
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Nanofibrillated cellulose (NFC) has been successfully esterified by lactic acid (LA) in the presence of HCl catalyst in an aqueous medium using a simple microwave heating process. The degree of substitution (DS) of ester groups on modified NFC (mNFC) was quantified through a systematic characterization consisting of titration, NMR, and XPS, revealing a consistent trend in the levels of DS in mNFC. The reaction parameters of the microwave heating process including the ratio between NFC and LA (1:10), amount of catalyst (5 wt%), energy input and time, have been optimized, achieving a DS of 0.66 in mNFC with a typical power of 800 Watts in 1 min only. The TEM and XRD results confirmed that the structure and characteristics of the nanofibrillated fibers were preserved following the process. Finally, the improved dispersibility of mNFC with high DS in low polarity solvents and polylactic acid (PLA) matrix was validated.
Graphical Abstract
1. Introduction
Cellulose, a linear carbohydrate polymer made up of glucopyranose repeating units, stands out as one of the most abundant renewable and non-toxic biomaterials on Earth [Citation1]. Its exceptional mechanical properties make it a highly attractive candidate for use as a filler and reinforcement in composites and nanocomposites. Among the various forms of cellulose nanofibers, nanofibrillated cellulose (NFC) is of particular interests due to its high aspect ratio, with a typical diameter ranging from 4 nm to 20 nm and lengths of approximately 500–2000 nm [Citation2]. Enhanced properties of NFC/polymer nanocomposites at low filler contents have been widely reported without adversely affecting other properties including material transparency [Citation3]. Nevertheless, the main obstacle to the utilization of NFC in nanocomposite technology arises from the poor dispersibility of the polar cellulose nanofibers in typically hydrophobic polymer matrices. Chemical modification is often required to increase compatibility of the two phases of composites and, hence, the dispersion of NFC [Citation4].
The hydroxyl groups presented in NFC offer excellent sites for modification. Therefore, NFC can be chemically modified to provide tailored dispersibility but also new functional properties. A wide variety of chemical reactions can be utilized to modify the surface hydroxyl groups of nanocellulose including oxidation, carbamation, etherification, and esterification [Citation5]. Among these reactions, esterification is the most commonly used modification due to its simplicity and versatility. This reaction introduces an ester functional group onto the surface of cellulose through the condensation of esterifying reagents with hydroxy groups. The mechanism involves the classic Fischer esterification reaction, wherein the carbonyl group on the carboxylic acid is activated using a strong acid as catalyst [Citation6].
In terms of processing, current research includes the esterification of NFC utilizing the reflux method at 80 °C for 2–8 h, with a degree of substitution (DS) between 0.2 and 1 [Citation7,Citation8]. The choice of organic solvents and esterifying agents has a significant impact on both the NFC surface derivatization and its dispersibility in solvents. Dry milling is a non-solvent processing method that involves directly (ball) milling solid cellulose with an esterifying agent. Although no organic solvent is involved in this approach, the energy consumption of this processing is often considerably intensive [Citation9]. Another route to esterify NFC surfaces involves sonication, which can achieve a maximum DS of 0.5 in modified NFC (mNFC) [Citation10,Citation11]. Unfortunately, it was demonstrated that the sonication process can induced undesirable damage to the cellulose nanofiber structures due to its extremely high shear stress, while the scaling up of the sonication process also poses difficulties in many applications [Citation10].
The use of microwave irradiation as a nonconventional heating source has been reported to achieve higher yields and product purities, as well as reduced reaction times due to improved selectivity in the esterification of various carboxylic acid types. Satgè et al. [Citation12] and Semsarilar et al. [Citation13] have reported the esterification of cellulose with acyl chloride derivatives in N,N-dimethylacetamide/lithium chloride and N,N-dimethylaminopyridine as a catalyst using microwave irradiation, where the reaction time was reduced from several hours to minutes. The esterified cotton cellulose nanowhiskers (CNW’s) by vinyl acetate and vinyl cinnamate in dimethylformamide with potassium carbonate as catalyst under microwave activation were studied by Sèbe et al. [Citation14]. These studies demonstrated that microwave could be a time- and energy-saving method, allowing the completion of the esterification of nanocellulose in a single step. However, very limited studies can be found in using microwave-assisted processes for nanocellulose modification.
In this work, we used a microwave-assisted approach as an environmentally benign and energy efficient process to modify NFC via esterification reaction. A novel system consisting of lactic acid (LA) esterifying reagent, hydrochloric acid (HCl) catalyst and water as a solvent was developed for a rapid modification of NFC using microwave heating. We systematically studied the influences of the NFC:LA ratio and amount of catalyst as well as microwave’s power and reaction time on the DS value of the resultant mNFC samples. Furthermore, nanocomposite films of NFC/PLA and mNFC/PLA were developed to examine the dispersibility of present mNFC in a hydrophobic polymer matrix.
2. Materials and methods
2.1. Materials
NFC was purchased from Cellulose Lab (Canada) in the form of a slurry with a solid content of 3 wt%, prepared by a supermass colloider. LA (90%), ethanol (98%), and analytical grade HCl (37%) were purchased from Union Science (Thailand). PLA pellets (Ingeo 4043D) film grade was supplied by NatureWorks, LLC (USA). Chemical solvents (methanol, acetone, chloroform, and toluene) were purchased from RCI Labscan (USA). All chemicals were used as received without further purification.
