
ABSTRACT
Background
Idiopathic Non-Specific Interstitial Pneumonia (iNSIP) is a rare interstitial lung disease, diagnosed, by definition, on the basis of a multidisciplinary team discussion (MDD). Association with an autoimmune background has been suggested in iNSIP.
Aims
To test the feasibility of conducting a multinational MDD to review the diagnosis in iNSIP cases and to estimate the emergence of connective tissue disease (CTD) during follow-up.
Methods
Investigators from three expert centers (Denmark, Estonia and Norway) met and discussed cases of biopsy-proven iNSIP at an international MDD. The cases were previously diagnosed at a national level between 2004 and 2014. Based on clinical, radiographic and pathological data, the diagnosis of iNSIP was re-evaluated and a consensus diagnosis was made. Cases incompatible with iNSIP were excluded. Relevant data were registered comprising any development of CTD.
Results
In total, 31 cases were discussed and 23 patients were included with a diagnosis of iNSIP. The mean follow-up time was 57 months. None of the patients developed CTD according to the rheumatologic criteria during the follow up period. Four patients (17.4%) met the criteria for interstitial pneumonia with autoimmune features.
Conclusion
We found that an international MDD was a feasible and valuable tool in the retrospective diagnostic evaluation of iNSIP. Diagnosis was changed in a statistically significant number of patients by our international MDD team. None of the patients developed CTD during follow-up.
Introduction
In 2002, Non-Specific Interstitial Pneumonia (NSIP) was described as a provisional entity in the American Thoracic Society (ATS)/European Respiratory Society (ERS) classification of idiopathic interstitial pneumonias [Citation1]. In 2008, the idiopathic form (iNSIP) was categorized as a distinct clinical entity by an ATS working group [Citation2]. Beyond its idiopathic label, however, it has been suggested that iNSIP may have an autoimmune background appearing during follow-up [Citation3,Citation4].
A statement from 2015 defined a relatively large group of patients with interstitial lung disease and serologic or morphologic autoimmune features, who did not fulfill the classic rheumatologic criteria for connective tissue diseases (CTD), as interstitial pneumonia with autoimmune features (IPAF) [Citation5].
iNSIP is a rare entity. It occurs most frequently among middle-aged non-smoking women [Citation2]. The incidence of interstitial lung diseases (ILD) was 4.1/100,000/year with only 7% being iNSIP in a Danish ILD cohort [Citation6]. Research collaboration between ILD centres nationally and internationally may thus be an advantage to collect larger iNSIP cohorts.
NSIP exists in a cellular subtype and a more common fibrotic subtype [Citation2]. In general, it has a milder course of disease with a better prognosis and survival compared to idiopathic pulmonary fibrosis (IPF), often demonstrating a good response to immunosuppressive therapy [Citation2]. The radiologic and histopathologic patterns of NSIP are patterns often found in non-idiopathic interstitial pneumonia as for instance CTD, hypersensitivity pneumonitis (HP), occupational and drug-induced interstitial lung disease. Idiopathic NSIP is accordingly a diagnosis of exclusion. However, besides an NSIP pattern, other histological findings can give important clues suggesting specific entities, e.g. granulomas and peri-lymphatic distribution in HP, lymphocytes aggregates and coexisting organising pneumonia in CTD-related interstitial lung disease (CTD-ILD). It remains challenging to diagnose these patients accurately and multidisciplinary discussion (MDD) is recommended to establish a diagnosis for both iNSIP and other ILDs in accordance with the diagnostic guidelines [Citation7].
The agreement between different MDD teams is substantial for IPF and CTD-ILD, whereas the agreement for NSIP is just fair [Citation8]. This is illustrated in the ATS project which examined if iNSIP was a distinct entity, defined the role and characteristics of histopathology and what other disorders needed to be excluded in the diagnostic process. Here Travis et al. reviewed 305 submitted cases of suspected iNSIP. Out of these, 193 had sufficient data, did not contradict the NSIP diagnosis and underwent further evaluation. Only 67 patients were categorized as iNSIP indicating that thorough evaluation changed the diagnosis in a considerable subset of cases who were initially suspected to have iNSIP [Citation2].
This pilot study aimed at testing the feasibility of conducting a multinational MDD and to review the diagnosis in cases with iNSIP. The co-primary aim was to estimate the emergence of CTD during follow-up.
