
ABSTRACT
Background
Legionella pneumophila is water-based bacterium causing Legionnaires’ disease (LD). We describe the first documented case of nosocomial LD caused by L. pneumophila sequence type (ST) 461 and serogroup 6. The etiology of LD was confirmed by culturing the bronchoalveolar lavage sample retrieving L. pneumophila strain ALAW1. A 7-days treatment of the LD patient with Azithromycin and Levofloxacin allowed complete recovery.
Methods
In details, we sequenced the whole genome of the L. pneumophila ALAW1 using Illumina HiSeq platform. The sequence of ALAW1 was aligned with the genome sequence from the closely related reference strain Alcoy 2300/99 and a whole-genome phylogeny based on single nucleotide polymorphisms (SNPs) was created using Parsnp software. Also, the TYGS web-server was used in order to compare the genome with type strain.
Results
An analysis of the population structure by SNP and TYGS comparison clustered ALAW1 with the reference genome Alcoy 2300/99. Blastp analysis of the type IV secretion Dot/Icm system genes showed that these genes were highly conserved with (≤25%) structural differences at the protein level.
Conclusions
Overall, this study provides insights into detailed genome structure and demonstrated the value of whole-genome sequencing as the ultimate typing tool for Legionella.
Introduction
L.pneumophila is an opportunistic bacterial pathogen with widespread distribution in freshwater environments. This bacterial species is the main etiology of legionellosis worldwide except Australia and New Zealand [Citation1–3]. The term ‘legionellosis’ describes Legionnaires’ disease (LD), a severe form of atypical pneumonia, and a non-pneumonic febrile illness called Pontiac Fever. L. pneumophila has 15 serogroups (Sgs); Sg1 is the most common causative agent of LD, followed by Sg6 [Citation4,Citation5]. Many studies have demonstrated that an important source for LD is the drinking water distribution systems (DWDS) in large buildings like hospitals and hotels [Citation6–8]. The contamination of hospital water systems with L. pneumophila is considered to pose a high risk for patients, especially for vulnerable and immunocompromised people. To this end, it is well known that LD is an important cause of hospital-acquired pneumonia [Citation9]. The presence of L. pneumophila in DWDS could be a serious health risk to hospital staff and patients, but the magnitude of the problem is often unrecognized [Citation1,Citation9].
For the identification of possible sources of contamination/infection, high-resolution genotyping of new isolates is needed to correlate environmental isolates with clinical isolates. This is currently done using the standard molecular approach Sequence-Based Typing (SBT). SBT of L. pneumophila is done by sequencing a set of seven reference genes per isolate, providing a specific Sequence Type (ST) that can be matched with an International database [Citation10–12]. Another typing method is Multi Locus Variable numbered of tandem repeats Analysis (MLVA) allowing a somewhat higher resolution than SBT for most L. pneumophila strains [Citation13]. Both SBT and MLVA were used for typing environmental and clinical strains of the West Bank in previous studies [Citation7,Citation14,Citation15].
Studies on this ubiquitous water-based pathogen should be directed towards both sides: i) the clinical side from pneumonia patients, and ii) the environmental side from DWDS of hospitals and public buildings [Citation16]. The problem is apparent in the West Bank because awareness about the prevalence of L. pneumophila or LD is lacking, and data scarcity for such arid regions. The study by Sharaby et al [Citation17] described a set of genotyped clinical and environmental L. pneumophila strains retrieved from the closely geographic area Israel. They studied the pathogenicity potential (Ex: hemolytic activity and cytotoxicity toward amoebae and macrophages.) of both clinical and environmental strains. The authors concluded that understanding the virulence characteristics of L. pneumophila genotypes may improve the assessment of public health risks of L. pneumophila in DWDS [Citation17]. Furthermore, in the West Bank, there are no specific guidelines for L. pneumophila surveillance or protection from exposure in hospitals or public buildings [Citation18]. Thus, the main aim of studying entensively the genomics of ALAW1 clinical isolate from this LD case is to focus into genome details, to compare among L. pneumophila intra-clonal genomic characteristics portraying the importance of hospital water habitat.