2.2. Preparation of mNFC using microwave-assisted process
NFC was modified by using a commercial microwave (MS23K3513AW/ST, Samsung, Korea), according to the process illustrated in . 50 g of NFC suspension in water (0.5 wt%) was mixed with LA and HCl prior to homogenization and microwave treatment. LA contains a carboxylic functional group which served as the modifying agent, while HCl was used as a catalyst to increase the rate of esterification. The ratio of NFC:LA, amount of HCl as well as the microwave’s power and time were studied (see ) in order to determine the best processing conditions. Before microwave heating, the initial suspension’s weight was recorded. At the end of the reaction, the remaining suspension’s weight as well as temperature were measured and then mNFC was collected, freeze-dried, and kept in a sealed plastic bag for further analysis.
Figure 1. Schematic diagram of NFC esterification using simple microwave-assisted process.
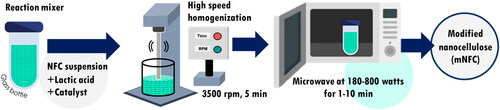
Table 1. Reaction conditions and parameters to modify NFC using simple microwave heating.
2.3. Preparation of PLA, PLA/NFC, and PLA/mNFC nanocomposite films
PLA pellets were dried in an oven at 60 °C overnight before use. The PLA and nanocomposite films were prepared by solution casting, obtaining a final thickness between 30 and 50 µm. Firstly, the PLA solution was prepared with 5 wt% polymer in chloroform solvent. NFC and mNFC were redispersed in chloroform solvent at 0.1 wt% concentration. Next, the PLA solution and nanocellulose suspension were blended together using a high-speed homogenizer (WISE TEG, HG-15D, Germany) at 3500 rpm for 5 min and then a sonication probe (VCX 750, Sonics & Materials Inc., Newtown, CT, USA) was used to enhance the dispersion of NFC and mNFC in PLA solution at the amplitude of 30% for 30 min at room temperature before casting in a glass Petri dish. The sample was left at room temperature for 24 h to evaporate chloroform. After that, the solidified film samples were further dried in an oven at 60 °C for 3 h before peeling off. The loading content of NFC and mNFC were fixed at 3 wt% in PLA polymer matrix.
2.4. Characterization
2.4.1. Determining DS by titration method
The freeze-dried mNFC sample (0.5 g) was added to 40 mL of aqueous ethanol (70%) and stirred with a magnetic bar at 400 rpm for 30 min. Stirring was continued for 48 h at 50 °C, after addition of 20 mL of a 0.5 M NaOH solution. Subsequently, any remaining unreacted NaOH was back-titrated with a 0.5 M HCl solution and the ester content (EC) was determined using the following equation:
where Va and Vb represent the volumes (mL) of a NaOH solution added to sample and blank, respectively. Vd and Vc represent the volumes (mL) of HCl added to sample and blank, respectively. Nb and Na stand for the concentration (in molarity) of NaOH and HCl solutions, respectively. Mw is the molecular weight of the acid reagent, while G signifies the weight (in gram) of the sample. DS1 was then determined by the following equation:
where 162.14 is the molecular weight of anhydroglucose monomer unit [Citation15].
2.4.2. Chemical structure analysis
Fourier transform infrared spectra (FTIR) of NFC and esterified NFC samples were acquired on a Thermo Scientific, Nicolet iS50 (USA) equipped with a Universal Attenuated Total Reflectance accessory. The study range spanned from 400 to 4000 cm−1, at a resolution of 8 cm−1. A total of 32 scans were recorded for each sample.
Liquid-state nuclear magnetic resonance (NMR) spectroscopy was used to detect the 1H-NMR spectra of mNFC. 5 mg of dried sample was dissolved in DMSO-d6 (99.8%). The experiment was conducted at 25 °C, using a Bruker ADVANCE NEO:500 MHz (Germany), equipped with a 5 mm probe operating at 399.959 MHz. The one-pulse sequence was used, with a 30° pulse, a relaxation delay of 60 s, a spectral width of 4500 Hz and 16K data points for acquisition. The positions of the peaks at chemical shift 2.49 ppm were referred to the residual solvent peak of DMSO-d6. Moreover, the DS from 1H-NMR (DS2) was evaluated using the following equation [Citation16]:
X-ray photoelectron spectroscopy (XPS) experiments were carried out using an AXIS ULTRA DLD, Kratos (UK) equipped with an aluminum Ka X-ray source (1486.6 eV) and operating at 15 kV under a current of 8 mA. Samples were placed in an ultra-high-vacuum chamber with electron collection by a hemispherical analyzer at an angle of 90°. XPS was performed on the dried powder of NFC and mNFC samples. From the XPS spectra, the DS can be calculated directly from the areas of the ester (C4 or C = O) peaks using the following equation [Citation17]:
where %C4 is the intensity of the signal attributed to O–C = O moieties, M(AGU) is the molecular weight of an anhydroglucose unit (162.14 g/mol), M(graft) is the molecular weight of the grafted moieties of lactic acid (90.08 g/mol) and M(C) is the molecular weight of one carbon atom.
2.4.3. X-ray diffraction (XRD)
XRD (X’Pert Pro MPD, UK) was used to determine the crystal structure and crystallinity of nanocellulose samples. The XRD patterns were recorded in a 2θ angle range of 5° to 45° at a step width of 0.02° with a scan speed of 2°/min. The crystallinity index (CrI) of the samples was calculated using Segal’s equation below:
where I002 is the maximum intensity of the 002 lattice diffraction at 2θ = 22° and Iam is the intensity of amorphous cellulose at 2θ =18°.