Methods
The study was a retrospective descriptive observational study. Investigators from three ILD expert centers in Norway, Estonia, and Denmark met in Denmark in August 2016 to form a multinational MDD. A MDD team in compliance with the international recommendations [Citation9–11] was set up and included four pulmonologists, one radiologist and one pathologist. The study protocol was approved by the national institutional ethics review boards in each participating country.
Study subjects
Cases of biopsy-proven iNSIP previously diagnosed at a national level between 2004 and 2014 were collected at the discretion of each ILD expert center. Cases were identified from diagnosis codes; the Danish patients were recruited from the ILD register [Citation6].
The MDD was planned for two days to allow up to 30 minutes for discussion of each case. Full HRCT images were available prior to the MDD and uploaded in the PACS system; pathology slides from surgical lung biopsies were brought to the MDD for simultaneous review. All images were simultaneously projected in the meeting room in Aarhus, Denmark.
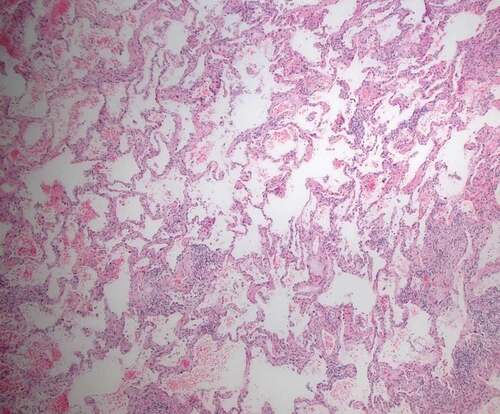
The MDD diagnosis was based on exhaustive clinical, demographic and physiological data from patient files and serology. Patients suspected to have CTD had been evaluated by a rheumatologist at a national level before being included. Basal predominance of bilateral ground-glass opacities, irregular reticulation with traction bronchiectasis/bronchiolectasis and subpleural sparing were considered typical HRCT abnormalities of NSIP (). The key histopathological features were interstitial inflammation and fibrosis with a uniform appearance, from cellular to predominantly fibrotic pattern ().
Figure 1. High-resolution computed tomography (HRCT) finding of one of the patients with non-specific interstitial pneumonia (NSIP)
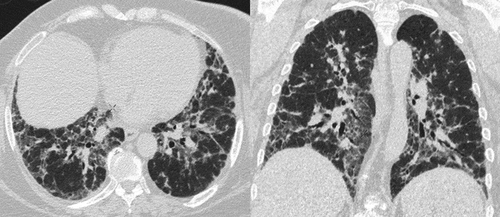
Figure 2. Surgical lung biopsy finding of one of the patients: Cellular and fibrotic non-specific interstitial pneumonia (NSIP) with interstitial inflammation and uniform collagen deposition. HE x 40
All authors participated in the MDD and a consensus diagnosis was reached based on all available data. Cases incompatible with iNSIP were excluded and an alternative diagnosis was suggested. The criteria for IPAF were applied in the included patients and fulfilling of these criteria did not result in exclusion.
The outcomes included the number of confirmed cases of iNSIP and the number of CTD diagnoses during follow-up. As this was a retrospective study, the follow-up period was not specified per protocol.
All data were summarized using descriptive statistics: For continuous variables, the number of patients, means with minima and maxima were described. For categorical variables, the number of patients, frequencies and percentages were used and Pearson’s chi-square test or Students t-test were applied to analyze the between-group differences. The agreement between the initial and the MDD-agreed diagnosis of iNSIP in our study population was assessed with McNemar’s test. P-values <0.05 were considered indicative of statistical significance. All analyses were performed using STATA statistical software version 14.2; StataCorp LLC, College Station, Texas, USA.
Results
A total of 31 retrospective cases of biopsy-proven iNSIP were discussed. The MDD was conducted over two days and all collected cases were reviewed. Time consumption was optimized during the 2 days and four patients were discussed per hour on average. Twenty-three cases were included in the iNSIP cohort. Eight out of the 31 cases (25.8%) were excluded due to inconsistency with the diagnosis of iNSIP, indicating that the diagnosis was significantly changed by our international MDD team (p = 0.013). The differential diagnoses were: hypersensitivity pneumonitis (n = 2), unclassifiable lung fibrosis (n = 2), cryptogenic organizing pneumonia (n = 1), familial interstitial pneumonia (n = 1), desquamative interstitial pneumonia (n = 1) and one case with the features of pulmonary Langerhans cell histiocytosis.
