
Abstract
Aim: Atypical teratoid rhabdoid tumor (ATRT) is a rare and highly aggressive primary CNS neoplasm, predominantly observed in children. The use of autologous stem cell transplantation (ASCT) in pediatric ATRT has shown promise; however, its utility in adult ATRT remains unclear. Patients & methods: This study presents the case of an adult patient with ATRT who is in remission after ASCT and reviews the literature on ASCT in adults with ATRT. Four cases of ATRT in adults who underwent ASCT were identified, with pertinent data summarized. Results: All five patients survived longer than the historical average survival rate, four of whom had no clinical or radiographic evidence of disease at the final follow-up. Conclusion: Based on limited data, there may be a role for ASCT in the treatment of adults with ATRT.
Atypical teratoid rhabdoid tumor (ATRT) is a rare and highly aggressive primary CNS neoplasm.
ATRT is predominantly observed in children and is exceedingly rare in adults.
There are currently no established treatment protocols specific to adult ATRT, management strategies for which are largely extrapolated from pediatric regimens.
Autologous stem cell transplantation (ASCT) can be used as a consolidative adjuvant in the treatment of pediatric ATRT.
This study presents the case of an adult patient with suprasellar ATRT who is in remission after ASCT in addition to reviewing the current literature on the use of ASCT in the treatment of adult ATRT.
A retrospective review identified four cases of adult (age >18 years) patients with ATRT who underwent ASCT—pertinent data (age, sex, tumor location, surgical intervention, chemoradiation regimen, ASCT conditioning, and follow-up details) were extracted and summarized alongside our case presentation.
All five patients received similar chemoradiation regimens ahead of ASCT and survived longer than the historical average survival rate.
Four patients had no clinical or radiographic evidence of disease at the final follow-up.
One patient experienced progressive disease and was deceased at the final follow-up.
The experience of the patient in the case described adds to a small body of literature suggesting a role for ASCT in the management of these rare and aggressive tumors.
1. Background
Atypical teratoid rhabdoid tumor (ATRT) is a highly malignant World Health Organization (WHO) grade 4 embryonal tumor primary to the CNS. It is defined by inactivating mutations in both copies of the tumor suppressor gene SMARCB1 or, rarely, SMARCA4, which are involved in chromatin remodeling in the SWI/SNF complex, a pathway mediating lineage specification. Biallelic inactivation is confirmed by immunohistochemical analysis demonstrating loss of INI1 or BRG1 expression, respectively [Citation1,Citation2]. The diagnosis may also be suspected based on histologic features which, although subject to wide variation, generally consist of some combination of rhabdoid cells (for which the tumor is named), poorly differentiated ‘small round blue cells’, and areas of mesenchymal/epithelial differentiation [Citation3]. Imaging findings may additionally prompt consideration of ATRT and typically include heterogenous enhancement of a hyperdense and hypo- to mixed-intensity mass, commonly with areas of cystic degeneration, necrosis, hemorrhage, or calcification; however, imaging alone is considered insufficient to distinguish ATRT from related neoplasms [Citation4].
While embryonal tumors are overwhelmingly seen in the pediatric population, ATRT is predominantly observed in infants or children less than 3 years of age. It is exceedingly rare in adults, with fewer than 100 cases reported in the literature since it was first described in 1987 [Citation5,Citation6]. Limited data pertaining to this disease poses a significant challenge to fully understanding its pathophysiology, prognostic factors, and effective treatment strategies. Recent studies examining inter-tumor heterogeneity among ATRTs via DNA methylation profiling and gene expression profiling have confirmed three distinct subgroups: ATRT-TYR, ATRT-SHH, and ATRT-MYC. Currently, there is no standard treatment regimen accepted for adults with ATRT, and the management of these patients is largely extrapolated from regimens used in the pediatric population. However, the success of this approach has been limited thus far, resulting in a persistently poor prognosis among adults with a median overall survival of just 15 months.