2.4.4. Thermal analysis
A differential scanning calorimeter (DSC) was used to analyze thermal properties of NFC and mNFC samples performing on a Mettler Toledo, DSC3+ (Spain). Around 5 mg of sample powder was introduced in an aluminum pan and then subjected to a heating ramp from room temperature to 275 °C (rate of 10 °C/min) under a nitrogen atmosphere to avoid thermo-oxidative reactions. A first heating/cooling cycle from room temperature to 100 °C (heating rate of 10 °C/min and cooling rate of 20 °C/min) was performed to clear the thermal history of the samples. The thermal transitions were recorded on the second heating-cooling cycle at 10 °C/min.
Thermal stability of nanocellulose samples were determined by a thermogravimetric (TGA) analyzer, Mettler Toledo, 851e (USA). The freeze-dried samples (about 10 mg) were placed in an aluminum pan. Each sample was run under an N2 atmosphere by using a flow rate of 10 mL/min, and heated from room temperature to 700 °C at 10 °C/min.
2.4.5. Dispersion behavior observation
NFC and mNFC in water suspension was exchanged with different solvents starting from methanol, ethanol, acetone, chloroform and toluene. To change the solvent, the mixture was homogenized for 5 min each time. The experiment was done with three replicates.
2.4.6. Fiber and film morphology
To observe cellulose nanofiber’s morphology, a 0.001 wt% suspension of both NFC and mNFC suspension in water was sonicated in a sonicating bath with power of 73 W (Crest Ultrasonics, 690HTAE, NY, USA) for 5 min, then a 2% uranyl acetate solution was added for contrast enhancement in transmission electron microscope (TEM) images. A drop of the suspension was deposited on a copper grid with Formvar film (200 mesh-Ted Pella) and allowed to dry in a desiccator for 24 h before examination at the acceleration voltage of 80 kV (Hitachi, HT7700, Japan).
The surface morphology and fractured surface (cross-section) of film samples was observed at an accelerating voltage of 5 kV by using FE-SEM (TESCAN, MIRA, USA). The films were fractured in liquid nitrogen for the cross-sectional images. All samples were coated with gold before being examined.
2.5. Statistical analysis
The results were obtained from experiments conducted in triplicate and are presented as the mean ± standard deviation (SD). Statistical analysis was performed using SPSS statistics software (IBM SPSS Statistics for Windows, Version 22.0. Armonk, NY, IBM Corp). Statistical significance was assessed using the Student’s t-test with a significance level set at p < 0.05.
3. Results and discussion
3.1. Optimizing the reaction conditions to modify NFC
For the heterogeneous reaction between NFC and LA, catalyzed by HCl, the influences of the NFC:LA ratio and amount of HCl on the DS values of mNFC was studied under constant microwave power and time, as shown in . The DS values were observed to increase as the ratio of NFC:LA increased from 1:5 to 1:10 (5LA-5HCl and 10LA-5HCl). Based on the esterification reaction mechanism, an increase in the feed ratio increases the concentration of the mixed acid anhydride in the reaction system, thereby increasing the probability of reacting with the hydroxyl groups present in NFC. However, when the NFC:LA ratio was set at 1:15 (15LA-5HCl), the DS value decreased. This reduction can be attributed to an excess of LA reacting with the hydroxyl groups of NFC, eventually reaching a saturation point, hampering further increases in DS [Citation16]. Furthermore, variations in the amount of catalyst were observed at 3, 5, and 7 wt%, leading to alterations in the DS values. When utilizing 3 wt% and 5 wt% of HCl as catalysts, the DS increased from 0.14 to 0.66. This enhancement underscores the fundamental role of the catalyst in expediting the chemical reaction. Conversely, at 7 wt% of HCl, the DS experienced a decrease. This decrease could be attributed to an excessive amount of acid catalyst, which could potentially disrupt the esterification reaction and lead to the occurrence of a transesterification reaction [Citation17]. The optimal NFC:LA ratio and catalyst amount were then identified as follows: NFC:LA molar ratio of 1:10 and an HCl catalyst of 5 wt% relative to the weight of LA.
Table 2. Effects of the NFC:LA molar ratio and amount of HCl on the degree of substitution (DS) of mNFC.