Case presentation
In January 2014, a 66-year-old woman was admitted to hospital F in Bethlehem – West Bank (Figure S1) (day 0) after evaluation in the emergency ward for high blood pressure and severe gastric pain (). The woman had been prescribed Arcoxia (NSAIDs) and Prednisolone (Corticosteroids) 6 days prior to admission (day −6) as treatment of joint inflammation. On day 4 after admission, the patient complained of dyspnea, chest pain, mild fever, and reproductive cough. A chest X-ray showed fluffy infiltrates in both lungs. Atypical pneumonia was suspected. The incubation period of LD is usually 2–10 days, but it has been recorded in some cases to be of up to 16 days. The severity of the disease ranges from a mild cough to a rapidly fatal pneumonia. Initial symptoms include fever, loss of appetite, headache, malaise, and lethargy. Some patients may also experience myalgia, diarrhea, and confusion [Citation19,Citation20]. On day 5, the patient had difficulty breathing with low oxygen and was transferred to the Intensive Care Unit (ICU) of a specialized hospital in East Jerusalem (hospital E) (Figure S1) where bronchoalveolar lavage (BAL) and sputum) were collected to be analyzed by cultivation for bacterial pathogens. On the same day, treatment with Azithromycin followed by Levofloxacin was administered. On day 9, bacteriological culture was negative, and the diagnosis was confirmed as an atypical pneumonia caused by a fastidious pathogen. On day 12, the patient recovered and was discharged ().
Figure 1. Timeline of events during investigation of a hospital acquired Legionnaires’ disease — in the West Bank, 2014. Abbreviations: NSAIDs: Non-steroidal anti-inflammatory drugs, ICU: Intensive Care Unit, BAL: Bronchoalveolar Lavage, Sg: Serogroup, ST: Sequence Type and AQU: Al-Quds University. Timeline represents days and the data presented are from one hospitalized patient. BAL sample was analyzed twice, at day 5 and day 10.
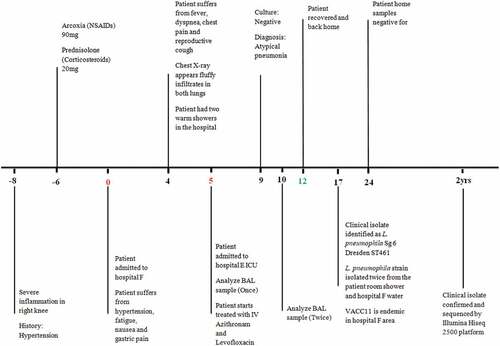
On day 17, the clinical isolate ALAW1 was retrieved from the BAL sample, and was identified as L. pneumophila Sg6 Dresden and ST 461 [Citation14]. According to Zayed et al [Citation7] ST 461 was endemic in the DWDS of hospital F in Bethlehem. During the patient’s stay in hospital F, she had two warm showers that may have caused the infection. On day 24 and at the request of the patient, a researcher from the Microbiology Research Laboratory collected and cultured samples from bath shower and faucets in the patients’ home, but the culture was negative for L. pneumophila (). The patient’s two stories family house appeared to be in good condition and was connected to a municipal water network rendering a nosocomial infection during her stay in hospital F in Bethlehem most likely.
Materials and methods
L. pneumophila clinical isolate ALAW1 and reference genomes
L. pneumophila clinical isolate ALAW1 was selected for this study. For a comparison, genome sequences of 15 L. pneumophila reference strains retrieved from NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/416/) were used as reference (Table S1). Details for all L. pneumophila isolates from the West Bank are in [Citation21].
Short read sequencing, genome assembly, and annotation
Whole-genome sequencing based on short reads was applied to this study. Therefore, the Illumina Hiseq 2500 was used, which is well established and produces low error rates [Citation22,Citation23]. DNA of the clinical isolate was provided for whole-genome sequencing to the Genome Analytics unit (GMAK) of the Helmholtz Center for Infection Research (HZI).
In detail, a DNA library for the Illumina sequencer was prepared using the NEBNext Ultra kit according to the manufacturer’s instructions (NEB, Ipswitch, MA, USA), and sequenced using 100 bp paired end runs on the Illumina Hiseq 2500. Processed sequence reads of the clinical isolate were assembled using Velvet version 1.2.10 with k-mer size of 61.
