
Abstract
The catalytic reduction of nitrogen oxides (NOx) under lean-burn conditions represents an important target in catalysis research. The most relevant catalytic NOx abatement systems for Diesel engine vehicles are summarized in this short review, with focus on the main catalytic aspects and materials. Five aftertreatment technologies for Diesel NOx are reviewed: (i) direct catalytic decomposition; (ii) catalytic reduction; (iii) NOx traps; (iv) plasma-assisted abatement; and (v) NOx reduction combined with soot combustion. The different factors that can affect catalytic activity are addressed for each approach (e.g. promoting or poisoning elements, operating conditions, etc.). In the field of catalytic strategies, the simultaneous removal of soot and NOx using multifunctional catalysts, is at present one of the most interesting challenges for the automotive industry.
Introduction
Air pollution from mobile sources, such as cars and trucks, contributes to a great extent to air quality problems and induces health risks in rural, urban, and industrialized areas in both developed and developing countries. About 60 million cars are produced every year and over 700 million cars are used worldwide. Moreover, the vehicle population is expected to grow to almost 1300 million by the year 2030.Citation1
Most vehicle transport relies on the combustion of gasoline and Diesel fuels, and hence the emission of carbon monoxide (CO), unburned hydrocarbons (HC), nitrogen oxides (NOx), and particulates matter (PM) is of particular concern.Citation2–4 The incomplete combustion of fuels causes the emission of partial oxidation products, such as alcohols, aldehydes . As a result of the thermal cracking reactions that occur in the flame, especially for incomplete combustion, hydrogen, as well as different hydrocarbons from those present in the fuel, are formed and emitted. Therefore, the total conversion of engine-out emissions into CO2, N2, and H2O using effective catalytic devices remains one of the most pressing challenges in the automotive industry.Citation5–8
In the case of Diesel engines, the lean-burn conditions that are found in the combustion chamber lead to the following average composition of the emissions: CO2 2–12%, H2O 2–12%, O2 3–17%, and N2 balance. However, the features of the Diesel fuel itself, and of the Diesel engine operating conditions (air-to-fuel ratios greater than 22) lead to the formation of both gaseous (NOx, CO, HC) and solid/liquid (PM) pollutants.Citation2 These Diesel engine emissions may originate from the incomplete combustion of fuel, from operating conditions that favor the formation of particular pollutants, or from the oxidation of nitrogen- and sulfur-containing compounds present in the fuel which are not hydrocarbons.Citation3, 4 A common engine management strategy to control NOx emissions is Exhaust Gas Recirculation (EGR). In this strategy, part of the exhaust (with O2, N2, H2O and CO2) is recycled back to the combustion chamber. In fact, since the heat capacity of CO2 in the recirculated exhaust gas is about 20–25% higher than that of O2 and N2, the energy released from the fuel combustion results in a lower temperature rise, and hence a lower peak cycle temperature, with consequent lower NOx levels. However, this strategy alone is not sufficient to meet the recent NOx regulations throughout the world.
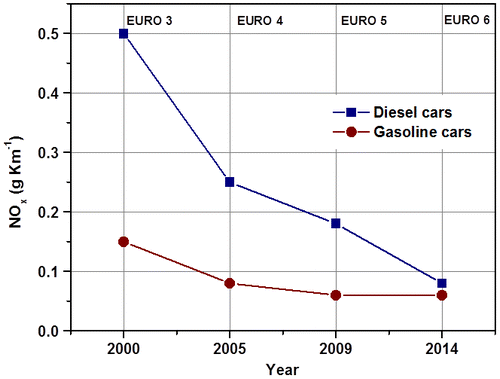
The application of strict measures to control Diesel engine emissions has been the main reason for the reductions in emissions in western European countries. The introduction of new vehicle technologies (e.g. cooled EGR) and stringent inspection systems related to Euro standards (Table ) have led to a progressive reduction in road traffic emissions, such as NOx, since 1990, despite the increase in fuel consumption.Citation9 Since NOx emission regulations have become more stringent over the last few years (Fig. ), several catalytic DeNOx approaches have been investigated for lean-burn conditions, such as the direct decomposition of NOx, selective catalytic reduction (SCR) using different reducing agents (e.g. ammonia/urea, hydrocarbons), and NOx storage-reduction Citation10–16. On the other hand, modern three-way catalysts cannot reduce NOx in the presence of excess oxygen, because high levels of oxygen suppress the necessary reducing reactions. Consequently, there is a strong driving force to develop multifunctional catalysts capable of reducing NOx to N2 and of oxidizing PM, HC, and CO to CO2 and H2O under lean conditions. In the present paper, the role of aftertreatment catalysis in Diesel NOx abatement is discussed comprehensively.
Table 1 European emission standards for passenger cars, g/km
Figure 1 Trend of the European emission NOx limits for both Diesel and gasoline cars
Direct catalytic NO decomposition
Engine-out NOx emissions mainly consist of NO (~90–95%) and, to a lesser extent, of NO2. The main source of NO formation is the thermal (Zeldovich) mechanism. This mechanism takes place through a chain of high temperature reactions (greater than 1600 °C) and it is responsible for more than 90% of NOx emissions from road transport.Citation9 During flame combustion, the reaction between N2 and O2 is thermodynamically favored (∆H298 K° = 180.6 kJ mol−1), thus resulting in the formation of NO, according to the reaction:(1)
Direct NO decomposition (the reverse of equation (1)) has received considerable attention in the field of environmental catalysis, since the overall process is thermodynamically favored below 1000 °C, and the use of reducing agents is not required.Citation17, 18 In the catalytic decomposition of NO, the exhaust containing NO is made to flow over a solid catalyst, where the NO compound is split into its elements (2NO → N2 + O2). The main concern is to find a material that is both active and oxidation-resistant.Citation19 Therefore, since the early 1990s, several materials have been investigated for direct NO decomposition.
Noble metals
Direct NO decomposition is assumed to occur through four elementary stepsCitation18:(2)
(3)
(4)
(5)
where NO*, N*, and O* are the adsorbed species on the catalytic active sites (*). Overall, the NO decomposition reaction consists of two parts: (i) adsorption and dissociation of the NO species (equations (2) and (3)); (ii) recombination and the removal of N2 and O2 from the catalyst surface (equations (4) and (5)).
Many studies have been conducted regarding NO decomposition over Pt–Al2O3 catalysts.Citation17, 20, 21 Freund et al.Citation20 have investigated the role of different adsorption and reaction sites on a structurally well-defined Pd/Al2O3 catalyst. They found that atomic nitrogen and oxygen species adsorb preferentially at sites in the vicinity of edges and defects of solid surfaces. The presence of these atomic species critically controls the NO dissociation activity. In other words, the direct decomposition of NO was found to be dominated by particle edges, steps, defects, and (100) sites rather than by (111) facets. Similarly, Ge and NeurockCitation21 noted an exceptional low energy barrier for the dissociation of NO adsorbed on Pt (100) surfaces.