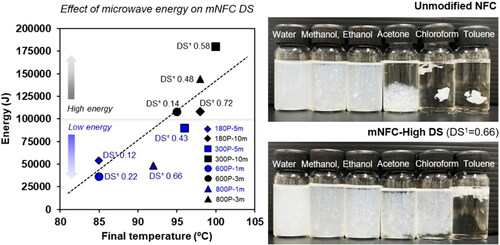
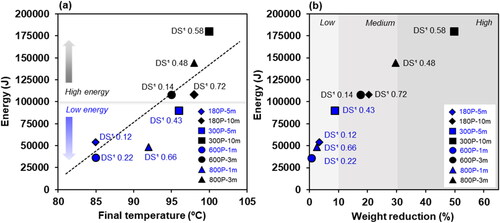
Another important parameter for the mNFC esterification reaction is the microwave’s power and time. Microwave radiation induces temperature changes for a chemical reaction depending on the microwave energy applied, which can be regulated by adjusting the microwave power and duration. In this study, the final temperature of the mNFC suspension in the reaction bottle after microwave irradiation linearly increased with the increasing microwave power, as expected (). The minimum energy 600 P-1m condition led to the final temperature of around 85 °C and was raised to 100 °C in the maximum energy 300 P-10m condition. Most research reported that an increasing temperature during the esterification tends to increase the substitution efficiency [Citation18–20]. Previous studies have noted that electromagnetic waves exhibit selective heating characteristics, primarily affecting highly polar molecules, such as water molecules [Citation21]. In the lower energy range, below 100,000 J (180 P-5m, 600 P-1m, 800 P-1m, and 300 P-5m), the gradual increase in heat can be attributed to the incremental vibration of water molecules at various positions, leading to the gradual release of low-pressure energy during the reaction. In this low energy range, the weight reduction of mNFC suspension following the process, from evaporation of water and other small molecules, remained relatively low (less than 10%) (). The DS of the mNFC prepared with low microwave energy range (below 100,000 J) was in a range of 0.12 to 0.66. When increasing the energy level to over 100,000 J (180 P-10m, 600 P-3m, 800 P-3m and 300 P-10m), the weight reduction of mNFC suspension exceeded 10% and ranged up to 50%. This can be attributed to the rapid vibration of water molecules, which causes a higher pressure release, even explosive, ejecting the solid nanocellulose from the reaction bottle. Nonetheless, the DS of the obtained mNFC was in the range of 0.14 to 0.72. These DS values were not increased with a further increasing of the energy input. The challenges arising from steam explosion, leading to the loss of mNFC products, made it considerably intricate to manage the chemical reaction rate as well as to maximize the DS of mNFC. Furthermore, upon completion of the high energy process (above 100,000 J), some black spots of degraded NFC were observed in certain areas of the reaction bottle. This suggests that the application of high energy in microwave processing can lead to an inadequate homogenization of the mNFC esterification in combination with the loss of nanocellulose sample. Next, to better investigate the impact of varying DS levels in the resulting mNFC, three samples from low energy conditions were chosen as follows: mNFC-Low DS (DS1 0.22; 600 P-1m), mNFC-Med DS (DS1 0.43; 300 P-5m), and mNFC-High DS (DS1 0.66; 800 P-1m).
Figure 2. The Relationship between microwave energy and final reaction’s temperature (a) and weight reduction of mNFC suspension after microwave heating (b); Low weight reduction (less than 10%) denotes the loss of solely water molecules, but medium to high weight reduction (>10%) indicates the loss of water molecules as well as solid mNFC ejected from the reaction bottle due to steam explosion.
FTIR was utilized to qualitatively ascertain the chemical structure of mNFC and confirm the success of the microwave-assisted esterification of NFC. From the FTIR spectra of mNFC-Low DS, mNFC-Med DS, and mNFC-High DS (), the emergence of a carbonyl (C=O) stretch at 1740 cm−1 is evident, a feature absent in unmodified NFC. Obviously, the highest intensity of carbonyl is shown in the mNFC-High DS sample with the highest DS for the ester side chain.1H liquid state NMR spectroscopy was as well used to examine the structure of mNFC. The NMR spectra of mNFC-High DS and its proposed modified chemical structure are shown in . The signals exhibiting chemical shifts at 4.66, 3.77, 3.55, and 3.05 ppm correspond to the peak positions of hydroxyl protons and methylene protons in the AGU (Anhydroglucose Unit) segments of unmodified NFC. Meanwhile, the signals at 5.46, 5.40, and 4.31 ppm are attributed to the protons of residual hydroxyl in NFC [Citation22]. Notably, the strong signal peak of mNFC-High DS was observed at 1.25 ppm, which corresponds to the terminal methyl protons (Hb) of the lactic acid side chain, and the peaks at 4.20 ppm were assigned to the terminal methine protons (Ha) of the lactic acid side chain [Citation23]. These characterizations collectively support the surface esterification of the cellulose nanomaterial. However, it is important to mention that the FTIR technique offers a depth of analysis superior to 1 mm, while the 1H liquid-state NMR spectra indicate only peaks from a very small sample, around 0.5 mg [Citation24].
Figure 3. Chemical structure analysis: NFC and mNFC with different DS levels (mNFC-Low DS, mNFC-Med DS, and mNFC-High DS) via; (a) FTIR, (b) 1H liquid-state NMR, and (c-d) XPS spectra.
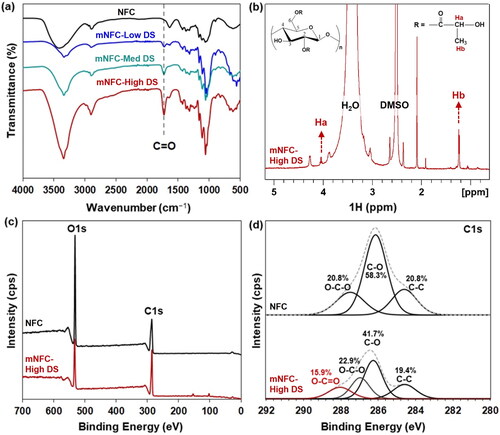
To directly verify the surface grafting on mNFC, XPS was employed. displays the XPS wide and high-resolution spectra of NFC and mNFC-High DS after grafting. In all cases, the main peaks are detected at 285 and 532 eV, corresponding to C and O atoms, respectively [Citation25]. The reduction in the intensity of the O atom peak in the mNFC-High DS sample clearly indicates the attachment of the alkyl or oligomer chain of lactic acid onto the NFC structure [Citation26]. Additionally, the deconvolution results of the C1s signals for unmodified NFC and mNFC-High DS are presented in . The peaks at 284.6, 286.1, and 287.4 eV in unmodified NFC were attributed to the C–C, C–O, and O–C–O structures, respectively. Notably, a new peak at 288.9 eV corresponding to O-C = O was observed in the mNFC-High DS sample, constituting 15.9% of the content. The presence of this peak indicates the generation of ester groups on the NFC surface as a result of esterification. Moreover, there was a slight reduction in the C–O content of mNFC-High DS to 41.7% as compared to NFC (58.3%). These results further confirm the successful esterification reaction between NFC and LA. The observed significant shift in the values of O-C-O functional group is probably attributed to differences in the chemical environment of the O-C-O functional group. The electron density surrounding this functional group appeared to be influenced by neighboring atoms, particularly the O-C = O groups, thereby giving rise to variations in the electron binding energy. Additionally, it is worth noting that the distinct electronegativities of functional groups within the mNFC structures may be contributed to shifts in binding energies [Citation27–30].
