
ABSTRACT
Background
Airway epithelial cells and lung fibroblasts play an important role in the development of chronic lung disease, but the exact mechanisms responsible have not been clarified. Our objective was to investigate the involvement of these cells in the inflammatory response associated to chronic lung disease.
Methods
Human lung fibroblasts and airway epithelial cells were challenged with Interleukin-1β and hypoxia, and with inhibitory (simvastatin) stimuli of the inflammatory response. Expression of markers of local inflammation ((IL-8, monocyte chemoattractant protein-1 (MCP-1), factor-κB1 (NF-κB1)), systemic inflammation ((C-reactive protein (CRP) and serum amyloid A (SAA)) and proteases matrix metalloproteinase (MMP) 9 and 12 were assessed by PCR and ELISA. Apoptosis/necrosis was analyzed by flow cytometry.
Results
Our results showed that the lung fibroblasts had a higher expression of local and systemic inflammation and protease activity markers when they were treated with IL-1β compared to airway epithelial cells. Under hypoxic conditions, we observed a decrease in systemic inflammation in lung fibroblasts, which was further attenuated by simvastatin.
Conclusion
The lung fibroblasts seem to be the main initially stimulated cells that could potentially trigger the inflammatory response, and be responsible for the eventual onset of chronic lung disease. The involvement of IL-1ß stimulation in systemic inflammatory and proteinase imbalance biomarkers is higher in lung fibroblasts. Apoptosis is not a predominant mechanism in these cells.
Introduction
Airway remodeling occurring in chronic lung disease is a multicellular response to mucosal injury, which results in epithelial cell state changes, enhanced extracellular deposition and the expansion of profibrotic myofibroblast populations. Previous evidence suggests that obstructive lung diseases have certain triggers, inflammatory responses and features of remodeling in common [Citation1] and that the inflammation which appears in the respiratory system precedes the onset of bronchial obstruction [Citation2,Citation3]. In this regard, recent research efforts have focused on the evaluation of early chronic obstructive pulmonary disease (COPD) [Citation4].
One relevant question involves determining where the inflammatory process that triggers the bronchial obstruction begins. The inflammatory processes involve multiple cell types, but specifically epithelial cells and lung fibroblasts seem to play a central role. Epithelial cells are a first barrier protecting the airway from exposure to the environment, and their alteration makes the airway more vulnerable and alters the signaling pathways involved in inflammatory processes and repair [Citation5]. Lung fibroblasts could play an essential role in inflammatory responses within the respiratory system [Citation6]. Collectively, inflammation can influence the epithelium and fibroblasts, leading to airway remodeling [Citation7]. The mechanistic basis underlying COPD, a group of progressive lung diseases that cause breathing difficulties, is complex and there are different biological pathways involved, including imbalances in proteases and their inhibitors and apoptosis [Citation8]. The role of apoptosis in cell physiology is probably related to particular presentations of the disease, in which there is an increased destruction of lung parenchyma, such as in emphysema. The rate of apoptosis is higher in epithelial cells because, during the pathogenesis of chronic lung disease, there is destruction of the lung tissue and development of pulmonary emphysema, while lung fibroblasts increase their proliferation and ability to produce fibrotic tissue [Citation9]. However, to date, no studies have assessed cell behavior under pro- and anti-inflammatory conditions as a starting point for research into chronic lung disease pathogenesis.
Interleukin (IL)-1β, a typical innate immune cytokine that has been correlated to COPD, plays an important role in initiating and maintaining airway inflammation [Citation10]. IL-1β is a key driver of neutrophil airway inflammation in COPD that can induce IL-8 and IL-6 in normal human airway epithelial cells, with two key cytokines promoting neutrophil recruitment and activation in the airways [Citation11]. The fact that IL-1β stimulation is not always associated with increased protein levels of pro-inflammatory mediators is also interesting [Citation12].
In previous studies, statins were characterized by their immunomodulatory effects and anti-inflammatory properties and their ability reduce inflammatory markers such as CRP, IL-6, IL-8 or TNF-α [Citation13,Citation14] in serum samples. A recent systematic review identified the clinical efficacy of statin therapy in COPD. Although statin treatment was associated with improvements in exercise capacity, lung function and health status, the authors found no associations with inflammatory markers [Citation15]. The effects of statins occur on several levels: first, by inhibiting the canonical target HMGCR (3-hydroxy-3-methylglutaryl-CoA reductase) and second, through off-target or non-canonical effects by directly inhibiting other enzymes such as leukocyte function antigen-1 (LFA-1) [Citation16]. Statins also exhibit pleiotropic effects by inhibiting the peroxisome proliferator activated receptor (PPAR) (i.e. functional inhibition) [Citation17].