One of the more recent advances to treatment protocols used for pediatric ATRT is the addition of autologous stem cell transplantation (ASCT), often alongside multimodal therapy with surgery followed by chemotherapy and radiation therapy (RT) [Citation7,Citation8]. However, evidence supporting the use of ASCT in adults with ATRT continues to be scarce, with only four cases currently described in the literature [Citation9–12]. Therefore, the role of ASCT in the management of adults with ATRT remains unclear.
Herein we present the case of an adult with ATRT treated at our institution who underwent ASCT and review the existing literature regarding the use of ASCT in the management of adults with ATRT. We also describe the results of our literature review on other published cases of adults with ATRT who underwent ASCT.
2. Methods
A retrospective review of the literature was conducted to identify cases of adult (age ≥18 years) patients with ATRT who underwent ASCT. PubMed was queried using the key term ‘atypical teratoid rhabdoid tumor’ along with the MeSH term ‘adult.’ This yielded 126 results that were subsequently filtered by language, resulting in the exclusion of four non-English articles. The remaining 122 studies were carefully evaluated, with only those reporting cases of ATRT in adult patients who received ASCT considered for further analysis. A total of four such cases were ultimately identified. The most salient aspects of each case, including age, sex, tumor location, surgical intervention, chemotherapy regimen, RT regimen, ASCT conditioning, and follow-up details, were extracted and summarized alongside the case of an adult treated at our institution. Appropriate consent was obtained from our patient in adherence to established ethical guidelines and regulations governing patient privacy and data confidentiality.
3. Case presentation
We present the case of a 46-year-old female who presented with a complaint of two weeks of intermittent headache and right-sided pressure-like retroorbital pain accompanied by one week of diplopia. Initial examination revealed a complete right abducens nerve palsy. MRI brain demonstrated a heterogeneously enhancing right anterior pituitary mass, measuring 1.9 cm × 1.2 cm × 1.1 cm with mild to moderate suprasellar extension and involvement of the right cavernous sinus. Neurosurgery was consulted, and outpatient follow-up was planned for further evaluation of a presumed pituitary macroadenoma later the same week.
Repeat examination at her follow-up appointment was unchanged from the previous; however, two days later, she developed acutely worsening retroorbital pain and visual acuity in her right eye coupled with new-onset photophobia, lacrimation, and right facial numbness. MRI sella pituitary at this time redemonstrated an enhancing sellar mass with right cavernous sinus invasion, with interval growth to 2.5 cm × 1.5 cm × 1.3 cm. The scan further clarified grade 3A right cavernous sinus invasion with superiorly displaced optic chiasm, grade 1 invasion in the left cavernous sinus, and a compressed pituitary infundibulum. Her laboratory results were significant for mildly elevated prolactin at 39.2 ng/ml and reduced IGF-1 at 105 ng/ml but were otherwise within normal limits. Surgical resection was planned; however, three days later, she experienced worsening headache, a new inability to open her right eye, right facial pain in a V2/V3 distribution, and near-complete right oculomotor nerve palsy with a relative afferent pupillary defect. Due to rapidly worsening symptoms, a recommendation for urgent surgical intervention was made.