Besides the titration technique, the more advanced techniques of 1H liquid state NMR and XPS, used to quantify the DS value of the prepared mNFC, are reported in . The most common standard technique used is wet chemistry or titration (acidification and saponification). This classic technique is considered straight forward and inexpensive; however, this method is time-consuming and the results are prone to be influenced by variable end-points during the titration [Citation24]. Therefore,1H liquid state NMR and XPS, were also chosen to evaluate the DS of mNFC in this study. It was found that the DS results obtained from the NMR and XPS techniques presented a similar trend of low to high DS values as compared to those DS values from the titration technique. The different DS values obtained from each technique, however, are not surprising given the differences in the technique principles, sample preparation and formulations to calculate DS value, among others [Citation22,Citation25].
Table 3. Comparing the DS values of mNFC assessed from three different techniques.
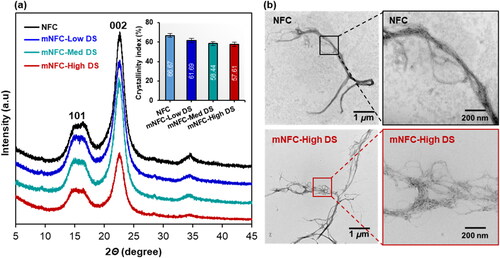
In , all samples displayed two main X-ray diffraction peaks at 2θ of 15.3° and 22.6°, corresponding to the typical diffraction patterns of cellulose type I [Citation20]. The crystallinity index of NFC was 69.8%. After modification, a decrease in the crystallinity index was observed in mNFC samples with different DS levels: 61.1% for mNFC-Low DS, 58.4% for mNFC-Med DS, and 57.6% for mNFC-High DS. This reduction can be attributed to the ester substitution disrupting the hydrogen bonding and diminishing the crystalline structure of NFC [Citation31]. Observing the cellulose nanofiber’s morphology through TEM (), native NFC exhibited tightly entangled nanofibrils due to intensive hydrogen bonding interactions. The nanofibrils in the mNFC-High DS sample exhibited a more fibrillated or separated morphology, likely due to reduced interactions among the fibrils as a result of the ester substitution, without compromising much the fibrils length. According to TEM image measurements, both NFC and mNFC-High DS have similar lengths in several micrometers but differ in diameter. NFC has an average diameter of 47.6 ± 14.8 nm and mNFC-High DS is 16.89 ± 9.95 nm. Sonication-assisted modification process in a prior study [Citation10], showed that the mNFC (maximum DS of 0.5) had been broken up into small fragments as a result of the high shearing stress of sonication. This suggests that the modification of NFC through microwave processing not only enables an effective substitution of ester groups but also largely preserves the crystal structure and nanofiber characteristics of mNFC.
Figure 4. (a) X-ray diffraction patterns and crystallinity index (embedded in top right corner) of NFC and mNFC; (b) TEM images of NFC and mNFC-High DS.
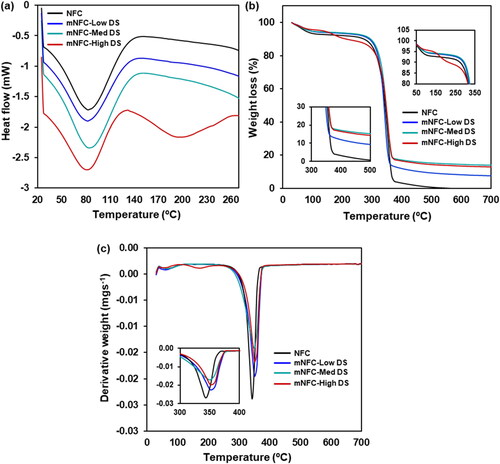
Aside from the structural changes, the physicochemical properties of the NFC were modified after esterification, as confirmed by DSC analysis (). The spectra of NFC, mNFC-Low DS, and mNFC-Med DS exhibited a single endothermic peak around 80–100 °C, attributed to the gradual evaporation of water absorbed by the nanofibers possibly during handling and storage in a plastic zip bag. Nanocellulose displays a strong affinity for hydroxyl-bearing systems, particularly small molecules like water, which aggregate and absorb onto the external surface of nanofibrils [Citation18]. Conversely, in the case of mNFC-High DS, two distinct peaks were observed. The initial endothermic shift, located at 80 °C, corresponds to the evaporation of adsorbed water molecules, analogously to NFC. The second peak visible in the DSC spectrum is likely associated with the thermal degradation of the ester side chain. This hypothesis is supported by the TGA analysis results (). A two-stage degradation pattern was discernible in the thermal degradation of NFC, mNFC-Low DS, and mNFC-Med DS. The initial stage entails moisture evaporation, followed by the decomposition of the chemical structure at around 300 °C. In contrast, the mNFC-High DS demonstrates a three-stage degradation pattern. It initiates with the dehydration of aliphatic hydroxy groups around 100 °C, followed by the cleavage of ester unit linkages around 200 °C, culminating in the decomposition of the ester oligomers [Citation32]. In comparison, the weight loss of all mNFC samples surpasses that of unmodified NFC. Previous research elucidated that after esterification, the char residue increased greatly due to the grafted ester units, indicating improved flame-retardant behavior of the esterified samples [Citation10].