Assembled contigs of isolate ALAW1 were annotated using Prokka v1.11 [Citation24]. Prokka connects several software tools in order to predict the coordinates of CDS, tRNAs, rRNAs, CRISPRs, and other genomic features encoded on contigs and chromosomes. In this case, Prokka was applied using a genus database (–usegenus) based on the high-quality, manually annotated L. pneumophila strain Corby [Citation25]. With that, 2997 Coding DNA Sequence (CDS) annotations could be successfully transferred to isolate ALAW1.
The Genome Sequence of isolate ALAW1 was uploaded to NCBI GenBank, Acc. No. JALDWK000000000. Furthermore, short read sequencing data is provided in NCBI Sequence Read Archive under BioProject ID PRJNA817377.
Genome analysis
For phylogenomic identification and phylogenomic tree construction, GenBank files of L. pneumophila clinical isolate ALAW1 and 15 reference genomes were submitted to the Type Strain Genome Server (TYGS) (https://tygs.dsmz.de) (Table S1). Hereby, a comparison and clustering of genome data was performed based on the established digital DNA – DNA hybridization (dDDH) threshold of 70% [Citation26]. Subspecies clustering was based on a 93% dDDH threshold as previously introduced [Citation27].
The software Parsnp was used for SNP analysis in order to construct a phylogenomic tree for the core genome of already published L. pneumophila genomes [Citation28]. Parsnp results were visualized using Gingr [Citation28]. Gingr provides an interactive display of multi-alignment variants and phylogenetic trees estimated from the core genome alignment.
Finally, BLASTp was applied to align the amino acid sequences against the Virulence Factors of Pathogenic Bacteria (VFDB) database [Citation29,Citation30]. Amino acid sequences with 75% match identity was chosen and the description of the best hit was assigned as the annotation of predicted gene compared to L. pneumophila str. Philadelphia1 as default bacteria on the webpage.
Results and discussion
Herein, we report, for the first time, that a strain representing L. pneumophila ST461 was isolated from a BAL sample from a patient hospitalized with LD in January 2014.
General features of the genome of L. pneumophila clinical isolate ALAW1
summarizes the main genome features of the L. pneumophila clinical isolate ALAW1. The genome of the L. pneumophila clinical isolate ALAW1 was sequenced with an approximately 105× coverage using the Illumina Hiseq platform. 35 contigs greater than 200 bp could be assembled in total summed up to a total genome size of 3,368,497 bp, 3041 genes and 2997 CDS were identified. It contained genomic islands of a total length of 161,380 bp representing 4.8% of the genome.
Table 1. Main features of L. pneumophila clinical isolate used in the study.
Identification of L. pneumophila clinical isolate ALAW1 based on SBT and MLVA
The clinical isolate belonged to Sg 6 Dresden and ST 461. By MLVA, ALAW1 is identified as MLVA-genotype (Gt) 9(92). Isolates of ST 461 of the West Bank were split up into three MLVA-genotypes, i.e. Gt 9(92), Gt 10(93) and Gt 10(141). These MLVA genotypes form the Clonal Complex 11 (VACC11), a clonal complex first described for the West Bank [Citation7]. Detailed ST and MLVA patterns and genes are seen in (Table S2).
Comparison of taxonomic resolution of STs and SNP analysis
Identification of L. pneumophila clinical isolate ALAW1 based on Illumina Hiseq Sequencing was performed using the TYGS webserver. Phylogenomic tree of L. pneumophila clinical isolate revealed correct taxonomic task to L. pneumophila. Hereby, the recommended dDDH values of 91.3–94.7% were computed against the type strain L. pneumophila str. Philadelphia1 as default reference strain the web page and thereby fulfill the criteria for bacterial species identification [Citation26]. A phylogenomic tree based on Illumina Hiseq sequencing was constructed using the TYGS web server4 and ALAW1 isolate cluster in the same clade with the reference strains Alcoy 2300/99 and Corby (, for the full TYGS phylogenomic tree see Figure S2). According to the TYGS recommended results of similarity according to the webpage. ALAW1 is most similar to L. pneumophila str. Alcoy 2300/99 with a dDDH value of 93.2% and a G+C content difference of 0.18% ( and Table S3)
Figure 2. A) Zoomed in Phylogenomic tree of L.pneumophila clinical isolate (Alaw1_cl_ps) and 15 L. pneumophila reference genomes and subspecies delineation based on the GBDP phylogenetic analyses retrieved from the TYGS website. the dark green squares showed ALAW1 clustered together with the two reference genomes Alcoy 2300/99 and Corby. The branch lengths are scaled in terms of GBDP distance formula d4 (the whole phylogenomic tree of Legionella species and subspecies in supplementary materials (Figure S2). B) Gingr visualization of 15 L. pneumophila reference genomes and L.pneumophila clinical isolate aligned with Parsnp. The leaves of the reconstructed phylogenetic tree (left) are paired with their corresponding rows in the multi-alignment. L. pneumophila str. Alcoy 2300/99 is the reference genome. The constructed tree share 72% of the core genome. The gray region isn’t a part of core genome. The white region is a part of core genome but no SNPs accumulation. The heatmap on the button of the figure indicates more reddish meaning more SNPs accumulation..