The purpose of the present study was to examine the roles of different epithelium associated cells, lung fibroblasts and airway epithelial cells, in the main pathways involved in the development of chronic lung disease. By treating these cells with inflammatory cytokine, IL-1β, and hypoxia, we explored the potential involvement of these cells in the pathogenesis of chronic lung disease. In addition, we investigated the inhibitory effect of statins (simvastatin) on the response of these cells to both stimuli (i.e. inflammatory cytokine and hypoxia). The results of these experiments will help reveal the alterations in biological pathways in these cell types, which should lay the basis for future research to further our understanding of the pathological development of chronic lung disease.
Material and methods
Cell culture
MRC-5 (human lung fibroblast cell line) cells were obtained from the European Collection of Authenticated Cell Cultures (ECACC, Salisbury, UK) and NuLi-1 (human airway epithelial cell line) cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were cultured following the manufacturer’s protocol. These cell line cultures have intrinsic alterations compared to cells in humans and to cells in patients with an ongoing pathogenesis.
Cell stimulation and inhibition
Hypoxia
The cells were grown to 80% confluence, washed and incubated with serum-free medium for 24 hours, and incubated in hypoxic conditions (1% O2) with or without 30 µM simvastatin sodium salt (Calbiochem, Darmstadt, Germany) for 24 and 48 h. We used cells in normoxic conditions (5% O2) for control. Altogether, there were 2 groups of experiments per cell type: hypoxia, and simvastatin+hypoxia. Each experiment was repeated six times.
IL-1 β
The cells were grown to 80% confluence, washed and incubated with serum-free medium for 24 hours, and then incubated with 10 ng/mL IL-1β (R&D Systems, Minneapolis, MN, USA) for 24 and 48 h with or without 30 µM simvastatin sodium salt (Calbiochem, Darmstadt, Germany). The IL-1β was dissolved in culture medium and we used the same solution in the control and the sample. The concentrations of IL-1β and simvastatin were chosen by preliminary concentration-response studies. The simvastatin treatment was started 45 min prior to cytokine treatment, as described previously [Citation18]. We used untreated cells as controls for the IL-1β and simvastatin experiments. Altogether, there were 3 groups of experiments per cell type: IL-1β, simvastatin, simvastatin+IL-1β. Each experiment was repeated six times.
Selection of biomarkers
We selected markers involved in the pathogenesis of chronic lung disease: IL-8, monocyte chemoattractant protein-1 (MCP-1) and nuclear factor-κB1 (NF-κB1), as markers of local inflammation; C-reactive protein (CRP) and serum amyloid A (SAA), as markers of systemic inflammation; and finally, matrix metalloproteinase (MMP) 9 and 12 as markers of proteases.
Gene expression
Gene expression was measured by reverse transcription quantitative polymerase chain reaction (RT-qPCR). The total RNA was isolated from the cultured cells using the High Pure RNA isolation kit (Roche Mannheim, Germany). RNA was reverse transcribed using the iScript™ cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). RT-qPCR was performed on an Applied Biosystems® 7900 Real-Time PCR System (Life Technologies, Grand Island, NY, USA). Expression of 18s rRNA was used as an internal control for the normalization of target gene expression. For the SAA, all SAA1, SAA2, and SAA4 genes were studied. The list of primers used to amplify the genes of interest appears in . Relative gene expression values were evaluated with the 2−∆∆Ct method [Citation19].
Table 1. Primers used for reverse transcription quantitative PCR.
Protein expression
The media of the cell cultures were collected, centrifuged at 2750 g for 5 min, and then stored at −80°C until further measurements were taken. We measured the cytokine levels using ELISA (Quantikine®, R&D Systems for IL-8, MCP-1, and MMP-9; Anogen, Mississauga, Ontario, Canada for SAA; and Abcam, Cambridge, UK for CRP), following the manufacturer’s protocol.
Apoptosis and necrosis analysis
For apoptosis analysis, the cells were trypsinized and washed twice with phosphate-buffered saline. Two hundred thousand cells were stained with Annexin V-CF Blue and propidium iodide (PI), following the manufacturer’s instructions (Immunostep, Salamanca, Spain). The percentage of annexin V-positive and/or PI-positive cells was analyzed using a Fortessa II flow cytometer (Becton Dickinson; Erembodegem, Belgium).