The confirmed cases of iNSIP (n = 23) included 11 (47.8%) females. Baseline characteristics are shown in . The patients, who retained the diagnosis of iNSIP after MDD, more often had dyspnoea and presented more frequently with reticulation, traction bronchiectasis and basal distribution on HRCT than did the non-iNSIP patients ().
Table 1. Baseline characteristics of the patients
Extrapulmonary symptoms were present at diagnosis in 10 (43.5%) of the iNSIP cases. Serology was positive in six patients (26.1%) of whom four had extrapulmonary symptoms (). The mean follow-up time was 57 months. One patient (4.3%) died during follow-up. None of the patients developed CTD that fulfilled the classic rheumatologic criteria during the observation period. Four patients (17.4%) fulfilled the criteria for interstitial pneumonia with autoimmune features (IPAF) [Citation5] ().
Table 2. Clinical symptoms, serology data and the presence of interstitial pneumonia with autoimmune features (IPAF) and connective tissue disease (CTD) among the 23 patients with idiopathic non-specific interstitial pneumonia confirmed by our international multidisciplinary discussion team. SSA: Anti-SSA antibody; SSB: anti-SSB antibody; ANA: antinuclear antibody; RF: rheumatoid factor; Ku: anti-Ku antibody
Discussion
Arranging a multinational MDD was feasible and also found valuable with reclassification of 25.8% of patients due to iNSIP inconsistency. We found no evidence of later development of CTD in the cases included in the cohort, though 17.4% fulfilled the IPAF criteria.
MDD is considered the diagnostic reference standard in interstitial lung disease [Citation7]. A recent study by Richeldi et al. [Citation12] evaluated diagnostic practices of ILD of centers worldwide and found that the MDD approach was widely implemented. Academic centers had a larger caseload and included more histopathology and rheumatologist expertise than non-academic centers [Citation12]. The composition of the MDD in ILD varied among centers and heterogeneity in the organization and information level was a general observation. Most MDDs include a radiologist, a pulmonologist and a pathologist, whereas rheumatologists only participate routinely in a minority of MDD meetings [Citation9,Citation13]. No standard setup for MDD has been defined, but criteria have been suggested [Citation9,Citation13]. Besides a pulmonologist and a radiologist, guidelines recommend the participation of a pathologist as histopathology, if a lung biopsy is performed, plays a crucial role in the diagnosis of ILD [Citation10,Citation11].
Flaherty et al. found that in idiopathic interstitial pneumonias, MDDs improved the diagnostic inter-observer agreement, and academic physicians in a MDD had better diagnostic agreement compared to community physicians [Citation14]. De Sadeleer et al. reported that a MDD provided a specific diagnosis in 80.5% comprising mainly various ILDs (n = 938) [Citation15]. In 191 patients (41.9% of patients with a pre-MDD diagnosis), a MDD changed the diagnosis [Citation15].
The number of patients reclassified in our study is considerably smaller than that reported by Travis et al., who excluded 238 of 305 (78.0%) patients with NSIP [Citation2]. Even though the cases in our study were previously diagnosed at expert ILD centers using the MDD approach, a significant percentage of patients were excluded. This reflects the findings by Walsh et al. that interobserver agreement on diagnostic likelihood among ILD experts was only fair for iNSIP with a kappa value of 0.25 [Citation8]. The different compositions of the MDD team may affect the final diagnosis and we cannot exclude that the participation of a rheumatologist in our multinational MDD would have changed the outcome. However, we find this unlikely, as all patients with symptoms, clinical findings or serology suggestive of a CTD-ILD had previously been evaluated by a rheumatologist at their home center; only four patients developed an IPAF, but not a CTD during follow-up.