It has been shown that NO adsorption onto a solid surface is the kinetically controlling step, and the following kinetic equation has been proposed:(6)
where r is the reaction rate, N is the Avogadro number, k is the kinetic constant for NO adsorption, and K is the equilibrium constant for O2 adsorption. This kinetic equation has been used to describe the catalytic behavior of several noble metal-containing catalysts. The main problem related to the use of noble metals for direct NO decomposition is likely that such materials are easily oxidizable and therefore not very active in the presence of oxygen. In fact, the high K value takes into account the inhibiting effect of oxygen on the kinetics of this reaction.Citation22
The results obtained by Hamada et al.Citation23 with Pt, Rh, Pd, and Ru on Al2O3 (noble metal content = 0.5 wt.%) had shown that the NO decomposition activity increased with a decrease in the affinity of the metals toward oxygen. Thus, Pt is the most active metal, and this is followed by Rh, Pd, and Ru. Ogata et al.Citation24 observed that Pd exhibits more activity when it is dispersed on oxides containing Mg2+ ions (e.g. MgAl2O4, MgO, Mg2Si3O8, MgZrO3) than when it is supported on Al2O3. The Pd–Mg interactions significantly increase the catalytic activity, since the two components are in intimate contact with each other on an atomic scale.Citation25 Thus, interactions among active sites may lead to beneficial effects (synergies) for catalysis.
On the other hand, Wu et al.Citation26 have shown the beneficial effect of small amounts of Au or Ag on the catalytic activity of Pd/Al2O3 catalysts (namely 0.5% Pd - 0.03% Au/Al2O3 and 0.5% Pd - 0.06% Ag/Al2O3) obtained by co-precipitation, due to an easier reducibility of the active phase.
Frank et al.Citation27, 28 have observed good performances for Pt–Mo-based materials. Specifically, the catalyst with the best activity exhibited Pt–Mo–Co components, thus suggesting cooperation phenomena among several active centers.Citation27. The latter catalyst decomposed ca. 60% NO at 150 °C (in the absence of CO). However, when CO was increased to 0.6%, the maximum NOx conversion decreased to ca. 40% at 220 °C. The authors explained this catalytic behavior by showing that NOx abatement begins when CO is fully oxidized and they argued that CO does not participate in NOx reduction.
Metal oxides
Several metal oxides have been studied as catalysts for direct NO decomposition, as can be seen in Table .Citation29 Among these, cobalt oxide (Co3O4) is one of the most active compounds. The high activity of Co3O4 seems to be related to the relatively weak Co–O bonds, which lead to an easy desorption of lattice oxygen (β-species), especially at low temperature. Co3O4 shows a spinel structure with Co2+ and Co3+ cations. During the direct NO decomposition reaction, Co2+ may partially oxidize to Co3+ and form a Co2O3-like phase on the surface. The latter is not stable and decomposes to Co3O4, thus favoring NO dissociation.Citation30.
Table 2 Catalytic activity of metal oxides for the decomposition of NO. Reaction conditions: PNO = 2.0%; W/F = 0.5 g s cm−3
Iwamoto et al.Citation31 have found that promoting Co3O4 catalysts with small amounts of Ag results in a significant increase in the catalytic activity. Moreover, the decrease in the NO decomposition activity with an increase in the O2 concentration was less marked when Co3O4 was doped with Ag. With the addition of either Ag or Na, the activity in fact enhances primarily due to the excess electron density on the surface.Citation30
The NO decomposition activity of metal oxides is closely related to the bond strength between the metal and the oxygen in the lattice.Citation17 NO usually dissociates on the metal oxides, although NO dissociation depends on several factors, including the temperature, surface coverage, crystal planes, and surface defects.Citation17, 20, 30 The catalytic activity of metal oxides towards the NO decomposition is usually lower than that of noble metals.Citation29, 30 Therefore, the operating temperature must be kept at quite high values (up to 1000 °C), which can lead to catalyst sintering, and it is of no interest for Diesel exhaust applications. As a whole, the inhibiting effect of oxygen is lower for metal oxides than for noble metals.Citation18, 29, 30. Moreover, the oxygen “self-poisoning effect” towards NO decomposition can be much lower on ceria-based materials, as a consequence of oxygen spillover phenomena.Citation32, 33 Zhang et al.Citation34 doped La2O3 catalysts with Sr (namely 4% Sr/La2O3), thus promoting NO decomposition activity under different operating conditions. Other authorsCitation35 have performed studies on CeO2–ZrO2 mixed oxides, which exhibit interesting activities in NOx decomposition.
Perovskites
Perovskite-type catalysts have been widely investigated for the direct decomposition of NO, as reported in Table .Citation29 Mixed oxides with an ABO3 chemical composition, in which the A cation is either a rare earth or an element of the II group (e.g. La, Sr, Ba, Y, etc.) and B is a transition 3d metal, belong to this category of catalysts. The substitution of A or B ions with other metals creates oxygen defects in the lattice, which are generally related to the catalytic activity, although the nature of these interactions has not yet been fully understood.Citation36 However, it is known that oxygen-deficient perovskites adsorb large amounts of oxygen, and that the nature and reactivity of the adsorbed oxygen, which are more weakly bonded with metal cations, are quite different from the oxygen in the lattice.Citation17 The order of the desorption temperature for oxygen is correlated to the NO decomposition activity: the weaker the adsorption of oxygen on the catalyst surface, the greater the mobility of the oxygen, and hence the greater the activity toward NO decomposition.Citation37 This suggests that surface oxygen species may act as catalytic active sites.Citation38 On the other hand, oxygen-deficient compounds, such as the YBa2Cu3O7−x perovskite, have been proved to be active for NO decomposition,Citation39 especially when supported on MgO. In perovskites, the complexity of surface defects (e.g. oxygen vacancies) is at least one order to magnitude higher than that of binary metal oxides and metals, because of the presence of ions and cations, which can assume a variety of charged states.
Table 3 Catalytic activity of perovskite-type oxides for the decomposition of NO. Reaction conditions: PNO = 2.0%; W/F = 0.5 g s cm−3
The effects of dopants in BaMnO3 perovskites on the direct NO decomposition activity have been investigated by Iwakuni et al.Citation40 The authors observed that the activity of NO decomposition increased by an order of Mg > Zr > Fe > Ni > Sn > Ta for the Mn-site dopant, and La > Pr for the Ba site.