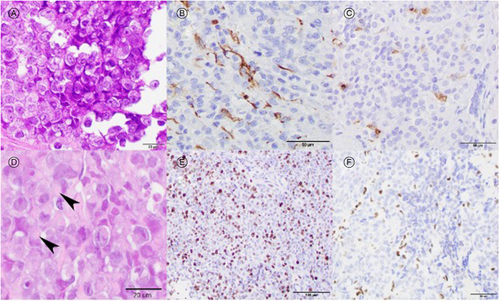
Preoperative MRI sella pituitary confirmed an enlarging mass measuring 3.0 cm × 1.5 cm × 2.0 cm with edema in the optic chiasm (). She underwent transsphenoidal endoscopic resection with abdominal fat grafting. Surgical pathology revealed a malignant tumor of the pituitary gland consisting of large cells with rounded to slightly irregular nuclei, prominent nucleoli, and abundant clear cytoplasm with occasional cells showing eccentric nuclei and eosinophilic intracytoplasmic inclusions with rhabdoid morphology. Immunostaining demonstrated polyimmunophenotypic protein expression with diffuse reactivity for vimentin, focal GFAP reactivity, focal EMA reactivity, and complete loss of INI-1 protein expression in malignant cells. As a result, poorly differentiated metastatic neoplasms, choroid plexus carcinoma, and adult non-CNS mesenchymal neoplasms were excluded. Diagnostic considerations of primary CNS neoplasms with INI1 loss include ATRT, cribriform neuroepithelial tumors, and very rare examples of high-grade gliomas or transformed glioneuronal tumors [Citation13–16]. The presence of prominent rhabdoid cytology, polyimmunophenotypic protein expression, lack of prominent GFAP expression, and absence of cribriform architecture confirmed the diagnosis of ATRT (). The ki67 proliferative index was ∼70%. Molecular sequencing confirmed biallelic inactivation of SMARCB1 and ATRT-SHH subgroup features. ATRT-SHH are characterized by overexpression of proteins in the SHH pathway in addition to compound heterozygous SMARCB1 point mutations, as seen in this case.
Figure 1. MRI brain T1 post-contrast axial images of the patient demonstrating a heterogeneously enhancing right anterior pituitary mass with mild to moderate suprasellar extension and involvement of the right cavernous sinus (yellow arrows). Images at presentation (A), at readmission four days later (B), and nine days after initial presentation pre-operatively (C).
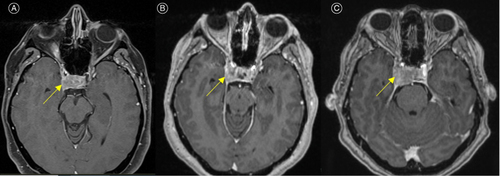
Figure 2. Histologic examination showing an undifferentiated malignant embryonal neoplasm comprised of large pale epithelioid cells with prominent nucleoli (A, H&E 600×). Neoplastic cells showing polyimmunophenotypic protein expression with focal GFAP staining (B, 400×) and focal EMA staining (C, 400×). Malignant cells have occasional characteristic rhabdoid cytologic features with eccentric nuclei and dense eosinophilic fibrillary inclusions (D, arrowheads, 600×). The ki67 proliferative index was ∼70% (E, 200×). Staining for INI-1 protein showing loss of nuclear immunoreactivity in malignant cells indicating inactivation of SMARCB1 (F, 400×).
The postoperative period was complicated by diabetes insipidus and central hypothyroidism, which were treated with desmopressin and levothyroxine, respectively. Postoperative MRI revealed small-volume residual disease within the sella and cavernous sinus.
Within a week, she developed a worsening headache, left vision loss, and left facial pain. MRI revealed an interval increase in thickness of enhancing soft tissue along the right dorsum sellae and prepontine cistern, consistent with disease progression. A planned chemotherapy regimen consisting of four cycles of high-dose methotrexate (3.5 mg/m2), vincristine (1 mg/m2), cisplatin (75 mg/m2), and cyclophosphamide (1.5 g/m2) with concurrent administration of dexamethasone was initiated.
She experienced symptomatic improvement over her chemotherapy cycles. However, her chemotherapy course was complicated by pulmonary emboli, multiple episodes of neutropenic fever with negative infectious workup, and peripheral neuropathy that resulted in the discontinuation of vincristine after cycle three. Prior to stem cell collection, she underwent complete CNS imaging showing no evidence of definitive residual disease or metastases, and cerebrospinal fluid analysis was normal including negative cytology. Stem cells were collected and focal RT (54 Gy in 30 fractions) was initiated. Following the successful completion of chemoradiation therapy, she underwent ASCT over two sessions on days 0 and 1, with preceding administration of high-dose etoposide (500 mg/m2) and thiotepa (300 mg/m2) on days -6, -5, and -4.