Statistics
The means and standard errors for the continuous variables and frequencies and the percentages for the categorical variables were estimated. The gene expression data were compared according to the treatment, using the U-Mann Whitney test. All the testing hypotheses were performed at a 5% level of significance (two-tailed), using the statistical analysis software, IBM SPSS Statistics version 20.0 (IBM Corporation, Armonk, NY, USA).
Results
Stimulation with IL-1β
Local inflammation markers
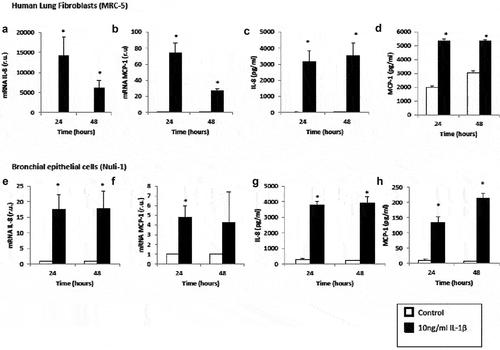
Cells stimulated with IL-1β for both 24 h and 48 h significantly increased mRNA expression and protein levels of IL-8 and MCP-1 compared with untreated cells (). Interestingly, lung fibroblast cells presented a higher gene expression of IL-8 compared to airway epithelial cells (p = 0.029). On the other hand, lung fibroblast cells presented a higher gene and protein expression for MCP-1. We found no significant differences in gene and protein expression for NF-κB1 (data not shown).
Figure 1. Effect of IL-1β on inflammatory mediator expression in lung fibroblasts and airway epithelial cells. mRNA expression of IL-8 (panel A), mRNA expression of MCP-1 (panel B), protein levels of IL-8 (panel C) and protein levels of MCP-1 (panel D) produced by human lung fibroblasts without stimulation (control, white bars) and stimulated with IL-1β (10 ng/mL, black bars). mRNA expression of IL-8 (panel E), mRNA expression of MCP-1 (panel F), protein levels of IL-8 (panel G) and protein levels of MCP-1 (panel H) produced by human airway epithelial cells without stimulation (control, white bars) and stimulated with IL-1β (10 ng/mL, black bars). r.u. = Relative units versus control; *p < 0.05 versus control. Each bar in the figure is mean± SE n = 6.
Systemic inflammation markers
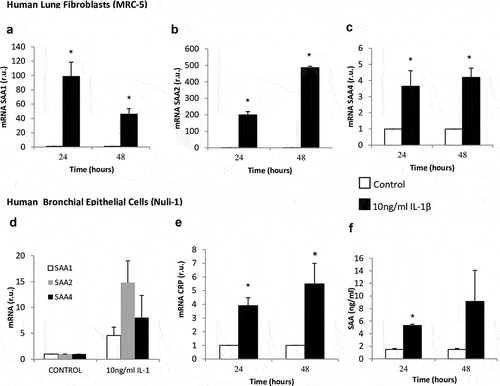
The lung fibroblasts stimulated with IL-1β significantly increased SAA mRNA expression compared with the control cells (), 98-fold for SAA1, 200-fold for SAA2, and 3.6-fold for SAA4. The levels of CRP mRNA, as well as CRP and SAA protein, did not change in lung fibroblast cells treated with IL-1β compared with untreated cells. The epithelial cells stimulated with IL-1β significantly increased their CRP mRNA expression compared with unstimulated cells (). However, we found no significant differences in the CRP protein level in this cell type (data not show). The levels of SAA protein were significantly higher in cells stimulated with IL-1β for 24 h compared with unstimulated cells (p = 0.029) (). Nevertheless, we found no significant differences in SAA1, SAA2, and SAA4 mRNA expression ().
Figure 2. Effect of IL-1β on SAA and CRP expression in lung fibroblasts and airway epithelial cells. mRNA expression of SAA1 (panel A), SAA2 (panel B), and SAA4 (panel C) produced by human lung fibroblasts without stimulation (white bars) and stimulated with IL-1β (10 ng/mL, black bars). mRNA expression of SAA (panel D), CRP (panel E) and SAA protein levels (panel D) produced by human airway epithelial cells without stimulation (white bars) and stimulated with IL-1β (10 ng/mL, black bars). r.u. = Relative units versus control; *p < 0.05 versus control. Each bar in the figure is mean± SE n = 6.