Teraoka et al.Citation41 considered the effect of the preparation method and catalyst composition on the activity toward NO decomposition for several perovskites with a general formula LaxSr1−xXO3 (X = Co, Cr, Mn, Fe, Ni and Cu).
Kim et al.Citation42 observed that under realistic conditions, La1−xSrxCoO3 catalysts exhibit higher NO-to-NO2 conversions than commercial Pt-based catalysts.
Complex perovskite-type catalysts have been prepared by Monceaux et al.Citation43 The authors have found that the substitution of a small quantity of Pt for Mn or Co makes it possible to prevent sulfur poisoning and to increase the catalytic performances of these materials toward several oxidation reactions. Perovskites exhibit high thermal stability, compared to metal oxides, and do not suffer to any great extent from the oxygen “self-poisoning effect.” Since they can easily desorb oxygen, these materials should be good candidates for purifying exhaust gas from Diesel engines, particularly for the removal of soot and for NO abatement. The most significant disadvantage of NO decomposition on perovskite catalysts is the high reaction temperature required to achieve a high NO decomposition activity. Other significant drawbacks are their low surface areas and pore volumes. The ceramic solid–solid reaction and co-precipitation methods, which are commonly used for the synthesis of perovskite-type materials, involve a high reaction temperature (>900 °C) and hence yield perovskite-type oxides with low surface areas (<2 m2 g−1), due to their sintering.Citation44 Research should therefore be addressed to the development of new preparation techniques, aimed at obtaining perovskite structures with better textural properties.
Zeolites
Several types of metal-ion-exchanged zeolites, including X- and Y-faujasite, mordenite, ferrierite, and ZSM-5-type zeolites with different Si/Al molar ratios, have been studied for direct NO decomposition.Citation45, 46 Among zeolite-based catalysts, the Cu/ZSM-5 catalyst has been paid much attention, due to its superior activity and N2 selectivity in a wide temperature range.Citation47 However, the Cu/ZSM-5 has been found to deactivate readily during high temperature hydrothermal treatment.Citation48 It has been reported that the primary causes for the thermal deactivation, such as agglomeration of the active metal, the migration of the reaction active sites, and the dealumination of zeolite support, can be obviously suppressed by the introduction of a second metal.Citation49, 50 In particular, the addition of Ce greatly improves the hydrothermal stability of the Cu/ZSM-5 catalysts, since Ce species stabilize the dispersion of CuO and suppress the bulk-type CuO crystallites formation.Citation51, 52
As a whole, the NO decomposition over Cu-ZSM-5 proceeds via a redox-type mechanism.Citation30 In other words, Cu+ ions created through a thermal pre-treatment can be oxidized to Cu2+ by gaseous oxygen. Thus, Cu2+ acts as an adsorption center for NO, as follows:(7)
NO molecules can be chemisorbed as NO+, NO−, and species on the zeolite surface. A fraction of the Cu2+ ions is reduced to Cu+ through the desorption of O2, whereas catalyst re-oxidation with NO restores Cu2+ sites, thus forming N2.
The catalytic behavior of Cu-ZSM-5 is negatively affected by the relatively high oxygen concentration in the feed, thus limiting their use for applications in Diesel exhausts. For instance, a maximum conversion rate of 6% has been obtained at 500 °C for a feed similar to Diesel exhausts (1000 ppm NO and 10 vol.% O2), which is far too low to be acceptable.Citation29 Moreover, this material is not stable in water vapor conditions for long periods of time and it has proved to be sensitive to SO2 poisoning.Citation29 As a whole, the presence of water vapor has an inhibition (although reversible) effect on the decomposition of NO, whereas SO2 poisons the catalyst surface.
On the other hand, Weisweiler et al.Citation53 have observed that Pt-ZSM-5 catalysts may/are able to adsorb NO under controlled dynamic conditions (simulation of a driving cycle), although real NO abatement cannot be attained at low temperatures (below 180 °C).
Researchers are usually critical about the use of zeolite-type catalysts for direct NO decomposition, since they have shown low hydrothermal stability and low SO2 resistance.Citation54, 55 On the other hand, since the operating temperatures of these catalysts are usually higher than those required for the catalytic reduction of NO, the decomposition route, even though very attractive, is actually not very interesting for application in aftertreatment exhaust devices.
Selective catalytic reduction (SCR) of NO
Since the ideal solution of direct catalytic NO decomposition has not been successful for the control of Diesel engine emissions, researchers have begun to investigate alternative approaches, such as the selective catalytic reduction (SCR) of NO.
The catalytic reduction of NO has been studied using a number of reductants, of which ammonia, urea, and hydrocarbons are the most frequently reported.Citation4, 16 Catalytic reduction with ammonia (or urea) is usually referred to as NH3-SCR, while reduction with hydrocarbons is often referred to HC-SCR.
Several catalytic materials have been developed for the SCR of NOx since the early 1970s.Citation56 The first generation of commercial SCR catalysts for mobile applications was monoliths containing V2O5/WO3/TiO2, which were similar to the V2O5-based catalysts used for NH3-SCR in stationary applications.Citation2 However, the stringent legislations on NOx emissions, the necessity of new materials to extend the temperature operation window, and the toxicity of vanadium have driven the research toward the development of more effective catalysts.Citation30
Use of ammonia/urea as a reductant
The reduction of NOx using ammonia is a widely commercialized technology for large stationary combustion plants (e.g. power plants, heaters and boilers in the process industry). In Japan, USA, and Europe, large-scale applications of SCR have been introduced over the last few decades.Citation57–59 Ammonia is commonly used as a reductant agent in large commercial scale SCR. NH3 is supplied to the SCR process using a gaseous solution (anhydrous form), an aqueous solution, or a solution of urea. The choice depends on the economic and safety issues involved in the handling of the preferred solution, that is, anhydrous ammonia. As a whole, a high efficiency of NOx removal can be obtained with the NH3-SCR process (namely 70–98%).Citation3 The uniqueness of this reaction with NH3 is that it can occur in the presence of excess O2. Thus, this technology has received a great deal of attention for Diesel-engine vehicles.