Currently, she remains in stable condition with no clinical or radiographic evidence of disease progression 20 months post-ASCT and 27 months since her initial diagnosis.
4. Discussion
Given its rarity in the adult population, treatment of adults with ATRT is unfortunately limited to non-standard regimens adapted from those used in the pediatric population. Based on a small number of trials, multimodal therapy with surgery, chemotherapy, and/or RT is generally considered the most effective approach. While the relatively higher incidence of ATRT among children has allowed for the more formal investigation of therapeutic strategies in this population, there is not yet enough evidence for a consensus to be reached. Chemoradiation regimens remain varied, especially with regard to the use of adjuvant ASCT. To date, only a handful of prospective trials have incorporated ASCT [Citation17].
First done in 2008 by Gardner et al., the Head Start (HS) trial consisted of two variations of the same regimen [Citation18]. This regimen, common to both HS I and II, included induction with five cycles of cisplatin, vincristine, cyclophosphamide, and etoposide, followed by consolidation with carboplatin, thiotepa, etoposide, and, finally, ASCT. The regimen used by HS II differed only in its addition of high-dose methotrexate to the induction regimen. A total of 13 pediatric patients, with ages ranging from 4 to 52 months, were enrolled. Three of those in HS II achieved long-term survival – unfortunately, none of those in HS I ultimately survived. HS III, published in 2014 by Zaky et al. as another variation of the original HS I regimen, incorporated RT post-consolidation [Citation19]. A total of 19 pediatric patients, with ages ranging from 0 to 32 months, were enrolled. Two of the three patients who completed induction and proceeded to consolidation were alive at the time of publication. The three-year overall survival for the group was 26 ± 10%.
More recently, ACNS0333, a Phase III clinical trial published in 2020 by Children's Oncology Group, similarly employed a multimodal approach that involved treatment with surgical resection followed by chemoradiation therapy [Citation20]. A total of 70 pediatric patients (ages 2–165 months) were enrolled, with 65 ultimately being evaluable. The chemotherapy regimen had significant overlap with that originally described in HS I and consisted of two cycles of vincristine, methotrexate, etoposide, cyclophosphamide, and cisplatin followed by consolidation with three cycles of carboplatin, thiotepa, and ASCT. Following consolidation, focal RT at either 50.4 Gy or 54 Gy was administered, with higher doses reserved for those >36 months of age. Four-year overall survival was 43%.
A year later SJMBO3, another Phase III clinical trial published by Gajjar et al. added further nuance to these treatment regimens [Citation21]. A total of 22 patients, with ages ranging from 3 to 21 years, were enrolled. They were risk-stratified into different regimens according to the presence of metastasis and the size of residual disease post-resection. Patients received craniospinal irradiation (CSI) along with focal RT to both primary and metastatic sites, followed by four cycles of cisplatin, vincristine, cyclophosphamide, and ASCT. Average-risk patients had a five-year overall survival of 81.8 ± 11%, significantly higher than the 18.2 ± 9.5% seen in high-risk patients.
Given the absence of a well-established standard of care for this rare, aggressive cancer and only after careful discussion with the patient in consultation with our institutional radiation oncologists and transplant physicians, our patient opted to pursue ASCT in the latter stage of the regimen following focal RT in order to coordinate stem cell collection and allow more recovery time from chemotherapy toxicity. She remains alive and well 20 months after undergoing high-dose chemotherapy and ASCT and 27 months post-diagnosis.