Metalloproteinases
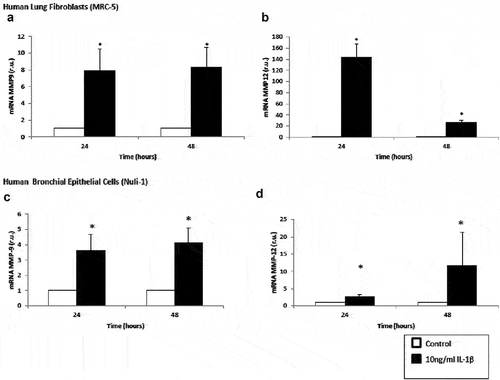
MMP9 and MMP12 mRNA expressions increased significantly in lung fibroblasts and airway epithelial cells incubated with IL-1β for 24 and 48 h compared with the control cell (). Interestingly, at 48 hours, MMP12 mRNA expression decreased in lung fibroblasts (), and conversely, this expression increased in epithelial cells (). Nevertheless, we found no significant differences in MMP9 protein level (data no shown).
Figure 3. Upregulation of matrix metalloproteinase mRNA expression by IL-1β in lung fibroblasts and airway epithelial cells. Expression of MMP9 (panel A) and MMP12 (panel B) mRNA by human lung fibroblasts without stimulation (white bars) and stimulated with IL-1β (10 ng/mL, black bars). Expression of MMP9 (panel C) and MMP12 (panel D) mRNA by human lung airway epithelial cells without stimulation (white bars) and stimulated with IL-1β (10 ng/mL, black bars). r.u. = Relative units versus control; *p < 0.05 versus control. Each bar in the figure is mean± SE n = 6.
Apoptosis/necrosis
We found no significant differences between unstimulated cells and cells treated with IL-1β with respect to cell death (data not show).
Culture in hypoxic conditions
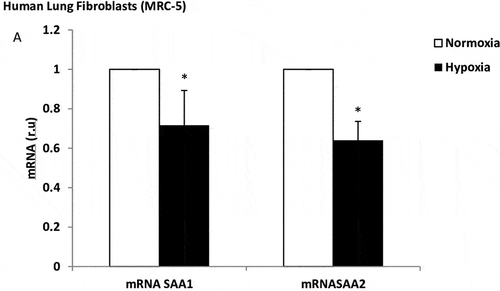
The cells cultured under hypoxic conditions for 24 h or 48 h did not alter their expression of local inflammation markers, metalloproteinase markers, or apoptosis/necrosis rates (data not show). However, the lung fibroblasts under hypoxic conditions decreased their SAA1 and SAA2 mRNA expression (3.7-fold, p = 0.014) (). We did not examine the effect of long-term hypoxia.
Figure 4. Effect of hypoxia on SAA expression in lung fibroblasts. mRNA expression of SAA1 and SAA2 produced by human lung fibroblasts, in normoxia (white bars) and stimulated with hypoxia (black bars). r.u. = Relative units versus control; *p < 0.05 versus normoxia. Each bar in the figure is mean± SE n = 6.
Inhibition with simvastatin
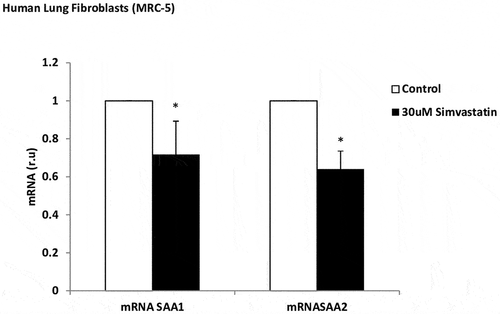
Simvastatin alone, in airway epithelial cell, did not downregulate gene expression or protein levels with any of the biomarkers, compared to the untreated cells (data not show). Simvastatin decreased SAA1 and SAA2 mRNA in human lung fibroblasts (1.2 and 1.6-fold, respectively, versus untreated cells) under hypoxic conditions (). Simvastatin did not show a decrease in the inflammatory parameters after stimulation with IL-1ß (data not show).
Figure 5. Effect of simvastatin on SAA expression in lung fibroblasts. mRNA expression of SAA1 and SAA2 produced by human lung fibroblasts, without stimulation (white bars) and stimulated with simvastatin (30 µM, black bars). r.u. = Relative units versus control; *p < 0.05 versus control. Each bar in the figure is mean± SE n = 6.