The overall NH3-SCR reaction isCitation60:(8)
The role of oxygen is to donate one electron to the redox process.Citation17 Thus, oxygen enhances the rate of the NO–NH3 reaction.Citation57 A complex reaction network can be observed on the catalyst surface. The main prevailing reactions areCitation60:(9)
(10)
(11)
(12)
(13)
These reactions are inhibited by water, which can be present in the exhaust gases. However, other reaction pathways may occur and, as a result, undesired products can be formed. These reactions may include a partial reduction of NOx, which leads to N2O (equations (14)–(16)), or the direct oxidation of NH3, which forms NO (equations (17)–(18))Citation60:(14)
(15)
(16)
(17)
(18)
Particular temperature conditions (100–200 °C) may lead to the formation of NH4NO3, which is explosive and deposits in the cavities of the catalytic material, causing temporary deactivation.Citation61 One possible way of reducing the ammonium nitrate or other byproducts is to tailor the reductant injection with different amounts rather than stoichiometric with respect to NOxCitation60:(19)
The molar ratio of ammonia to NOx is set below one (sub-stoichiometric) to minimize ammonia slip. The typical SCR process operates using/with an oxidation catalyst (e.g. V2O5–WO3/TiO2) downstream from the SCR, which prevents the unreacted ammonia from leaving the reactor. The oxidation catalyst may also favor the oxidation of CO and HC emissions.Citation60 On the other hand, an increase in N2O and NO in the exhaust gases may occur due to the oxidation of ammonia.Citation63 Similarly, the catalyst may enhance SO oxidation and hence cause an increase in sulfate emissions. Since ammonia poses health and practical problems (NH3 is a toxic gas that has to be stored under pressure), an alternative source of NH3 has been developed, in the form of urea, especially for non-stationary Diesel engines.Citation14, 30 Urea, which is a solid that is highly soluble in water, can be injected, as an aqueous solution, into the exhaust gases, where it decomposes, according to equation (20), at about 200 °C:(20)
Equation (20) is the result of the following two stepsCitation62:
(i) Thermal decomposition (hydrolysis)(21)
(ii) Isocyanic acid reaction with water(22)
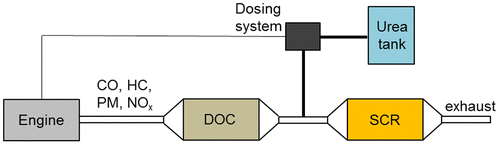
The layout of an SCR process for mobile Diesel engines, fueled with urea, is generally structured as an open-loop control, namely the amount of injected urea follows a pre-determined route of the NOx emissions as a function of the engine operating conditions. This technology has been shown to yield above 80% conversion of the engine-out NOx.Citation2, 14 The urea solution is injected into the exhaust line upstream from the SCR catalyst. The atomization allows the solution that has been tailored to obtain a good mixing with the exhaust gasses to evaporate quickly, a process that can be assisted through the use of static mixers. A uniform distribution of the flow in the catalytic converter is necessary to reach high conversion efficiencies.Citation64 The SCR is usually placed after the Diesel oxidation catalyst (DOC), which is used to oxidize CO, HC, and part of the NO (Fig. ). The DOC oxidizes the NO to NO2, and this compound is more reactive and extends the operating temperature window for the SCR process. In this way, the catalyst can take advantage of the “fast SCR” (equation (13)) to significantly enhance the DeNOx efficiency at low temperatures.Citation10, 14 The SCR catalyst can be fouled and deactivated due to the deposition of ammonium sulfate and disulfate, resulting from the oxidation SO2, with the subsequent formation of H2SO4 in the DOC, and the reaction with NH3 in the SCR. The SCR deactivation occurs at temperatures below 250 °C; hence, at low temperatures (150–250 °C), the urea injection can be interrupted to prevent SCR catalyst deactivation. Urea has mainly been selected as the best ammonia source, due to its low toxicity, safety, availability, and low cost. However, 32.5% urea solutions have freezing temperatures of −11 °C, which is not acceptable for winter conditions in cold climates. Thus, the use of ammonium formate (HCO2NH4) has been proposed for SCR applications in cold climates (a 40% aqueous solution of HCO2NH4 has a freezing point of −35 °C), but it has a lower NH3 content than urea. An alternative reductant supply method is to use solid urea rather than aqueous solutions.
Figure 2 Schematization of DOC and SCR systems in Diesel vehicles
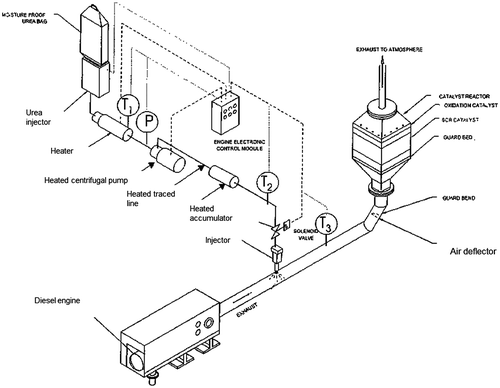
An interesting approach has been introduced by Tarabulski et al.Citation65 in which urea, or another reducing agent, is employed in the SCR process in a solid state (Fig. ). Aqueous solutions of urea or other reagents are not required in the Tarabulski process. The solid reagent is fed to a gas generator that produces a reactant gas through heating; the latter gas is rich in NH3 and can therefore be added to the exhaust gas on an as-needed basis for NOx abatement. Using urea or another solid reducing agent can cause nozzle plugging and fouling of the catalyst. This technology offers several advantages, including the realization of significant savings in energy, which would otherwise be necessary to vaporize the water, and savings on the cost of antifreeze additives. The temperature that must be reached for urea gasification is about 400 °C and, to reduce the vessel volume, it is possible to use solid catalysts, such as platinum, palladium, oxides of vanadium, titanium, and chromium. However, the SCR systems that have been proposed for dosing solid urea appear more complex than those that utilize urea water solutions, which are now the most common applications for Diesel engines.
