodies[Citation7].
In recent years, treatment regimes with RTX as initial therapy to AS-ILD have emerged. Sem M et al. [Citation19] reported some success with RTX in nine AS-ILD patients. However, their follow-up period was limited to 6 months after treatment initiation. Two of our patients received RTX as initial therapy with favourable response. They both continued on RTX as maintenance therapy with one still being stabilised after a follow-up period of 7 years and another with a mild progression to a NSIP pattern during the 9-year follow-up. Two other patients were started on RTX as maintenance therapy and one of them stabilised. The two patients with UIP pattern on HRCT (case 4 and 11) showed no response to treatment. This is in line with lacking treatment responses seen in other patients with connective tissue disease and UIP pattern, especially rheumatoid arthritis associated UIP [Citation20]. It could be speculated that a subgroup of AS patients with non-UIP pattern could be treated successfully with RTX as the first choice of treatment, however more studies are needed.
There are obvious limitations to our study due to the small patient number. Not all AS-ILD patients may have been included because we did not include arthritis codes in our inclusion criteria. Cases with ILD and arthritis, who had not presented with myositis yet, may have been missed. The strength of our study is the multidisciplinary diagnosis and the well-characterised cohort with long follow-up
In summary, our cohort of AS-ILD patients showed clinical characteristics in accordance with previous reports. The clinical course and the response to treatments were variable. Complete remission in AS-ILD is rare and long-term immunosuppressive therapy is usually required. The optimal treatment regime is not yet defined and well-designed prospective, multicentre studies are warranted. However, promising studies for the treatment of ILD-associated connective tissue diseases are ongoing [Citation21]. Our cohort is limited by the small sample size, but contributes to the general knowledge of AS. Hopefully, improved sub-classification of AS-ILD by specific anti-ARS, ILD patterns and symptomatology will lay the foundation for more specific and personalised treatment strategies.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Notes on contributors
Mads Lynge Jensen
Mads Lynge Jensen, MD, is a specialist registrar at The Department of Respiratory Diseases and Allergy, Aarhus University Hospital, Denmark. He is a 5th year registrar in pulmonary diseases.
Anders Løkke
Anders Løkke, MD, is a consultant at The Department of Respiratory Diseases and Allergy, Aarhus University Hospital, Denmark. He is a specialist in pulmonary diseases, and his main research focus is COPD.
Ole Hilberg
Ole Hilberg, MD, DMSc, is a consultant at The Department of Respiratory Diseases, Vejle Hospital, Vejle, Denmark. He is a specialist in pulmonary diseases with several research areas.
Charlotte Hyldgaard
Charlotte Hyldgaard, MD, PhD, is a speciality registrar at The Diagnostic Center, Silkeborg Hospital, Silkeborg, Denmark. She is a specialist in pulmonary medicine, and her main research focus is interstitial lung diseases.
Dan Tran
Dan Tran, MD, is a consultant at The Department of Respiratory Diseases, Sydvestjydsk Regional Hospital, Esbjerg, Denmark. He is a specialist in pulmonary diseases.
Elisabeth Bendstrup
Elisabeth Bendstrup, MD, PhD, is a consultant at The Department of Respiratory Diseases and Allergy, Aarhus University Hospital, Denmark. She is a specialist in pulmonary diseases, and her main research focus is interstitial lung diseases.
References
- Cavagna L, Nuño L, Scirè CA, et al. AENEAS (American and European Network of Antisynthetase Syndrome) collaborative group. Serum Jo-1 autoantibody and isolated arthritis in the antisynthetase syndrome: review of the literature and report of the experience of AENEAS collaborative group. Clin Rev Allerg Immunol. 2017;52:71–7.
- Satoh M, Tanaka S, Ceribelli A, et al. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allerg Immunol. 2017 Feb;52(1):1–19.