Figure 5. Thermograms of NFC and mNFC with Low DS, Med DS, and High DS; (a) DSC, (b) TGA, and (c) DTG.
The dispersion of mNFC is of critical importance for its successful modification. The results could be seen in , all nanocellulose samples were well dispersed in water, methanol, and ethanol. The very strong polarity and hydrophilicity of NFC made them easily dispersed in polar solvents. On the other hand, NFC was poorly dispersed in acetone, chloroform, and toluene, whereas mNFC increased dispersion stability with increasing DS levels (High DS > Med DS > Low DS). This could be due to larger numbers of ester groups on the surfaces of these mNFC samples.
Figure 6. Dispersion of NFC and mNFC with different DS levels in solvents ranging in the order of decreasing polarity.

3.2. Nanocomposite film of PLA/mNFC-High DS
In order to investigate the potential of the mNFC as nanofiber reinforcement, nanocomposite films based on PLA, one of the most commonly used biopolymers, were prepared. The dispersibility of NFC and mNFC-High DS in PLA solution and the subsequent casted films were first observed (). It was obvious that mNFC-High DS dispersed in PLA solution and PLA matrix much better than unmodified NFC. This underline that the agglomeration of NFC filler in a hydrophobic polymer matrix can indeed be overcome by suitable chemical modification of NFC. After film casting and drying, the pure PLA film was highly transparent, showing the typical characteristic of amorphous PLA films [Citation33]. On the other hand, in both nanocomposite films, a reduction in transparency was clearly noticed, particularly in the PLA/NFC film. This suggested that NFC was strongly agglomerated, forming visible white spots, in the PLA matrix because of their differences in chemical structure and nature [Citation34]. The dispersion of mNFC-High DS in PLA, however, was greatly improved as well as the nanocomposite film’s consistency.
Figure 7. Photographs of PLA solution (pure PLA) and PLA/NFC solution and PLA/mNFC-High DS solution (a) and the subsequent casted films of the aforementioned solutions with thickness of 30–50 µm (b).

The surface morphology of films observed by SEM () showed that the pure PLA film’s surface was smooth and homogeneous. In contrast, the PLA/NFC film had a very rough surface, particularly in the areas where NFC agglomerates were present. The more hydrophobic mNFC-High DS exhibited an improved compatibility with the PLA matrix, showing a smoother surface and no visible mNFC-High DS on the nanocomposite film’s surface. Furthermore, large NFC agglomerates and voids were discovered inside the fractured surface of the PLA/NFC film due to their poor compatibility (). The fractured surface of the PLA/mNFC-High DS film was not as smooth as that of pure PLA, but mNFC-High DS could disperse within the PLA matrix, forming less voids. However, based on visual inspection of the solution and SEM results of the PLA/mNFC-High DS film, there is still a high possibility of mNFC agglomeration in some locations. It suggests that the DS levels of each mNFC sample prepared in this study may vary. This is most likely due to uneven heating generated by the oscillating pattern of microwave irradiation [Citation35], which results in inhomogeneous esterification of NFC at different locations of the reaction bottle.
Figure 8. SEM images of (a) film surface and (b) fractured surface morphology of pure PLA film and nanocomposite films of PLA/NFC and PLA/mNFC-High DS.

Conclusions
Nanofibrillated cellulose have been efficiently esterified with lactic acid (esterifying agent) and HCl (catalyst) in water using a simple microwave heating. In this study, the optimal reaction conditions for modifying NFC were found to be as follows: NFC:LA ratio, catalyst amount, microwave power, and time are all 1:10, 5% based on LA, 800 Watts, and 1 min, respectively. The optimized reaction led to mNFC with a maximum degree of substitution (DS) of 0.6. The use of high microwave energy (>100,000 J) resulted in steam explosion, which ejected solid mNFC from the reaction bottle, resulting in a significant loss of sample. The mNFC prepared using low microwave energy (<100,000 J), with three DS levels, mNFC-Low DS (0.2), mNFC-Med DS (0.4) and mNFC-High DS (0.6), were further analyzed using 1H NMR and XPS. Unlike mNFC prepared by other processing (e.g. sonication), TEM and XRD results confirmed that the nanofiber characteristics and crystal structure of the present mNFC were mostly preserved after microwave processing, while presenting a more fibrillated morphology. From DSC and TGA analysis, thermal properties of mNFC-High DS changed noticeably, owing to the high density of ester groups present on the surface of this mNFC. The mNFC-High DS sample could be sufficiently dispersed in low polar solvents (i.e. acetone and chloroform) and PLA matrix, verifying the success and efficiency of the NFC’s modification via simple microwave heating. However, in each modified sample, the resultant mNFC may exhibit a slightly varied DS levels due to uneven microwave irradiation. Our future research will focus on improving the homogeneity in nanocellulose modification, as well as the preparation and characterization of improved nanocomposite films.