Figure 3 Experimental plant developed by Tarabulski et al. to test solid urea as a reducing agent for NOx abatementCitation65
Platinum, vanadium oxide and zeolites
Two important features of an SCR catalyst are that the material is active as an oxidation catalyst, and that materials which are effective in partial oxidation, when supported on TiO2 (namely anatase), are usually good SCR catalysts.Citation3 Such materials can be based on V2O5–WO3 on TiO2 or V2O5–MoO3 on TiO2, although other materials (e.g. zeolites) have been considered. The main reason for such dominance is that they offer excellent performances, yet at the same time they are very tolerant toward poisons in the flue gasses.Citation2–4 The anatase form of TiO2 is the preferred support, mainly because SO2 poisoning has a lower influence on the TiO2 surface.Citation68
The first SCR technology that was developed was based on a Pt-containing catalyst. However, NOx reduction over the Pt surface is only effective at temperatures below 250 °C (Fig. ).Citation2 In fact, at temperatures between 225 and 250 °C, the oxidation of NH3 to NO and H2O (equation (18)) becomes dominant and, as a result, poor selectivity toward N2 can be achieved/observed. On the other hand, low temperatures (150–200 °C) may lead to the above-mentioned NH4NO3 formation, which entails a very narrow range of available working conditions.Citation2 Moreover, Pt catalyzes the reduction of NOx to N2O, which is a powerful greenhouse gas. Pt has the benefit of generally being insensitive to SO2 and possesses good thermal stability, but it may favor the formation of SO3.Citation2
Figure 4 Operating temperature windows for different NH3-SCR catalysts
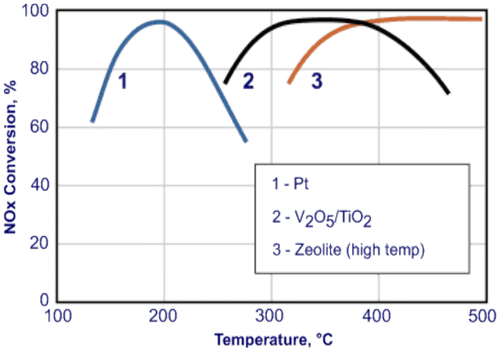
Vanadium oxide catalysts act well in a wider and upper temperature range, from 260 °C up to 450 °C, with the best SCR performances taking place between 300 and 400 °C.Citation67 This range is optimal for both light-duty (lower limit) and high duty (upper limit) applications. Catalytic materials, such as V2O5/TiO2 or V2O5–WO3/TiO2, are capable of NOx reduction in excess of 90%, and they are most probably the best candidates to meet severe NOx reduction goals.Citation14 Despite discrepancies concerning the detailed nature of the active centers, there is general consensus in experimental analyses that the SCR reaction involves both Brønsted (V–OH) and Lewis (V=O) acidic sites, and hence the presence of water has significant effects on the SCR process.Citation14 The terminal V5+=O groups of V2O5 appear to be essential in carrying out this reaction, since they are the energetically favored sites and, in addition, are accessible for the formation of Brønsted acidic sites. Thus, ammonia can readily adsorb on V4+–OH species to form an NH4+ intermediate, which subsequently reacts with co-adsorbed NO to form the adsorbed (NH3–NHO)+ intermediate. The N–H bond in the ammonium intermediate is broken as it reacts with NO (from the gas-phase). The proton which transfers from the NH4+ to the V2O5 surface during this initial step is subsequently transferred back to the NO molecule from the V2O5 (redox cycle). Ultimately, V5+=O groups can be restored via proton transfer. This produces gas-phase NH2NO, which can be converted into N2 and H2O through subsequent reactions over V2O5.Citation3, 17
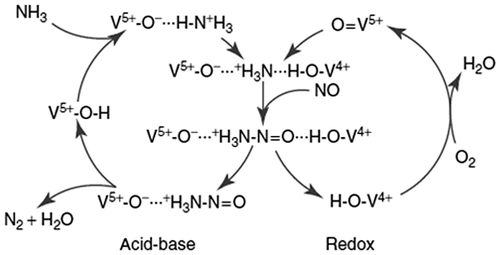
Several studies have confirmed an Eley–Rideal-type mechanism (ER) for the SCR reaction, where ammonia adsorbs onto the V2O5 surface and NO molecules react from the gas-phase (or as weakly adsorbed species) on the solid surface. Although ER is the prevalent mechanism in most of the operating conditions, at low temperatures (<200 °C), the reaction seems better described as a Langmuir–Hinshelwood-type mechanism (LH), thus suggesting that the SCR process takes place between adsorbed NH3 and NO species on the solid surface.Citation3 Moreover, Dumesic et al.Citation69 proposed an SCR catalytic cycle, which consists of two cycles interacting with each other (namely acid-base and redox cycles), thus confirming the complexity of this catalytic system. V-species can in fact act simultaneously as Lewis/Brønsted acidic and redox centers (Fig. ).
Figure 5 Catalytic cycle of the SCR reaction over a V2O5/TiO2 catalyst
Over the last few years, reports on health issues concerning vanadium emissions from SCR catalysts in mobile applications,Citation70 V2O5 having been classified as possibly carcinogenic to humans (group 2B),Citation71 have limited its further exploitation for mobile systems.
Therefore, zeolites containing transition metal ions (Cu, Fe, Cr, Mn, Ni, Ce, etc.) have been investigated extensively for mobile applications.Citation14 Zeolite-type catalysts have been considered for special applications, in particular for low-sulfur fuels. Unfortunately, however, zeolites are relatively expensive and thus are not suitable for extruded monoliths, although they are suitable for coated monoliths.Citation3
Zeolitic materials have a relatively wide temperature range of application (300–450 °C), and the most frequently studied material for the SCR reaction is Cu-exchanged ZSM-5.Citation14, 15, 29, 30, 50 This catalyst does not oxidize NH3 to NOx at high temperatures, and the upper temperature limit for the SCR process depends on its structural stability.Citation72
Low-temperature zeolites have also been synthesized, with the aim of widening the temperature range at which they are operative, and moderate NOx to N2 conversion efficiencies of between 200–400 °C have been reached.Citation73 Metal-exchanged zeolites have been found to adsorb considerable amounts of NH3 under certain operating conditions, and this results in rather slow responses to temperature variations or changes in NOx concentration. The NH3 adsorption capacity of catalysts depends to a great extent on the temperature; unwanted ammonia slip occurs for the increases of fast temperature.Citation61
Moreover, it has been observed that the SCR reaction shows considerable sensitivity to the nature of the support, and hence comparative studies on different zeolite supports (ZSM-5, A, beta, FAU, ferrierite, CHA, Linde type L) have been performed. From these studies, it emerged that Fe- or Cu-exchanged ZSM-5 zeolite showed good activity and selectivity for N2 production.Citation14 However, they demonstrate a lack of hydrothermal stability at temperatures above 700 °C. In particular, Cu-CHA catalysts have become the state of the art in NH3-SCR catalysis for Diesel vehicles, due to their excellent low temperature activity and high hydrothermal stability,Citation74, 75 which makes it a valuable candidate in the functionalization of DPFs with NH3-SCR catalysts, in order to increase the operating temperatures of the SCR catalysts by being close-coupled with the DOC instead of in underfloor position, in the so-called “SCR on filter” concept. Indeed, compared to ZSM-5 or beta-type zeolites, the chabazite zeolite contains small-sized pores and can coordinate isolated Cu2+ species, which are more resistant to hydrothermal aging.Citation72
Many other NH3-SCR catalysts have been investigated over the years. Among these, it is worth mentioning Fe2O3, Fe-containing mixed oxides, and Fe-exchanged materials that may show good SCR performances.Citation30, 73 Similarly, Mn-oxides have received much/ a great deal of interest for the SCR reaction because of their low-temperature activity, although they exhibit low selectivity for N2 production. In particular, MnCr2O4 spinel-Oxide Catalysts have shown promising catalytic results, as they can reach an NO conversion of 96% and selectivity to N2 of 97% at 125 °C.Citation76
Use of hydrocarbons as reductants
An alternative to the use of ammonia as a reductant agent is the employment of hydrocarbons, namely the HC-SCR process. The latter is also known as DeNOx or lean-NOx process. According to this approach, the hydrocarbons can be oxidized by the oxygen present in NO, as follows:(23)
The reaction of equation (23), which leads to N2, CO2, and H2O, is not the only path involved in the NO reduction, since undesired products, such as N2O, can also be obtained.