- Marie I, Josse S, Hatron PY, et al. Interstitial lung Disease in anti-Jo-1 patients with antisynthetase syndrome. Arthritis Res Ther. 2013 May;65(5):800–808.
- Marie I, Josse S, Decaux O, et al. Comparison of long-term outcome between anti-Jo-1 and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmun Rev. 2012;11:739–745.
- Dobloug C, Garen T, Bitter H, et al. Prevalence and clinical characteristics of adult polymyositis and dermatomyositis; data from a large and unselected Norwegian cohort. Ann Rheum Dis. 2015;74:1551–1556.
- Smoyer-Tomic KE, Amato AA, Fernandes AW. Incidence and prevalence of idiopathic inflammatory myopathies among commercially insured, Medicare supplemental insured, and Medicaid enrolled populations: an administrative claims analysis. BMC Musculosketal Disord. 2012 Jun;15(13):103.
- Pinal-Fernandez I, Casal-Dominguez M, Huapaya JA, et al. A longitudinal cohort study of the anti-synthetase syndrome: increased severity of interstitial lung disease in black patients and patients with anti-PL7 and anti-PL12 autoantibodies. Rheumatology (Oxford). 2017 Jun 1;56(6):999–1007.
- Poveda Gomez F, Merino JL, Maté I, et al. Polymyositis associated with anti-Jo1 antibodies: severe cardiac involvement as initial manifestation. Am J Med. 1993;94:110–111.
- Frost NA, Morand EF, Hall CL, et al. Idiopathic polymyositis complicated by arthritis and mesangial proliferative glomerulonephritis: case report and review of the literature. Br J Rheumatol. 1993;32:929–931.
- Marguerie C, Bunn CC, Beynon HL, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J med. 1990;77:1019–1038.
- Zamora AC, Hoskote SS, Abascal-Bolade B, et al. Clinical features and outcomes of interstitial lung disease in anti-Jo-1 positive antisynthetase syndrome. Respir Med. 2016 Sep;118:39–45.
- Bauhammer J, Blank N, Max R, et al. Rituximab in the treatment of Jo1 antibody-associated antisynthetase syndrome: anti-Ro52 positivity as a marker for severity and treatment response. J Rheumatol. 2016 Aug;43(8):1566–1574.
- Cavagna L, Nuño L, Scirè CA, et al. AENEAS (American, European Network of Antisynthetase Syndrome) collaborative group. Clinical spectrum time course in anti-Jo-1 positive antisynthetase syndrome. Results from an international retrospective multicenter study. Med August. 2015;94(32):1–7.
- Mileti LM, Strek ME, Niewold TB, et al. Clinical characteristics of patients with anti–jo-1 antibodies: a single center experience. J Clin Rheumotolol. 2009;15(5):254–255.
- Hyldgaard C, Hilberg O, Muller A, et al. A cohort study of interstitial lung diseases in central Denmark. Respir Med. 2014;May:108(5):793–799.
- Passadore I, Ladarola P, Di Poto C, et al. 2-DE and LC-MS/MS for a comparative proteomic analysis of BALf from subjects with different subsets of inflammatory myopathies. J Proteome Res. 2009 May;8(5):2331–2340.
- Cavagna L, Carapoli R, Abdi-Ali L, et al. Cyclosporine in anti-Jo1- positive patients with corticosteroid-refractory interstitial lung disease. J Rheumatol. 2013;49(4):483–492.
- Schnabel A, Reuter M, Biederer J, et al. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum. 2003;32(5):273–284.
- Sem M, Molberg O, Lund MB, et al. Rituximab treatment of the anti synthetase syndrome – a retrospective case series. Rheumatology. 2009;48:968–971.
- Kim EJ, Collard HR, King TE Jr. Rheumatoid arthritis-associated interstitial lung disease. The relevance of histopathologic and radiographic pattern. Chest Nov. 2009;136(5):1397–1405.
- Saunders P, Tsipouri V, Keir GJ, et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials. 2017 jun 15;18(1):275.