Discussion
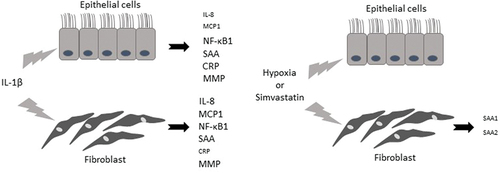
The present study shows an overall assessment of the cellular responses of airway epithelial cells and lung fibroblasts after inflammatory stimuli associated with chronic lung disease. Our results show that 1) IL-1β stimulates different biological pathways involved in chronic lung disease (IL-8 and MCP-1), except apoptosis and specific inflammatory pathway biomarkers; 2) The involvement of IL-1ß stimulation in systemic inflammatory and proteinase imbalance biomarkers is higher in lung fibroblasts; 3) Under hypoxic conditions, there is a systemic inflammatory decrease in lung fibroblasts; 4) simvastatin maintains this downregulation in lung fibroblasts; 5) in the final stage, the two cell types show a different inflammatory response. ().
Figure 6. Responses of airway epithelial cells and lung fibroblasts after inflammatory stimuli associated with chronic lung disease. IL-1β, interleukin 1β; IL-8, interleukin 8; MCP1, monocyte chemoattractant protein-1; NF-κB1, factor-κB1; CRP, C-reactive protein; SAA, serum amyloid A; MM9 and MMP12, proteases matrix metalloproteinase 9 and 12.
The evaluation of biological pathways that underlie cellular physiology represents a vital step in our understanding of the cellular mechanisms which may be involved in the initial pathophysiology of chronic lung diseases such as COPD. The cooperation between different epithelium associated cells forming the airway allows tissue homeostasis to be maintained with the normal physiology of human respiratory tract. These results highlight the initially stimulated biological pathways, which could potentially trigger the inflammatory response responsible for the eventual onset of chronic lung disease. Immediate anatomical and functional interactions between airway epithelial cells and lung fibroblast, known as the epithelial-mesenchymal trophic unit, are necessary for lung tissue to function properly. Therefore, its dysfunctionality is observed in respiratory disorders such as a chronic obstructive pulmonary disease (COPD). It is important to consider that for this study we used epithelium associated cells, which are involved in the remodeling of lung damage induced by inflammatory stimulus. Due to changes in these different pathways in patients who develop COPD [Citation20], the cells in the respiratory system of patients with this disease may respond differently.
Lung fibroblasts, together with protein imbalance, seem to be the main factors involved in the inflammatory response in chronic lung disease. However, we need to extend the study to confirm this assumption. According to our results, both airway epithelial cells and lung fibroblasts participate, each in a different way, in the inflammatory and remodeling process. The inflammatory process begins in the bronchial epithelium with the release of IL-8 and MCP-1 in airway epithelial cells and lung fibroblasts, and of CRP and SAA in airway epithelial cells. Subsequently, the activated macrophages release metalloproteinase 9 and 12, which contribute to the degradation of elastin in the lung parenchyma. However, this inflammatory process in lung fibroblasts could be attenuated by simvastatin. Interestingly, those biomarkers associated with a disease-specific response or with more advanced disease were not significantly changed.
Systemic inflammation in COPD, defined as increased levels of inflammatory markers from different biological pathways [Citation21], has become a new target for chronic lung disease-related research and its origin is a challenge for researchers. Not only may proteins originating from the lung exert systemic effects, but there is also a lack of correlation between airway cytokine concentrations and those in the circulation [Citation22]. However, researchers have been unable to find an association between the inflammatory load of induced sputum and plasma [Citation23]. This may be due to hepatic hyperstimulation during COPD, and this lung-liver feedback should be explored further. Serum amyloid A (SAA), an acute phase protein that occurs in high levels in the blood during infection, is produced by hepatocytes and secreted into serum [Citation24]. However, one study demonstrated that SAA mRNA [Citation25] is normally expressed in the epithelial components of a variety of human organs and tissues, and could be released locally in certain organ-specific diseases [Citation26]. Moreover, SAA can also be produced by macrophages and other extrahepatic cells as well as in the lung [Citation27]. Our previous findings show that SAA expression was different between pulmonary cell types [Citation28]. Our results show SAA mRNA expression but no protein expression in lung fibroblasts. In contrast, we observed a protein expression of SAA, but no SAA mRNA expression in airway epithelial cells. This may be due to differences in the posttranscriptional regulation depending on the cell type.