It is thought that the HC-SCR reaction proceeds competitively with the combustion reaction of hydrocarbons, and the selectivity to N2 determines the feasibility of the HC-SCR process. The selectivity can be defined as the ratio between the amount of hydrocarbon oxidized by NO, with subsequent N2 formation, and the total reacted hydrocarbon. Thus, the HC promoted SCR reaction of the NO reduction and the simple HC combustion areCitation60:(24)
(25)
Consequently, catalyst selectivity is the key parameter that has to be optimized using suitable catalytic materials, as well as a suitable reducing agent, HC/NOx ratio, temperature range, and so on.
As highlighted by/in Zelenca et al.,Citation77 the research should be conducted in three main directions:
| • | Choice, characterization, and improvement of suitable catalytic materials, which should be industrially available; | ||||
| • | Determination of the most efficient hydrocarbon through the injection of different hydrocarbon compounds into the synthetic gas ahead of the catalyst to increase NOx conversion rates; | ||||
| • | Optimization of the different parameters that affect the catalyst performance, in order to improve its use with vehicles. | ||||
An important parameter for NOx abatement is the HC to NOx ratio; generally, a two to fourfold surplus of hydrocarbons (expressed as ppm-HC) relative to the NOx concentration (ppm-NOx) is necessary to reach ca. 80% NOx conversion.Citation78–80. As such a surplus of hydrocarbons is not usually present in Diesel exhaust gases, hydrocarbons or Diesel fuel would have to be added to the Diesel exhaust gases.Citation81–83 There are two main possibilities of HC enrichment, that is, low or high pressure injection of Diesel fuel ahead of the catalyst (“active DeNOx”) and utilization of unburned HC emissions directly from the engine exhaust gas (“passive DeNOx”). In “active DeNOx” systems, the increase in the HC amount, and the control of their concentration to optimize the SCR catalyst can be realized by two means: the injection of hydrocarbons, preferably Diesel fuel, into the exhaust system upstream of/from the catalyst, or late in-cylinder injection in a common rail fuel system. The DOC system, which has a classical CO/HC emission reduction function, can be positioned downstream from the DeNOx system, in order to also undertake the role of preventing possible HC emissions.
On the other hand, “passive DeNOx” should be a simpler and cheaper option, since no additional injection equipment would be necessary. However, since the HC concentration in the exhausts is dependent on the engine points and it is somewhat limited, low conversion efficiencies can be reached. In order to achieve the necessary HC concentrations, engine modifications should be undertaken, in order to obtain higher hydrocarbon emissions. As a result, active DeNOx systems offer higher NOx conversion efficiency, but at the cost of increased system complexity and a fuel economy penalty.
A wide variety of metal oxides, alumina-based catalysts, zeolites, and perovskites have so far been tested for this promising technology.Citation84 The first catalyst screening, which shows the catalytic behavior of several metals (Pt, Au, Cu, Co and Ag) supported on Al2O3 and ZSM-5, was published by Obuchi et al.Citation85 Then, Ag/alumina have been found to be promising catalysts for the selective catalytic reduction of NOx to N2 by hydrocarbons in laboratory tests as well as in full-scale diesel engine operation.Citation86
The addition of small amounts of H2 can promote the HC-SCR activity of Ag-based catalysts for low temperature NOx reduction. Over the years, several explanations have been proposed for this beneficial “H2-effect”, including the enhancement of the partial oxidation of the reducing agent, the formation of reactive N species (NCO-like groups or gas phase radicals) from the reducing agent, the easier formation of active cationic Ag clusters, and the destabilization of surface nitrates blocking active Ag sites. Hence, H2 promotes the HC-SCR activity of Ag-based catalysts through multiple roles, involving morphological, chemical, and kinetic changes.Citation87, 88
Appropriate reductants, such as C3H6, CH4, CH3OH and C2H5OH (which are efficient reductant agents), were used. The catalytic performances for each catalyst are summarized in Table . The most promising materials are based on platinum, although the high emission of N2O is a difficult problem to solve. For this reason, the most suitable catalyst seems to be the Co/Al2O3 one, with the addition of an oxidation catalyst downstream to prevent the leakage of non-reacted or incompletely oxidized reductants. De SoeteCitation89 has published an interesting work that shows the reduction rate expressions of NO to N2 in the HC-SCR reaction over Cu/ZSM-5. The author has found that C2H4 and C6H14, when used as reductants, show different catalytic behavior: the reaction order in C2H4 is negative, whereas it is positive for C6H14. In fact, ethylene is less active in NO reduction than n-hexane, although HC oxidation is lower.
Table 4 Catalytic performances of several materials towards the HC-SCR reaction
Many studiesCitation82, 89 have shown that hydrocarbons do not participate directly in the reduction of NO, but they are first partially oxidized to active intermediates (e.g. aldehydes) which then can react with NO to form N2 and O2. On the other hand, Bell et al.Citation90–93 reported that, for HC-SCR over Co-, Mn-, Fe- and Pd-ZSM-5, the highly active species are CN groups which react with NO2 to form N2 and CO2.