Authors’ contributions
Supattra Klayya: Investigation, Methodology, Visualization, Writing - original draft, Writing - review & editing. Patcharee Pripdeevech: Formal Analysis, Validation, Writing - original draft, Writing - review & editing. Han Zhang: Methodology, Validation, Writing - review & editing. Emiliano Bilotti: Methodology, Validation, Writing - review & editing. Nattakan Soykeabkaew: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing - original draft, Writing - review & editing.
Acknowledgments
The authors are grateful for the Fundamental Fund (grant no. 672401035) supported by Thailand Science Research and Innovation (TSRI). We also would like to thank the Office of Postgraduate Studies and the Scientific and Technological Instruments Center, Mae Fah Luang University (Thailand) for the additional financial supports and facilities used in this research.
Disclosure statement
The authors report there are no competing interests to declare.
Additional information
Notes on contributors
Supattra Klayya
Supattra Klayya is a PhD student in Materials Innovation Program at the School of Science, Mae Fah Luang University, Thailand.
Patcharee Pripdeevech
Patcharee Pripdeevech is an Associate Professor in Chemistry at the School of Science, Mae Fah Luang University, Thailand. Her research focuses on essential oils, volatile chemicals, and active biodegradable films in the fields of chemistry and chemical biology.
Han Zhang
Han Zhang is a Senior Lecturer in Materials Science at the School of Engineering and Materials Science, Queen Mary University of London. His current research mainly focuses on the sustainable development of advanced composites and nanocomposites, including energy efficient sustainable manufacturing, integrated health monitoring and easy repairing functionalities for extended components’ life.
Emiliano Bilotti
Emiliano Bilotti is a Senior Lecturer in Multifunctional Polymer Composites at the Department of Aeronautics, Imperial College London. His recent research has been focused on on smart polymer composites (sensors, pyro-resistivity for self-regulating heater, actuation/shape programming), energy harvesting (organic thermoelectric, ferroelectric and piezoelectric) and energy storage (capacitors).
Nattakan Soykeabkaew
Nattakan Soykeabkaew is an Assistant Professor in Materials Science at the School of Science, Mae Fah Luang University, Thailand. Her current research focuses on cellulose-based material, nanocellulose, bionanocomposite, biomass utilization, and sustainable packaging. She currently serves as the director of Mae Fah Luang University’s Center for Innovative Materials for Sustainability (iMatS).
References
- Brinchi L, Cotana F, Fortunati E, et al. Production of nanocrystalline cellulose from lignocellulosic biomass: technology and applications. Carbohydr Polym. 2013;94(1):154–169. doi: 10.1016/j.carbpol.2013.01.033.
- Missoum K, Martoïa F, Belgacem MN, et al. Effect of chemically modified nanofibrillated cellulose addition on the properties of fiber-based materials. Ind Crops Prod. 2013;48:98–105. doi: 10.1016/j.indcrop.2013.04.013.
- Rhim JW, Park HM, Ha CS. Bio-nanocomposites for food packaging applications. Prog Polym Sci. 2013;38(10–11):1629–1652. doi: 10.1016/j.progpolymsci.2013.05.008.
- Cui Y, Kumar S, Kona BR, et al. Gas barrier properties of polymer/clay nanocomposites. RSC Adv. 2015;5(78):63669–63690. doi: 10.1039/C5RA10333A.
- Ghasemlou M, Daver F, Ivanova EP, et al. Surface modifications of nanocellulose: from synthesis to high-performance nanocomposites. Prog Polym Sci. 2021;119:101418. doi: 10.1016/j.progpolymsci.2021.101418.
- Joseph T, Sahoo S, Halligudi SB. Brönsted acidic ionic liquids: a green, efficient and reusable catalyst system and reaction medium for fischer esterification. J Mol Catal A Chem. 2005;234(1–2):107–110. doi: 10.1016/j.molcata.2005.03.005.
- Peng SX, Chang H, Kumar S, et al. A comparative guide to controlled hydrophobization of cellulose nanocrystals via surface esterification. Cellulose. 2016;23(3):1825–1846. doi: 10.1007/s10570-016-0912-3.
- Daud JB, Lee KY. Surface modification of nanocellulose. Handb Nanocel Cellul Nanocomp. 2017;1:101–122.
- Huang P, Wu M, Kuga S, et al. Aqueous pretreatment for reactive ball milling of cellulose. Cellulose. 2013;20(4):2175–2178. doi: 10.1007/s10570-013-9940-4.
- Klayya S, Tawichai N, Intatha U, et al. Tailoring nanofibrillated cellulose through sonication and its potential use in molded pulp packaging. Nanocomposites. 2021;7(1):109–122. doi: 10.1080/20550324.2021.1949517.
- Sethi J, Oksman K, Illikainen M, et al. Sonication-assisted surface modification method to expedite the water removal from cellulose nanofibers for use in nanopapers and paper making. Carbohydr Polym. 2018;197:92–99. doi: 10.1016/j.carbpol.2018.05.072.
- Satgè C, Verneuil B, Branland P, et al. Rapid homogenous esterification of cellulose induced by microwave irradiation. Carbohydr Polym. 2002;49(3):373–376.
- Semsarilar M, Perrier S. Solubilization and functionalization of cellulose assisted by microwave irradiation. Aust J Chem. 2009;62(3):223–226. doi: 10.1071/CH08491.