4.1. Literature review of ASCT in adults with ATRT
To further evaluate the use of ASCT in such a scenario, we reviewed the literature and ultimately identified four additional cases of adults with ATRT treated with ASCT as consolidation therapy, as outlined in . Ages ranged from 19 to 35 years with an equal proportion of males and females. All four tumors were supratentorial, with three located in the sellar region and one in the left frontal lobe. The extent of resection ranged from subtotal to complete for the three who underwent surgical intervention. Chemotherapy regimens predictably had significant overlap with those detailed above and placed particular emphasis on alkylator and vinca alkaloid agents during both induction and consolidation. Focal RT and/or CSI were delivered at variable doses. All four patients were long-term survivors, three of whom had no clinical or radiographic evidence of disease at final follow-up. One patient experienced progression and was deceased at the final follow-up; however, it is worth noting that she achieved a remarkable 10-year survival, representing a significant prolongation of life for a disease associated with a five-year survival rate of just 32.2% [Citation22].
Table 1. Case details of adult atypical teratoid rhabdoid tumor patients treated with autologous stem cell transplantation , summarized.
It is important to note that adults tend to experience increased toxicity with high-dose and myeloablative chemotherapy compared with young adults and children, though generally this increase in toxicity is felt to be an acceptable risk in conditions for which efficacy has been established. The patient in the case described experienced several toxicities due to both high-dose chemotherapy and myeloablative chemotherapy with ASCT, including peripheral neuropathy which she still has not fully recovered from 20 months postcompletion of her cancer treatment. Given the increased potential toxicity of high-dose chemotherapy in adults and the paucity of data on its efficacy in adults with ATRT, it is particularly important to thoroughly counsel patients about the risks of such treatment regimens. As a whole, this limited body of data suggests a potential future role for ASCT in adults. Ultimately, more rigorous study is needed to support this notion. The University of Minnesota is home to an ongoing trial exploring the use of ASCT after conditioning carboplatin and thiotepa in adults with CNS tumors, including ATRT [clinicaltrials.gov, NCT01505569]. With an estimated completion date of March 2025, this study may help us better define the current and potential future applications of ASCT for this rare and deadly disease.
5. Conclusion
Although a paucity of data exists regarding the use of ASCT in adults with ATRT, associated outcomes in children have been encouraging and have spurred continued examination of its potential utility in the adult population. The experience of the patient in the case described adds to a small body of literature suggesting a role for ASCT in the management of these rare and aggressive tumors.
Financial disclosure
The authors have no financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–820. doi:10.1007/s00401-016-1545-1
- Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro-oncology. 2021;23(8):1231–1251. doi:10.1093/neuonc/noab106
- Zin F, Cotter JA, Haberler C, et al. Histopathological patterns in atypical teratoid/rhabdoid tumors are related to molecular subgroup. Brain Pathol. 2021;31(5):e12967. doi:10.1111/bpa.12967
- Kanoto M, Toyoguchi Y, Hosoya T, Kuchiki M, Sugai Y. Radiological image features of the atypical teratoid/rhabdoid tumor in adults: a systematic review. Clin Neuroradiol. 2015;25(1):55–60. doi:10.1007/s00062-013-0282-2
- Broggi G, Gianno F, Shemy DT, et al. Atypical teratoid/rhabdoid tumor in adults: a systematic review of the literature with meta-analysis and additional reports of 4 cases. J Neurooncol. 2022;157(1):1–14. doi:10.1007/s11060-022-03959-z