Zeolite-type catalysts
As a whole, the HC-SCR activities of zeolite-type catalysts are better than those of metal oxides, such as alumina. The crystalline structure seems to contribute to the high activity of the zeolites. Thus, several zeolite-related compounds, such as metallo-silicates and silicoaluminophosphates, have been reported to be active for the HC-SCR reaction.Citation14, 30, 46
Many ion-exchanged zeolites, such as Cu-, Fe-, Pt-, Co-, Ga-, Ce- and H-exchanged zeolites, have been found to be active for this reaction. In particular, Cu- and Co-ZSM-5 have received a great deal of attention over the last few decades.Citation2, 3, 11, 17, 46 Several studies have revealed/pointed out the key role of reductant agents toward selectivity to N2. It has been observed, for Cu-ZSM-5, that some hydrocarbons (namely C2H4, C3H8, C4H8, and alcohols) behave as selective reductants, while other reductants (i.e. H2, CO and CH4) are non-selective toward N2 production.Citation94 Conversely, CH4 has been shown to be a selective reductant over Co-ZSM-5 and Ga- or In-ZSM-5 catalysts.Citation95, 96 Erkfeldt et al.Citation97 established that a C–C bond in the reducing agent is required for lean NOx reduction over Cu-ZSM-5. The influence of the hydrocarbon concentration on the activity of a Cu-ZSM-5 catalyst has been investigated by Konno et al.,Citation81 who reported that the reduction rate increases with the hydrocarbon concentration up to an HC/NO molar ratio of ca. 8. The presence of some co-exchanged cations (Ca, Sr, Fe, Co and Ni) had the effect of expanding the temperature range over which a Cu-ZSM-5 can be active for the HC-SCR process.Citation42, 98 Interestingly, lanthanum co-exchange resulted in an improvement in the NO adsorption capacity of the zeolite, both in the absence and in the presence of water.Citation99
Pt-ZSM-5 is more stable than Cu-ZSM-5: Iwamoto et al.Citation94 investigated the long-term stability of Pt-ZSM-5 under simulated and actual exhaust conditions, and found that its activity did not decrease in the presence of water vapor or SO2 in the reactant stream. Moreover, they observed that the catalyst activity barely changes after 1000 h exposure to water vapor.Citation100 On the other hand, the catalytic performances of Cu-exchanged zeolites are affected by the presence of SO2 (Fig. ).
Figure 6 Conversion of NO + O2 + C3H6 system into N2, with and without SO2. Catalyst Cu-MFI. Without SO2: PNO = 500 ppm, PO2 = 1.0%; PC3H6 = 990 ppm, W/F = 0.1 g cm−3; With SO2) PNO = 530 ppm, PO2 = 1.0%; PC3H6 = 1000 ppm, W/F = 0.1 g cm−3, PSO2 = 300 ppm
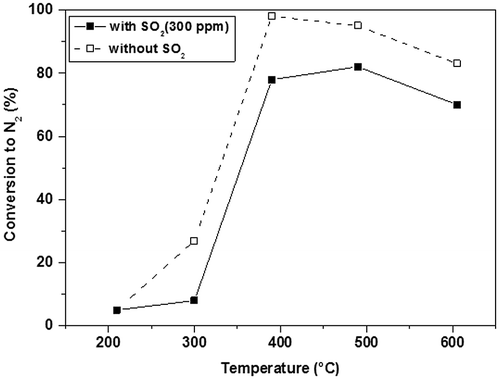
Falley et al.Citation101 have prepared a new catalytic material, based on a zeolite chosen from between β-Zeolite Y-Zeolite and ZSM-5, in which Cu, Co, and Fe have been incorporated as active species. They observed that a combination of these three metals tends to lower the temperature at which a Cu-containing catalyst reaches its optimum NOx conversion rate (Fig. ), particularly after aging (Fig. ). This trimetallic-based system lends stability to the catalytic material, so that NOx conversion rates after an accelerated ageing cycle are higher than those of comparative materials.
Figure 7 NOx conversion vs. temperature obtained using fresh ZSM-5-based catalytic material, with the addition of small amounts of Cu, Co and Fe: E-1 3.31% Cu, 2.27% Fe, 0.72% Co; R-1 3.22% Cu, 1.96% Fe; R-2 3.17% Cu, 3.13% Co; R-3 3.3% Co, 1.98% Fe
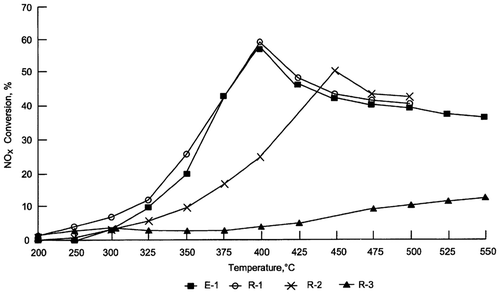
Figure 8 NOx conv. vs. T obtained using an aged (through exposure to a mixture of 10% steam in air for 5 h at 700 °C) ZSM-5-based catalytic mat. with addition of small amounts of Cu, Co and Fe: E-2 3.46% Cu, 2.03% Fe, 1.35% Co; R-1 3.22% Cu, 1.96% Fe; R-2 3.17% Cu, 3.13% Co
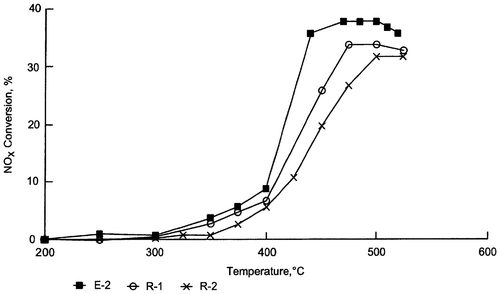
Although Cu is the exchange metal chosen for most studies, many other metals have been tested, either alone or as co-cations: interesting tests were carried out using Gallium and Cerium. Gallium exchanged ZSM-5 was tested in SCR with 1000 ppmv of NO and C3H6 by Yogo et al.,Citation96 who found that this catalyst exhibits similar catalytic behavior to that of Cu-based systems.
Over the last few years, new multicomponent catalysts have been designed to expand the lean NOx reduction capacity of zeolite-type catalysts. For instance, Deeba et al.Citation102 reported interesting NOx conversions over new four-way catalysts, which were active over a temperature range of between 150 and 320 °C. These materials allow hydrocarbons to be activated and stored; these hydrocarbons then reduce NOx selectively. Moreover, synergistic phenomena (e.g. cooperative adsorption) in the HC-SCR reaction can be observed for zeolite-type catalysts when different hydrocarbons (e.g. CH4 and C3H8) are used as reductant agents.Citation103, 104
Supported noble metals
Due to the low hydrothermal stability of zeolitic materials, several researchers have addressed their efforts to the development of noble metal catalysts.Citation105–108 An interesting work on the catalyst choice for the HC-SCR process was conducted by Nakatsuji et al.,Citation105 and remarkable results were shown for the Ag/Al2O3 catalyst. This catalyst can be active at 300 °C and exhibit good catalytic stability in the presence of SOx, which is further improved by the addition of WO3, MoO3, and Pt. The authors observed that catalyst activity can be improved by adding the aldehydes that form from the injected fuel in the exhaust gas. Thus, they tried to partially oxidize fuel in a catalytic oxidizing reactor before injecting it into flue gas. The results obtained using this system are very interesting: at 450 °C, the engine bench test exhibited an NOx conversion of 75%.