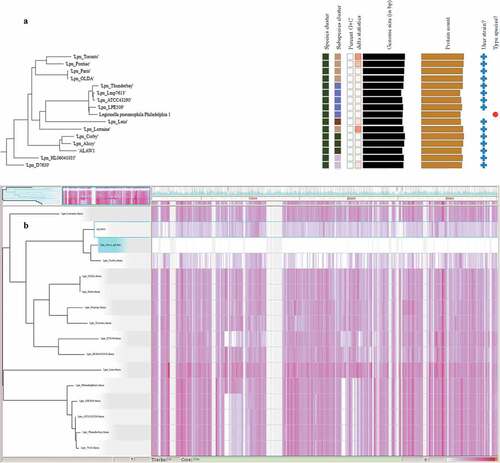
Additionally, a phylogenomic tree was constructed using SNP analysis of 15 reference genomes from GeneBank (Table S1) and our clinical L. pneumophila genome sequenced strain. The main branches or clusters were observed in the constructed phylogenomic tree (). ALAW1 is clustered with the two complete reference genomes (L. pneumophila strain Alcoy 2300/99 and Corby) ().
By ParSNP analysis L. pneumophila str. Alcoy 2300/99 was shown to be the closest reference strain so far with a complete genome available in the GenBank database. We found 11,674 SNPs compared to L. pneumophila clinical isolate. Indeed, L. pneumophila str. Alcoy 2300/99 showed 1,303 SNPs less than performing the complete ParSNP analysis with L. pneumophila str. Corby as a reference genome. Thus, the best result for our SNP-based phylogenomic tree was retrieved with L. pneumophila str. Alcoy 2300/99 as reference and documented in and . Overall, we observed a typical concordance between phylogenomic tree based on SNPs and phylogenomic tree based on TYGS webpage.
Table 2. Whole-genome SNP comparison of L. pneumophila isolates and reference strains.
Results of TYGS phylogenomic tree of L. pneumophila clinical isolate are identical with the constructed SNP phylogenomic tree. Comparing the phylogenomic tree obtained with these two molecular methods and mathematical algorithms, we found that ST is in concordance with the SNP genotyping, but it cannot cover the needed high resolution at intra-clonal level provided by WGS.
Actually, a threshold for the number of SNPs necessary to identify outbreak-associated L. pneumophila strains still has to be established. For example, more than 200 SNPs have been reported in phylogenetically closely related L. pneumophila outbreak strains [Citation31,Citation32]. Mercante et al [Citation33] showed that up to 20 core SNPs were identified in comparison of Philadelphia clade L. pneumophila isolates. In the present case study, 11,674 SNPs were identified in the same sub-species cluster with the reference strain Alcoy 2300/99 from Valencia, Spain. While SBT from the same sub-lineage in a phylogenomic cluster is different as L. pneumophila str. Alcoy 2300/99 is ST578 and ALAW1 is ST461. These results are in concordance with Khodr et al [Citation34]. They sequenced six ST1 genomes (four clinical and environmental isolates from a hospital and the other two were unrelated) and observed that geographically unrelated isolates differed by more than 1,500 SNPs ().