- Ostrom QT, Chen Y, M de Blank P, et al. The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001–2010. Neuro-oncology. 2014;16(10):1392–1399. doi:10.1093/neuonc/nou090
- Nicolaides T, Tihan T, Horn B, Biegel J, Prados M, Banerjee A. High-dose chemotherapy and autologous stem cell rescue for atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol. 2010;98(1):117–123. doi:10.1007/s11060-009-0071-6
- Gidwani P, Levy A, Goodrich J, Weidenheim K, Kolb EA. Successful outcome with tandem myeloablative chemotherapy and autologous peripheral blood stem cell transplants in a patient with atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol. 2008;88(2):211–215. doi:10.1007/s11060-008-9553-1
- Paolini MA, Kipp BR, Sukov WR, et al. Sellar region atypical teratoid/rhabdoid tumors in adults: clinicopathological characterization of five cases and review of the literature. J Neuropathol Exp Neurol. 2018;77(12):1115–1121. doi:10.1093/jnen/nly091
- Dardis C, Yeo J, Milton K, et al. Atypical teratoid rhabdoid tumor: two case reports and an analysis of adult cases with implications for pathophysiology and treatment. Front Neurol. 2017;8:247. doi:10.3389/fneur.2017.00247
- Nobusawa S, Hirato J, Sugai T, et al. Atypical teratoid/rhabdoid tumor (AT/RT) arising from ependymoma: a type of AT/RT secondarily developing from other primary central nervous system tumors. J Neuropathol Exp Neurol. 2016;75(2):167–174. doi:10.1093/jnen/nlv017
- De Amorim Bernstein K, Sethi R, Trofimov A, et al. Early clinical outcomes using proton radiation for children with central nervous system atypical teratoid rhabdoid tumors. Int J Radiat Oncol Biol Phys. 2013;86(1):114–120. doi:10.1016/j.ijrobp.2012.12.004
- Hasselblatt M, Oyen F, Gesk S, et al. Cribriform neuroepithelial tumor (CRINET): a nonrhabdoid ventricular tumor with INI1 loss and relatively favorable prognosis. J Neuropathol Exp Neurol. 2009;68(12):1249–1255. doi:10.1097/NEN.0b013e3181c06a51
- Gessi M, Japp AS, Dreschmann V, et al. High-resolution genomic analysis of cribriform neuroepithelial tumors of the central nervous system. J Neuropathol Exp Neurol. 2015;74(10):970–974. doi:10.1097/NEN.0000000000000239
- Kleinschmidt-DeMasters BK, Alassiri AH, Birks DK, Newell KL, Moore W, Lillehei KO. Epithelioid versus rhabdoid glioblastomas are distinguished by monosomy 22 and immunohistochemical expression of INI-1 but not claudin 6. Am J Surg Pathol. 2010;34(3):341–354. doi:10.1097/PAS.0b013e3181ce107b
- Bertrand A, Rondenet C, Masliah-Planchon J, et al. Rhabdoid component emerging as a subclonal evolution of paediatric glioneuronal tumours. Neuropathol Appl Neurobiol. 2018;44(2):224–228. doi:10.1111/nan.12379
- Alva E, Rubens J, Chi S, et al. Recent progress and novel approaches to treating atypical teratoid rhabdoid tumor. Neoplasia (New York, N.Y.). 2023;37:100880. doi:10.1016/j.neo.2023.100880
- Gardner SL, Asgharzadeh S, Green A, Horn B, McCowage G, Finlay J. Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatric Blood Cancer. 2008;51(2):235–240. doi:10.1002/pbc.21578
- Zaky W, Dhall G, Ji L, et al. Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: the Head Start III experience. Pediatric Blood Cancer. 2014;61(1):95–101. doi:10.1002/pbc.24648
- Reddy AT, Strother DR, Judkins AR, et al. Efficacy of high-dose chemotherapy and three-dimensional conformal radiation for atypical teratoid/rhabdoid tumor: a report from the Children's Oncology Group Trial ACNS0333. J Clin Oncol. 2020;38(11):1175–1185. doi:10.1200/JCO.19.01776
- Upadhyaya SA, Robinson GW, Onar-Thomas A, et al. Relevance of molecular groups in children with newly diagnosed atypical teratoid rhabdoid tumor: results from prospective St. Jude multi-institutional trials. Clin Cancer Res. 2021;27(10):2879–2889. doi:10.1158/1078-0432.CCR-20-4731
- National Cancer Institute. Atypical Teratoid Rhabdoid Tumor (ATRT) Diagnosis and Treatment. The Website of the National Cancer Institute. 2023. Retrieved September 4, 2023, from https://www.cancer.gov/rare-brain-spine-tumor/tumors/atrt