We found no development of CTD in our iNSIP cohort despite long-term follow-up. This is in contrast to findings in other studies. Sato et al. presented a retrospective analysis of 26 patients with underlying CTD. In six of these patients (23.1%), NSIP was present for more than six months before the CTD diagnosis [Citation16]. Kono et al. compared the clinical features of 72 patients with NSIP; of these, 35 patients (48.6%) were diagnosed as idiopathic and 37 (51.4%) with CTD-NSIP [Citation17]. They found no significant difference between the two groups with respect to clinical characteristics and survival. Seventeen percent of the patients with iNSIP developed CTD during a 5.5-year follow-up [Citation17]. Other authors found that in patients with iNSIP, 9.6% and 11.1%, respectively developed CTD during follow-up [Citation4,Citation18]. The follow-up period in our study of 57 months was comparable with several of the above mentioned cohorts [Citation4,Citation17] but we cannot exclude that a longer follow-up period would have resulted in more patients being diagnosed with a CTD-ILD. Furthermore, autoimmunity and CTD had developed within two years in more than 50% of cases [Citation3,Citation4,Citation18]. In our study autoantibodies were repeated only on clinical suspicion during follow-up and development of an asymptomatic CTD by routinely antibody testing cannot be ruled out.
Our cohort differed from the previous cohorts by having a slight male predominance that may contribute to the iNSIP phenotype. We found 10 patients (43.5%) with a cellular histological pattern, which is also different from the findings in other cohorts [Citation2,Citation4] but comparable to findings by Xu et al., who showed a cellular pattern in 50.0% at initial presentation in a group of iNSIP cases (n = 74). In this cohort, 6.8% developed CTD during a 45-month follow-up [Citation19].
The possibility of misdiagnosis in some of the cases due to histopathologic sampling error failing to identify UIP in biopsies exists [Citation20]. Still, the long follow-up period, the modest mortality, the mean age and the majority of patients being non-smokers speak against IPF as a crucial differential diagnosis.
A major limitation in our study included the retrospective nature of the data. Differences in standard diagnostic procedures between centers made complete comparison of clinical data from patient files difficult.
A reclassified confident diagnosis by a MDD may have a therapeutic impact for the patients. However, regional differences and availability of experts can be constraining and decide the constitution of the MDD. We sought to evaluate the value of a multinational MDD in the light of a rare condition such as iNSIP, and if collaboration between countries by a multinational MDD could improve the diagnostic confidence and strengthen the opportunity and power of research. A MDD of rare lung diseases such as iNSIP could have been conducted within different ILD centers at a national level with similar results, indicating that the international aspect in this study to some extend represent re-review of cases with broader input. However this would not be possible in small countries like Estonia and international collaboration would be expedient to collect larger iNSIP cohorts. Internationally targeted MDDs have been conducted within other areas of ILD, e.g. in inherited pulmonary fibrosis [Citation21]. Along with the increasing digitization of medicine and development of telecommunication, possibilities for virtual MDD meetings have emerged and are currently being studied in the ‘Starliner’ study [Citation22]. To our knowledge, this study was the first attempt to perform a cross-border multinational MDD specifically targeting iNSIP.
Conclusion
We found that a cross-border international MDD was a feasible and a highly valuable tool in the retrospective diagnostic evaluation of iNSIP. The diagnosis was significantly changed by our international MDD team as 25.8% of all cases appeared inconsistent with iNSIP. This highlights the importance of a multidisciplinary approach to establish a confident diagnosis. In our iNSIP cohort, four patients fulfilled the criteria for interstitial pneumonia with autoimmune features, but no patients developed neither CTD nor other ILD during follow-up, making the term ‘idiopathic’ more confident.
Disclosure statement
Janne Møller has received lecture fees from Roche and participated in advisory boards of Roche.
Alan Altraja has received lecture fees and sponsorships from Boehringer Ingelheim and Roche and research support from Boehringer Ingelheim, and has participated in advisory boards of Boehringer Ingelheim and Roche.
Elisabeth Bendstrup reports fees and grants from Boehringer Ingelheim, Hoffman la Roche, Galapagos, and Bristol-Myers-Squibb.
Additional information
Funding
Notes on contributors
Janne Møller
Janne Møller, MD, is a consultant and a PhD student at Department of Respiratory Diseases and Allergy, Aarhus University Hospital, Denmark. She is a specialist in pulmonary diseases, and her main research focus is sarcoidosis.
Alan Altraja
Alan Altaja is a Professor of Pulmonary Medicine at the University of Tartu, Faculty of Medicine, Department of Pulmonology. His research area includes asthma, COPD and interstitial lung disease.
Tone Sjåheim
Tone Sjåheim, is a consultant pulmonologist; MD, PhD at Department of Respiratory Medicine Oslo University Hospital. She is a specialist in pulmonary diseases, and her main research focus is interstitial lung diseases.