Strains of VACC11 are affiliated with ST461 and were classified as Sg 6 Dresden [Citation7]. ST461 was previously reported by the European Working Group for Legionella infections (EWGLI) [Citation7,Citation8,Citation35,Citation36], to be found in hospitals water in Poland [Citation35]. Moreover, ST461 was identified in Michigan, (USA) water systems and showed high capability to efficiently infect THP-1 macrophages [Citation36]. More recently, ST461 was identified in hotel water in southern Israel [Citation8] and in the West Bank hospital water systems [Citation7].
Identification of pore-forming genes mediating cytotoxicity in L. pneumophila clinical isolate ALAW1
Central to the pore-forming mediated cytotoxicity of L. pneumophila are the Dot/Icm loci, which taken together directly assemble to a type IV secretion system (T4SS) [Citation37,Citation38]. Also, the toxin rtxA plays an important role in the pore-mediated cytotoxicity [Citation39–41]. Although all L. pneumophila strains examined until today contain the complete Dot/Icm loci, sequence variations among the Dot/Icm genes among different L. pneumophila strains have been reported [Citation37,Citation42]. Eleven Dot/Icm T4SS genes (icmT [Citation43,Citation44], icmS [Citation45], icmR [Citation45], icmQ [Citation45], icmL/dotI [Citation46], icmK/dotH [Citation46], icmE/dotG [Citation46], icmC/dotE [Citation46], dotB [Citation46], dotA [Citation46] and icmW [Citation45]) and rtxA gene [Citation41] are responsible for the pore-forming mediated cytotoxicity of L. pneumophila. The 11 Dot/Icm T4SS pore-forming mediated cytotoxicity genes and the rtxA gene were identified by a BLASTp search against the Virulence Factor Database (VFDB) using L. pneumophila strain Philadelphia1 as default reference genome for the reference strain L. pneumophila str. Alcoy 2300/99 and L. pneumophila clinical isolate ALAW1, which had been genome sequenced ().
Table 3. Percentage of nucleotide identity of orthologous pore-forming activity genes with respect to the BLASTp search against the VFDB using L. pneumophila strain Philadelphia1 as default reference genome.
shows 75% to 100% similarity between the pore-forming mediated cytotoxicity genes. Morozova et al [Citation47] showed that the Dot/Icm genes are highly conserved in L. pneumophila strains. After WGS technology was available on the market, Gomez-Valero et al [Citation48,Citation49] confirmed the previous study showing high conservation (98%) among orthologs of the reference strains Corby, Paris, Philadelphia, and Lens with few exceptions in the dotA gene. The dotA gene is an essential gene for virulence activity of L. pneumophila strains since it encodes an integral membrane protein with eight domains. This explains why a dotA mutant of L. pneumophila strain Corby is being used as a negative control for all virulence assays [Citation50]. Costa et al [Citation51] analyzed 300 dotA gene sequences from L. pneumophila strains and demonstrated that pathogenic L. pneumophila strains belong to a subset of the genotypes existing in the environment. Khodr et al [Citation34] explained the high variation of the dotA gene of L. pneumophila by indicating that this gene is a target for host speciation and adaptive evolution to different hosts and environments. Dumenil et al [Citation52] showed that IcmR is a regulator gene for the IcmQ gene that possesses pore-forming activity. In addition, Gomez-Valero et al [Citation53] demonstrated that dotB, IcmS and IcmW are highly conserved genes. These facts are in accordance with our results () showing that IcmR (95%), IcmS and dotB (100%) and IcmQ (100%) were highly conserved gene while dotA had only a 78% gene similarity for our L. pneumophila clinical isolate ALAW1. More recently, Zayed et al studied the infectivity and cytotoxicity to all clonal complexes and their affiliated genotypes of the West Bank [Citation54]. Overall, the Dot/Icm system is a highly conserved and complex molecular system ().
Taken together, this detailed analysis suggests that the 12 genes studied out of more than 200 of the Dot/Icm components of L. pneumophila are representing a large repertoire of effectors, which are necessary for virulence [Citation48]. In general, both of the L. pneumophila strains studied shared the same Dot/Icm T4SS with less than 25% structural differences at the protein level.