In addition, our results could indicate an overlap in airway epithelial cells between SAA kinetics and the kinetics of the C-reactive protein, which would be in line with previous studies [Citation29].
The interactions between airway epithelial cells, lung fibroblasts and the extracellular matrix of the airway wall are intimately involved in a number of functions within the lung [Citation30]. MMPs play a key role in COPD pathogenesis, as well as in degrading matrix proteins, and their presence is required for normal matrix processing and lung repair [Citation31]. In addition, MMP-12 plays a pivotal role in the inflammatory process that leads to lung injury [Citation32], as well as in inflammatory lung disease and tissue remodeling [Citation33]. Hence, an increased expression of MMP-9 and MMP-12 may cause an unwanted degradation of lung tissue. According to our results, both cell types studied participate actively and differentially during the initial injury in chronic lung disease. MMP12 has a relevant function in chronic lung disease after an inflammatory stimulus, and its production and release occur earlier in lung fibroblasts than in the airway epithelial cells. Our findings suggest that the lung fibroblasts act as sentinel cells in the event of injury, orchestrating the remodeling process in the early phases by secreting MMP-12. In addition, lung fibroblasts could modulate the response of airway epithelial cells, which is in line with previous studies [Citation34]. These differences between MMPs may be of major importance when considering the role of a given MMP and, particularly, in selecting an MMP as a natural inhibitor instead of a synthetic inhibitor.
In addition, we explored the effect of hypoxia as a stimulus which was representative of acute pulmonary diseases, in which hypoxemia occurred due to the severity of the disease. Although current recommendations, to measure the severity of COPD, has been refined to separating the spirometric assessment from the evaluation of symptoms [Citation35], patients with COPD and secondary hypoxemia should be classified as particularly serious cases from a clinical perspective, and they have serious prognostic implications [Citation36]. Hypoxia and inflammation are intimately linked [Citation37]. Some studies have shown ‘in vitro’ evidence that hypoxia may aggravate airway inflammation through an effect on immune cells such as macrophages [Citation38] or monocytes [Citation39]. Our study suggests that inflammation before hypoxia does not worsen local inflammation near lung fibroblasts and airway epithelial cells. We observed that exposure of normal lung fibroblasts to hypoxia in culture resulted in decreased SAA mRNA expression. Nevertheless, patients with high SAA had greater dyspnea and more frequent interstitial lung disease [Citation40]. Therefore, our results suggest that lung fibroblasts can be considered a cellular model, used to analyze the underlying mechanisms in the pathogenesis of overlap syndrome (COPD and obstructive sleep apnea), or to improve hypoxia-induced inflammation, which could be facilitated by high altitude acclimatization. This expression of SAA mRNA remains downregulated after treatment with simvastatin. However, the effect of the statins depends on the underlying stimulus, mechanisms, microenvironmental conditions [Citation41] and cell type used in the experiment. The relationship between pulmonary disease and simvastatin is complex [Citation42]. A previous study has shown that statins may be considered as an adjunctive treatment in COPD patients because their anti-inflammatory effects include suppression of the upregulation of pro-inflammatory cytokines, chemokines, adhesion molecules and MMPs by inflammatory cells [Citation43], and their potential role in respiratory disease has been hypothesized [Citation44]. Interestingly, the impact of simvastatin on airway epithelial cell lines is less obvious, despite a few reports that simvastatin inhibits alveolar destruction [Citation45] and affects alveolar recovery [Citation46].
On the other hand, the role of increased apoptotic alveolar cells in the peripheral lung is relevant in severe stable COPD patients with pulmonary emphysema [Citation47]. Our results did not show any differences in the rate of apoptosis and necrosis when we compared cells stimulated with and without IL-1β. Therefore, our model was probably not ideal for studying emphysema.
Given these findings, our results should be replicated in the near future with primary cultures obtained from patients with chronic lung disease in order to clarify their involvement in the pathogenesis of the disease.
Conclusions
The present study is an overall assessment of the cellular behavior of airway epithelial cells and lung fibroblasts after inflammatory stimuli associated with chronic lung disease. We have shown that one of the different biological pathways involved in the remodeling and pathogenesis of chronic lung disease could increase after stimulation with IL-1β. These results highlight the initially stimulated biological pathways that could potentially trigger the inflammatory response which is responsible for the eventual onset of chronic lung disease. Future studies should replicate this approach with primary cell cultures obtained from patients with chronic lung disease, in order to clarify their involvement in the pathogenesis of the disease at more advanced stages.