- Sèbe G, Ham-Pichavant F, Pecastaings G. Dispersibility and emulsion-stabilizing effect of cellulose nanowhiskers esterified by vinyl acetate and vinyl cinnamate. Biomacromolecules. 2013;14(8):2937–2944. doi: 10.1021/bm400854n.
- Peydecastaing J, Girardeau S, Vaca-Garcia C, et al. Long chain cellulose esters with very low DS obtained with non-acidic catalysts. Cellulose. 2006;13(1):95–103. doi: 10.1007/s10570-005-9012-5.
- Guo Y, Liu Q, Chen H, et al. Direct grafting modification of pulp in ionic liquids and self-assembly behavior of the graft copolymers. Cellulose. 2013;20(2):873–884. doi: 10.1007/s10570-012-9847-5.
- Missoum K, Belgacem MN, Barnes JP, et al. Nanofibrillated cellulose surface grafting in ionic liquid. Soft Matter. 2012;8(32):8338–8349. doi: 10.1039/c2sm25691f.
- Wang X, Wang N, Xu B, et al. Comparative study on different modified preparation methods of cellulose nanocrystalline. Polymers (Basel). 2021;13(19):3417. doi: 10.3390/polym13193417.
- Wang K, Lu J, Tusiime R, et al. Properties of poly (l-lactic acid) reinforced by l-lactic acid grafted nanocellulose crystal. Int J Biol Macromol. 2020;156:314–320. doi: 10.1016/j.ijbiomac.2020.04.025.
- Ferdous K, Uddin MR, Uddin MR, et al. Preparation and optimization of biodiesel production from mixed feedstock oil. CES. 2013;1(4):62–66. doi: 10.12691/ces-1-4-3.
- Ding J, Xia Z, Lu J. Esterification and deacidification of a waste cooking oil (TAN 68.81 mg KOH/g) for biodiesel production. Energies. 2012;5(8):2683–2691. doi: 10.3390/en5082683.
- Wang Y, Wang X, Xie Y, et al. Functional nanomaterials through esterification of cellulose: a review of chemistry and application. Cellulose. 2018;25(7):3703–3731. doi: 10.1007/s10570-018-1830-3.
- Han G, Liu F, Zhang T, et al. Study of microwave non-thermal effects on hydrogen bonding in water by raman spectroscopy. Spectrochim Acta A Mol Biomol Spectrosc. 2023;285:121877. doi: 10.1016/j.saa.2022.121877.
- Van Hong TD, Thao NT, Kieu TDT, et al. Effects of reaction conditions on the degree of substitution in acetylated nanocellulose. Vietnam J Catal Adsorpt. 2020;9(4):22–28. doi: 10.51316/jca.2020.065.
- Abdul Hadi N, Wiege B, Stabenau S, et al. Comparison of three methods to determine the degree of substitution of quinoa and rice starch acetates, propionates, and butyrates: direct stoichiometry, FTIR, and 1H-NMR. Foods. 2020;9(1):83. doi: 10.3390/foods9010083.
- Qin C, Wang W, Li W, et al. Developing bagasse towards superhydrophobic coatings. Cellulose. 2021;28(6):3617–3630. doi: 10.1007/s10570-021-03743-8.
- Stevie FA, Donley CL. Introduction to x-ray photoelectron spectroscopy. J Vac Sci Technol A. 2020;38(6):3204.
- Greczynski G, Hultman L. X-ray photoelectron spectroscopy: towards reliable binding energy referencing. Prog Mater Sci. 2020;107:100591. doi: 10.1016/j.pmatsci.2019.100591.
- Khiari R, Rol F, Brochier Salon MC, et al. Efficiency of cellulose carbonates to produce cellulose nanofibers. ACS Sustainable Chem Eng. 2019;7(9):8155–8167. doi: 10.1021/acssuschemeng.8b06039.
- Cabrera IC, Berlioz S, Fahs A, et al. Chemical functionalization of nano fibrillated cellulose by glycidyl silane coupling agents: a grafted silane network characterization study. Int J Biol Macromol. 2020;165(Pt B):1773–1782. doi: 10.1016/j.ijbiomac.2020.10.045.
- Gao X, Liu H, Shuai J, et al. Rapid transesterification of cellulose in a novel DBU-derived ionic liquid: efficient synthesis of highly substituted cellulose acetate. Int J Biol Macromol. 2023;242(Pt 4):125133. doi: 10.1016/j.ijbiomac.2023.125133.
- Baraka F, Robles E, Labidi J. Microwave-assisted esterification of bleached and unbleached cellulose nanofibers. Ind Crops Prod. 2023;191:115970. doi: 10.1016/j.indcrop.2022.115970.
- Byun Y, Whiteside S, Thomas R, et al. The effect of solvent mixture on the properties of solvent cast polylactic acid (PLA) film. J Appl Polymer Sci. 2012;124(5):3577–3582. doi: 10.1002/app.34071.
- Kumar R, Kumari S, Rai B, et al. Effect of nano-cellulosic fiber on mechanical and barrier properties of polylactic acid (PLA) green nanocomposite film. Mater Res Express. 2019;6(12):125108. doi: 10.1088/2053-1591/ab5755.
- Chandrasekaran S, Ramanathan S, Basak T. Microwave food processing—A review. Food Res Int. 2013;52(1):243–261. doi: 10.1016/j.foodres.2013.02.033.