According to David et al [Citation55] a certain local micro-evolution could be observed if isolates from the same site of isolation have been obtained at different times. Such a case of micro-evolution could have occurred in the environmental isolates obtained from the DWDS of hospital F. Environmental strains of ST461 has had time to diversify by genetic drift since it was endemic in Bethlehem area (hospital F) and the clinical isolate originated from environmental source. Also, conditions in the water systems such as disinfection, temperature, and amoeba might have increased the virulence of the ST461 strains. A thorough genome-based comparison of the environmental strains and the clinical isolate ALAW1 might give insight into this development and the respective mechanisms.
Conclusions and future plans
This study of L. pneumophila clinical isolate ALAW1 highlights conserved genetical features that may be critical for pathogenesis in human lungs. Future plans aim to compare the clinical isolate genome with genome sequenced environmental isolates belonging to ST461 from hospital F hotspots. Moreover, complete genome sequencing of L. pneumophila ST461 strains is highly recommended to better understand the evolution and environmental adaptation within the VACC11 clade.
Patient consent
This study has consent form PHRC/HC/211/17
Supplemental Material
Download MS Word (735.3 KB)Acknowledgements
We thank Josefin Koch, Verena Maiberg and Suha Butmeh for their skilful technical support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
Data are available at GenBank accession numbers JALDWK000000000. Furthermore, short read sequencing data is provided in NCBI Sequence Read Archive under BioProject ID PRJNA817377.Full WGS methods and results are provided in the Supplementary Data.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/20018525.2023.2168346.
Additional information
Funding
References
- Yu VL, Stout JE. Hospital characteristics associated with colonization of water systems by Legionella and risk of nosocomial legionnaires’ disease: a cohort study of 15 hospitals. Infect Control Hosp Epidemiol. 2000;21(7):434–9.
- O’Connor BA, Carman J, Eckert K, et al. Does using potting mix make you sick? Results from a Legionella longbeachae case-control study in South Australia. Epidemiol Infect. 2007;135(1):34–39.
- Kenagy E, Priest PC, Cameron CM, et al. Risk factors for legionella longbeachae legionnaires’ disease, New Zealand. Emerg Infect Dis. 2017;23(7):1148–1154. DOI:10.3201/eid2307.161429
- Faccini M, Russo AG, Bonini M, et al. Large community-acquired legionnaires’ disease outbreak caused by legionella pneumophila serogroup 1, Italy, July to August 2018. Euro Surveill. 2020;25(20). DOI:10.2807/1560-7917.ES.2020.25.20.1900523
- Messi P, Bargellini A, Anacarso I, et al. Protozoa and human macrophages infection by Legionella pneumophila environmental strains belonging to different serogroups. Arch Microbiol. 2013;195(2):89–96.
- Buse HY, Morris BJ, Gomez-Alvarez V, et al. Legionella diversity and spatiotemporal variation in the occurrence of opportunistic pathogens within a large building water system. Pathogens. 2020;9(7). DOI:10.3390/pathogens9070567
- Zayed AR, Pecellin M, Salah A, et al. Characterization of legionella pneumophila populations by multilocus variable number of tandem repeats (MLVA) genotyping from drinking water and biofilm in hospitals from different regions of the West Bank. Pathogens. 2020;9(11). DOI:10.3390/pathogens9110862
- Yakunin E, Kostyal E, Agmon V, et al. A snapshot of the prevalence and molecular diversity of legionella pneumophila in the water systems of Israeli Hotels. Pathogens. 2020;9(6). DOI:10.3390/pathogens9060414
- Mojtahedi SY, Rahbarimanesh A, Noorbakhsh S, et al. Urinary antigene and PCR can both be used to detect Legionella pneumophila in children’s hospital-acquired pneumonia. Eur J Trans Myol. 2019;29(2):8120.
- Gong X, Li J, Zhang Y, et al. Molecular typing of Legionella pneumophila from air-conditioning cooling waters using mip gene, SBT, and FAFLP methods. J Microbiol Methods. 2017;139:1–7.
- Gaia V, Fry NK, Afshar B, et al. Consensus sequence-based scheme for epidemiological typing of clinical and environmental isolates of Legionella pneumophila. J Clin Microbiol. 2005;43(5):2047–2052. DOI:10.1128/JCM.43.5.2047-2052.2005
- Ratzow S, Gaia V, Helbig JH, et al. Addition of neuA, the gene encoding N-acylneuraminate cytidylyl transferase, increases the discriminatory ability of the consensus sequence-based scheme for typing Legionella pneumophila serogroup 1 strains. J Clin Microbiol. 2007;45(6):1965–1968.