Author contributions
Conceptualization, Elena Arellano-Orden and José Luis López-Campos; Data curation, Remedios Otero-Candelera; Formal analysis, Elena Arellano-Orden, Carmen Calero-Acuña, Cecilia López-Ramirez and Carmen Marín-Hinojosa; Funding acquisition, José Luis López-Campos; Methodology, Elena Arellano-Orden and Verónica Sánchez-López; Supervision, Remedios Otero-Candelera and José Luis López-Campos; Writing – original draft, Elena Arellano-Orden; Writing – review & editing, Carmen Marín-Hinojosa.
The authors declare no conflict of interest.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Kim SR, Rhee YK. Overlap between asthma and COPD: where the two diseases converge. Allergy Asthma Immunol Res. 2010;2(4):209–10.
- de Marco R, Accordini S, Marcon A, et al. Risk factors for chronic obstructive pulmonary disease in a European cohort of young adults. Am J Respir Crit Care Med. 2011;183(7):891–897.
- Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653.
- Borras-Santos A, Garcia-Aymerich J, Soler-Cataluna JJ, et al. Determinants of the appearance and progression of early-onset chronic obstructive pulmonary disease in young adults. A case-control study with follow-up. Arch Bronconeumol. 2019;55(6):312–318.
- Aghapour M, Raee P, Moghaddam SJ, et al. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol. 2018;58(2):157–169.
- Weidner J, Jarenback L, Aberg I, et al. Endoplasmic reticulum, Golgi, and lysosomes are disorganized in lung fibroblasts from chronic obstructive pulmonary disease patients. Physiol Rep. 2018;6(5):e13584.
- Eapen MS, Myers S, Walters EH, et al. Airway inflammation in chronic obstructive pulmonary disease (COPD): a true paradox. Expert Rev Respir Med. 2017;11(10):827–839.
- Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis. 2011;6:413–421.
- Demedts IK, Demoor T, Bracke KR, et al. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7:53.
- Baines KJ, Simpson JL, Wood LG, et al. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol. 2011;127(1):153–160, 160 e151–159.
- Khan YM, Kirkham P, Barnes PJ, et al. Brd4 is essential for IL-1beta-induced inflammation in human airway epithelial cells. PloS one. 2014;9(4):e95051.
- Titz B, Sewer A, Schneider T, et al. Alterations in the sputum proteome and transcriptome in smokers and early-stage COPD subjects. J Proteomics. 2015;128:306–320.
- Young RP, Hopkins RJ. The mevalonate pathway and innate immune hyper-responsiveness in the pathogenesis of COPD and lung cancer: potential for chemoprevention. Curr Mol Pharmacol. 2017;10(1):46–59.
- Lu Y, Chang R, Yao J, et al. Effectiveness of long-term using statins in COPD - a network meta-analysis. Respir Res. 2019;20(1):17.
- Zhang W, Zhang Y, Li CW, et al. Effect of statins on COPD: a meta-analysis of randomized controlled trials. Chest. 2017;152(6):1159–1168.
- Yeganeh B, Wiechec E, Ande SR, et al. Targeting the mevalonate cascade as a new therapeutic approach in heart disease, cancer and pulmonary disease. Pharmacol Ther. 2014;143(1):87–110.
- Zhou Q, Liao JK. Pleiotropic effects of statins. - Basic research and clinical perspectives. Circ J. 2010;74(5):818–826.
- Voleti B, Agrawal A. Statins and nitric oxide reduce C-reactive protein production while inflammatory conditions persist. Mol Immunol. 2006;43(7):891–896.
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108.
- Boue S, Fields B, Hoeng J, et al. Enhancement of COPD biological networks using a web-based collaboration interface. F1000Res. 2015;4:32.
- Garcia-Rio F, Miravitlles M, Soriano JB, et al. Systemic inflammation in chronic obstructive pulmonary disease: a population-based study. Respir Res. 2010;11:63.
- Vernooy JH, Kucukaycan M, Jacobs JA, et al. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med. 2002;166(9):1218–1224.
- Nunez B, Sauleda J, Garcia-Aymerich J, et al. Lack of correlation between pulmonary and systemic inflammation markers in patients with chronic obstructive pulmonary disease: a simultaneous, two-compartmental analysis. Arch Bronconeumol. 2016;52(7):361–367.