Finn Rasmussen
Finn Rasmussen, Associate Professor, MD, DMSc, is a consultant at Department of Radiology, Aarhus University Hospital, Denmark. His main research interests focus on interstitial lung diseases and lung cancer.
Line Bille Madsen
Line Bille Madsen, MD, is a consultant at Department of Pathology, Aarhus University Hospital, Denmark. Her main research interests focus on interstitial lung diseases and lung cancer.
Elisabeth Bendstrup
Elisabeth Bendstrup, Professor, MD, PhD, is a consultant at Department of Respiratory Diseases and Allergy, Aarhus University Hospital, Denmark. She is a specialist in pulmonary diseases, and her main research focus is interstitial lung diseases.
References
- International S, Consensus M. American thoracic society American thoracic society/European respiratory society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277–7.
- Travis WD, Hunninghake G, King TE Jr, et al. Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am J Respir Crit Care Med. 2008;177(12):1338–1347.
- Kinder BWCollard HR, Koth L, et al. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease?. Am J Respir Crit Care Med. 2007;176(7):691–697.
- Romagnoli M, Nannini C, Piciucchi S, et al. Idiopathic nonspecific interstitial pneumonia: an interstitial lung disease associated with autoimmune disorders?. Eur Respir J. 2011;38(2):384–391.
- Fischer A, Antoniou KM, Brown KK, et al. An official European respiratory society/American thoracic society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–987.
- Hyldgaard C, Hilberg O, Muller A, et al. A cohort study of interstitial lung diseases in central Denmark. Respir. Med. 2014;108(5):793–799.
- Travis WD, Costabel U, Hansell DM, et al. An official American thoracic society/European respiratory society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748. .
- Walsh SLF, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med. 2016;4(7):557–565.
- Jo HE, Corte TJ, Moodley Y, et al. Evaluating the interstitial lung disease multidisciplinary meeting: a survey of expert centres. BMC Pulm Med. 2016;16(1):1–6. .
- Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner society white paper. Lancet Respir Med. 2018;6(2):138–153.
- Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis an official ATS/ERS/JRS/ALAT clinical practiceguideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68.
- Richeldi L, Launders N, Martinez F, et al. The characterisation of interstitial lung disease multidisciplinary team meetings: a global study. ERJ Open Res. 2019;46(2):00209–02018.
- Furini F, Carnevale A, Casoni GL, et al. The role of the multidisciplinary evaluation of interstitial lung diseases: systematic literature review of the current evidence and future perspectives. Front Med. 2019;6:246.
- Flaherty KR, Andrei AC, King TE Jr, et al. Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis?. Am J Respir Crit Care Med. 2007;175(10):1054–1060.
- De Sadeleer LJ, Meert C, Yserbyt J, et al. Diagnostic ability of a dynamic multidisciplinary discussion in interstitial lung diseases: a retrospective observational study of 938 cases. Chest. 2018;153(6):1416–1423.
- Sato T, Yamadori I, Ohtsuki Y, et al. Non-specific interstitial pneumonia; as the first clinical presentation of various collagen vascular disorders. Rheumatol Int. 2006;26(6):551–555.
- Kono M, Nakamura Y, Yoshimura K, et al. Nonspecific interstitial pneumonia preceding diagnosis of collagen vascular disease. Respir Med. 2016;117(2016):40–47.
- Park IN, Jegal Y, Kim DS, et al. Clinical course and lung function change of idiopathic nonspecific interstitial pneumonia. Eur Respir J. 2009;33(1):68–76.
- Xu W, Xiao Y, Liu H, et al. Nonspecific interstitial pneumonia: clinical associations and outcomes. BMC Pulm Med. 2014;14:175.
- Flaherty KR, Travis WD, Colby TV, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164(9):1722–1727.
- Borie R, Kannengiesser C, Gouya L, et al. Pilot experience of multidisciplinary team discussion dedicated to inherited pulmonary fibrosis. Orphanet J Rare Dis. 2019;14(1):1–10.
- Wijsenbeek M, Bendstrup E, Valenzuela C, et al. Design of a study assessing disease behaviour during the peri-diagnostic period in patients with interstitial lung disease: the starliner study. Adv Ther. 2019;36(1):232–243.