- Sobral D, Le Cann P, Gerard A, et al. High-throughput typing method to identify a non-outbreak-involved Legionella pneumophila strain colonizing the entire water supply system in the town of Rennes, France. Appl Environ Microbiol. 2011;77(19):6899–6907. DOI:10.1128/AEM.05556-11
- Jaber L, Amro M, Tair HA, et al. Comparison of in situ sequence type analysis of Legionella pneumophila in respiratory tract secretions and environmental samples of a hospital in East Jerusalem. Epidemiol Infect. 2018;146(16):2116–2121. DOI:10.1017/S0950268818002340
- Pecellin M Structrure and Virulence of Legionella pneumophila Populations from Freshwater Systems in Germany and Middle East. PhD thesisTechnical University of Braunschweig, Germany. 2016.
- Iliadi V, Staykova J, Iliadis S, et al. Legionella pneumophila: the journey from the environment to the blood. J Clin Med. 2022;11(20). DOI:10.3390/jcm11206126
- Sharaby Y, Rodriguez-Martinez S, Pecellin M, et al. Virulence traits of environmental and clinical legionella pneumophila multilocus variable-number tandem-repeat analysis (MLVA) genotypes. Appl Environ Microbiol. 2018;84(10). DOI:10.1128/AEM.00429-18
- Zayed AR, Butmeh S, Pecellin M, et al. Biogeography and environmental drivers of legionella pneumophila abundance and genotype composition across the West Bank: relevance of a genotype-based ecology for understanding legionella occurrence. Pathogens. 2020;9(12). DOI:10.3390/pathogens9121012
- Zarogoulidis P, Alexandropoulou I, Romanidou G, et al. Community-acquired pneumonia due to Legionella pneumophila, the utility of PCR, and a review of the antibiotics used. Int J Gene Med. 2011;4:15–19.
- Patel H, Shelley P, Hatoum H. Hypertriglyceridemia and massive rhabdomyolysis in a patient with disseminated legionella. Respir Med Case Rep. 2021;32:101321.
- Zayed AR Ecology, virulence and genomics of Legionella pneumophila isolates from the West Bank, Palestine. PhD thesisTechnical University of Braunschweig, Germany. 2018.
- Quail MA, Smith M, Coupland P, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341.
- Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme J. 2012;6(8):1621–1624. DOI:10.1038/ismej.2012.8
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069.
- Steinert M, Heuner K, Buchrieser C, et al. Legionella pathogenicity: genome structure, regulatory networks and the host cell response. Int J Med Microbiol. 2007;297(7–8):577–587.
- Meier-Kolthoff JP, Goker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun. 2019;10(1):2182.
- Meier-Kolthoff JP, Hahnke RL, Petersen J, et al. Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci. 2014;9(2). DOI:10.1186/1944-3277-9-2
- Treangen TJ, Ondov BD, Koren S, et al. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome bio. 2014;15(11):524.
- Chen L, Yang J, Yu J, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33(Database issue):D325–8. DOI:10.1093/nar/gki008
- Chen L, Zheng D, Liu B, et al. VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 2016;44(D1):D694–7.
- Graham RM, Doyle CJ, Jennison AV. Real-time investigation of a Legionella pneumophila outbreak using whole genome sequencing. Epidemiol Infect. 2014;142(11):2347–2351.
- Reuter S, Harrison TG, Koser CU, et al. A pilot study of rapid whole-genome sequencing for the investigation of a Legionella outbreak. BMJ Open. 2013;3(1). DOI:10.1136/bmjopen-2012-002175
- Mercante JW, Morrison SS, Desai HP, et al. Genomic analysis reveals novel diversity among the 1976 philadelphia legionnaires’ disease outbreak isolates and additional ST36 strains. PLoS ONE. 2016;11(9):e0164074.
- Khodr A, Kay E, Gomez-Valero L, et al. Molecular epidemiology, phylogeny and evolution of Legionella. Infect Genet Evol. 2016;43:108–122.