- Ye RD, Sun L. Emerging functions of serum amyloid A in inflammation. J Leukoc Biol. 2015;98(6):923–929.
- Urieli-Shoval S, Cohen P, Eisenberg S, et al. Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. J Histochem Cytochem. 1998;46(12):1377–1384.
- Smith JW, Colombo JL, McDonald TL. Comparison of serum amyloid A and C-reactive protein as indicators of lung inflammation in corticosteroid treated and non-corticosteroid treated cystic fibrosis patients. J Clin Lab Anal. 1992;6(4):219–224.
- Lopez-Campos JL, Calero C, Rojano B, et al. C-reactive protein and serum amyloid a overexpression in lung tissues of chronic obstructive pulmonary disease patients: a case-control study. Int J Med Sci. 2013;10(8):938–947.
- Calero C, Arellano E, Lopez-Villalobos JL, et al. Differential expression of C-reactive protein and serum amyloid A in different cell types in the lung tissue of chronic obstructive pulmonary disease patients. BMC Pulm Med. 2014;14:95.
- Moran G, Carcamo C, Concha M, et al. Expression of the protein serum amyloid A in response to aspergillus fumigatus in murine models of allergic airway inflammation. Rev Iberoam Micol. 2015;32(1):25–29.
- Minoo P, King RJ. Epithelial-mesenchymal interactions in lung development. Annu Rev Physiol. 1994;56:13–45.
- Ishii T, Abboud RT, Wallace AM, et al. Alveolar macrophage proteinase/antiproteinase expression in lung function and emphysema. Eur Respir J. 2014;43(1):82–91.
- Aggarwal T, Wadhwa R, Rohil V, et al. Biomarkers of oxidative stress and protein-protein interaction in chronic obstructive pulmonary disease. Arch Physiol Biochem. 2018;124(3):226–231.
- Garbacki N, Di Valentin E, Piette J, et al. Matrix metalloproteinase 12 silencing: a therapeutic approach to treat pathological lung tissue remodeling? Pulm Pharmacol Ther. 2009;22(4):267–278.
- Semlali A, Jacques E, Rouabhia M, et al. Regulation of epithelial cell proliferation by bronchial fibroblasts obtained from mild asthmatic subjects. Allergy. 2010;65(11):1438–1445.
- Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Arch Bronconeumol. 2017;53(3):128–149.
- Brat K, Plutinsky M, Hejduk K, et al. Respiratory parameters predict poor outcome in COPD patients, category GOLD 2017 B. Int J Chron Obstruct Pulmon Dis. 2018;13:1037–1052.
- Wanderer AA. Hypoxia and inflammation. N Engl J Med. 2011;364(20):1976. author reply 1977.
- Imtiyaz HZ, Williams EP, Hickey MM, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010;120(8):2699–2714.
- Ghezzi P, Dinarello CA, Bianchi M, et al. Hypoxia increases production of interleukin-1 and tumor necrosis factor by human mononuclear cells. Cytokine. 1991;3(3):189–194.
- Lakota K, Carns M, Podlusky S, et al. Serum amyloid A is a marker for pulmonary involvement in systemic sclerosis. PloS one. 2015;10(1):e0110820.
- Zhu XY, Daghini E, Chade AR, et al. Disparate effects of simvastatin on angiogenesis during hypoxia and inflammation. Life Sci. 2008;83(23–24):801–809.
- Pinho-Ribeiro V, Melo AC, Kennedy-Feitosa E, et al. Atorvastatin and simvastatin promoted mouse lung repair after cigarette smoke-induced emphysema. Inflammation. 2017;40(3):965–979.
- Patyk I, Rybacki C, Kalicka A, et al. Simvastatin therapy and bronchoalveolar lavage fluid biomarkers in chronic obstructive pulmonary disease. Adv Exp Med Biol. 2019;1150:43–52.
- Hothersall E, McSharry C, Thomson NC. Potential therapeutic role for statins in respiratory disease. Thorax. 2006;61(8):729–734.
- Lee JH, Lee DS, Kim EK, et al. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. 2005;172(8):987–993.
- Takahashi S, Nakamura H, Seki M, et al. Reversal of elastase-induced pulmonary emphysema and promotion of alveolar epithelial cell proliferation by simvastatin in mice. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L882–890.
- Calabrese F, Giacometti C, Beghe B, et al. Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respir Res. 2005;6:14.