- Pancer K. Sequence-based typing of Legionella pneumophila strains isolated from hospital water distribution systems as a complementary element of risk assessment of legionellosis in Poland. Ann Agric Environ Med. 2013;20(3):436–440.
- Byrne BG, McColm S, McElmurry SP, et al. Prevalence of infection-competent serogroup 6 legionella pneumophila within premise plumbing in southeast michigan. MBio. 2018;9(1). DOI:10.1128/mBio.00016-18
- Segal G, Shuman HA. Characterization of a new region required for macrophage killing by Legionella pneumophila. Infect Immun. 1997;65(12):5057–5066.
- Berger KH, Isberg RR. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol. 1993;7(1):7–19.
- Zink SD, Pedersen L, Cianciotto NP, et al. The Dot/Icm type IV secretion system of Legionella pneumophila is essential for the induction of apoptosis in human macrophages. Infect Immun. 2002;70(3):1657–1663.
- Segal G, Shuman HA. Legionella pneumophila utilizes the same genes to multiply within Acanthamoeba castellanii and human macrophages. Infect Immun. 1999;67(5):2117–2124.
- Cirillo SL, Bermudez LE, El-Etr SH, et al. Legionella pneumophila entry gene rtxA is involved in virulence. Infect Immun. 2001;69(1):508–517.
- Broich M, Rydzewski K, McNealy TL, et al. The global regulatory proteins LetA and RpoS control phospholipase A, lysophospholipase A, acyltransferase, and other hydrolytic activities of Legionella pneumophila JR32. J bacteriol. 2006;188(4):1218–1226.
- Molmeret M, Alli OA, Zink S, et al. icmT is essential for pore formation-mediated egress of Legionella pneumophila from mammalian and protozoan cells. Infect Immun. 2002;70(1):69–78.
- Bitar DM, Molmeret M, Kwaik YA. Structure-function analysis of the C-terminus of IcmT of Legionella pneumophila in pore formation-mediated egress from macrophages. FEMS Microbiol Lett. 2005;242(1):177–184.
- Molmeret M, Bitar DM, Han L, et al. Cell biology of the intracellular infection by Legionella pneumophila. Microbes Infect. 2004;6(1):129–139.
- Kirby JE, Vogel JP, Andrews HL, et al. Evidence for pore-forming ability by Legionella pneumophila. Mol Microbiol. 1998;27(2):323–336.
- Morozova I, Qu X, Shi S, et al. Comparative sequence analysis of the icm/dot genes in Legionella. Plasmid. 2004;51(2):127–147. DOI:10.1016/j.plasmid.2003.12.004
- Gomez-Valero L, Rusniok C, Jarraud S, et al. Extensive recombination events and horizontal gene transfer shaped the Legionella pneumophila genomes. BMC Genomics. 2011;12:536.
- Gomez-Valero L, Rusniok C, Cazalet C, et al. Comparative and functional genomics of legionella identified eukaryotic like proteins as key players in host-pathogen interactions. Front Microbiol. 2011;2:208.
- Roy CR, Berger KH, Isberg RR. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol Microbiol. 1998;28(3):663–674.
- Costa J, Tiago I, Da Costa MS, et al. Molecular evolution of Legionella pneumophila dotA gene, the contribution of natural environmental strains. Environ Microbiol. 2010;12(10):2711–2729.
- Dumenil G, Montminy TP, Tang M, et al. IcmR-regulated membrane insertion and efflux by the Legionella pneumophila IcmQ protein. J Biol Chem. 2004;279(6):4686–4695.
- Gomez-Valero L, Rusniok C, Rolando M, et al. Comparative analyses of Legionella species identifies genetic features of strains causing Legionnaires’ disease. Genome bio. 2014;15(11):505. DOI:10.1186/PREACCEPT-1086350395137407
- Zayed AR, Pecellin M, Jaber L, et al. Cytotoxicity, intracellular replication, and contact-dependent pore formation of genotyped environmental legionella pneumophila isolates from hospital water systems in the West Bank, Palestine. Pathogens. 2021;10(4). DOI:10.3390/pathogens10040417
- David S, Rusniok C, Mentasti M, et al. Multiple major disease-associated clones of Legionella pneumophila have emerged recently and independently. Genome Res. 2016;26(11):1555–1564. DOI:10.1101/gr.209536.116