Several works have been published on platinum-group metals, mainly due to their relatively high surface stability.Citation107, 108 Bamwenda et al.Citation109 studied platinum-group metals (Pt, Pd, Rh and Ir) deposited on different supports (TiO2, ZnO, ZrO2 and Al2O3) for the SCR of NO in the presence of C3H6. They found that the alumina supported catalysts showed both the highest activity for NO conversion and the highest selectivity toward N2 formation. The catalytic activities followed the Pt/Al2O3 > Rh/Al2O3 > Pd/Al2O3 > Ir/Al2O3 order, whereas Rh/Al2O3 was found to have the highest selectivity toward N2 formation.Citation110
Other studies have been carried out for Ag supported on Al2O3Citation105–108 or saponite.Citation108 Miyadera et al.Citation105 have shown that 2 wt.% Ag/Al2O3 can reduce NO with ethanol (a higher molar ratio of C3H5OH/NOx than 1.25), even in the presence of water vapor, although several byproducts may appear (e.g. N2O, NH3, CH3CN and HCN). Cyanide is the dominating byproduct below 400 °C (the typical exhaust temperature of a Diesel engine), although its concentration decreases at higher temperatures.
The influence of the reducing agents (C1–C3 hydrocarbons) on the HC-SCR of NO in a Ga2O3–Al2O3 system has been studied by Miyahara et al.Citation110: the most efficient NO abatement was achieved with C2 hydrocarbons, whereas C3 hydrocarbons were less selective for N2 production. Thus, it has been proposed that only one carbon atom per molecule is used for/in the HC-SCR process, and the other carbons are consumed by the combustion reaction. However, under wet conditions, the NO conversion on the Ga2O3–Al2O3 catalyst decreased for all the hydrocarbons. The decrease in NO conversion caused by the presence of water is due to the preferential adsorption of water, which inhibits the HC adsorption on the catalyst surface (competitive adsorption).Citation110 Noble metals should be added to avoid the retarding effect of water vapor. For instance, Haneda et al.Citation109 have studied the catalytic behavior of an Indium supported catalyst (5 wt.% ln/TiO2-ZrO2) for the SCR of NO with C3H6 with or without the presence of water: a decrease in NO conversion occurred under wet conditions. However, the authors observed that small amounts of Pd (0.005–0.02 wt.%) may improve the “resistance” of In/TiO2–ZrO2 against the presence of water (up to ca. 400 °C).
Highly dispersed Au catalysts have been studied for the HC-SCR of NO in recent years.Citation111 These catalysts are active in the reduction of NO with C3H6 in the presence of O2 and moisture. The catalytic behavior of Au-species depends on the metal oxide support. In fact, an activity scale has been drawn up: α-Fe2O3 ∼ ZnO < MgO ∼ TiO2 < Al2O3. The highest conversion to N2 (around 70%) at 427 °C has been achieved over a 0.1–0.2 wt.% Au/Al2O3 catalyst.Citation111, 112 It is worth noting that the conversion of NO to N2 over Au/Al2O3 was increased slightly by the presence of water. Thus, in order to improve the performances of the latter catalyst, Mn2O3 was then mixed with Au/Al2O3.Citation113 This mechanical mixture exhibited interesting results for the HC-SCR of NO in the 250 and 500 °C temperature range. This catalyst has been considered one of the most effective for the NO emission control of lean-burn gasoline and Diesel engines.Citation111–113
NOx traps
NOx traps (or NOx adsorbers) constitute an interesting NOx control technology for gasoline direct injected engines and for Diesel engines. NOx traps are also referred to using different terms: Lean NOx traps (LNT), NOx adsorber catalysts (NAC), DeNOx traps (DNT), NOx storage catalysts (NSC), NOx storage/reduction (NSR) catalysts.
NOx traps, which are incorporated into the catalyst washcoat, chemically bind the NOx species and convert them into solid species (metal bonded nitrates). NOx accumulation is carried out during lean conditions, and proceeds until the adsorber capacity is saturated. Then, the NOx trap is regenerated, and the released NOx species are reduced to N2 when a rich air-to-fuel mixture is injected.Citation114
Since the rich mode of operation is not feasible for Diesel engines, periodic fuel injections are necessary. The amount of fuel and the periodicity of the injections, as well as the storage capacity of the materials, are the main parameters that need to be optimized to reduce the fuel penalty associated with this technology.
Among the main drawbacks, it should be pointed out that the supply of additional fuel to either the cylinder or directly to the exhaust pipe, causes PM, CO, and HC emissions, whose concentration must comply with the current legislation limits. Moreover, the regeneration step must be carried out as efficiently as possible in order to prevent it from having too much of an impact on the fuel economy of the vehicle. Finally, NOx adsorbers can be poisoned by sulfur compounds,Citation2, 115, 116 thus requiring both the use of low sulfur Diesel fuels and the development of efficient desulfation strategies. Therefore, low sulfur fuels favor NOx conversion levels and reduce the frequency of the desulfation step.
Operating principles
During operating conditions, the NOx reduction takes place according to a two-stage mechanism, as shown in Fig. . The NOx trap combines the effect of an oxidation catalyst (e.g. platinum), an adsorbent material (e.g. BaO), and a reduction catalyst (e.g. rhodium).
Figure 9 The basic concept of NOx storage and reduction mechanisms
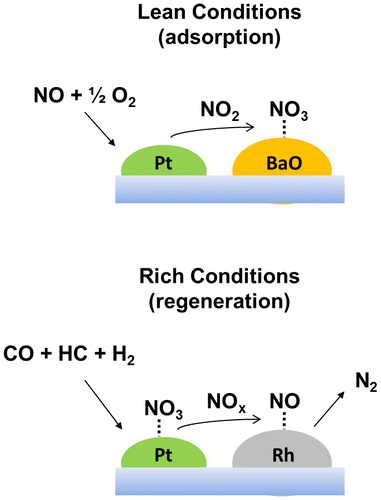
The exhaust is rich in NO (a thermodynamically stable species at high-temperatures), but traps are more effective toward NO2 entrapment. Therefore, the step immediately before NOx adsorption should be the NO oxidation to NO2 step. This operation is performed by means of an oxidation catalyst, such as a Pt-containing system, which is able to operate at the low exhaust temperatures of light-duty Diesel engines, as followsCitation60, 117:(26)
The NO2 formed on the solid surface is trapped on an adsorbent (BaO) in the form of a nitrate (e.g. Ba(NO3)2), which is chemically stable at the operating conditions.
Thus, the following reaction steps can occurCitation10, 60:(27)
(28)
(29)
The adsorption capacity of an NOx trap depends on the accessibility of the BaO sites, which must be regenerated when a certain NOx concentration is attained at the exit of the converter.
During the regeneration process, the oxygen concentration decays to almost zero, and reductant conditions for NO abatement are thus achieved.
The first regeneration step consists of the decomposition of the Ba(NO3)2 and the recovery of the BaO active phase (equation (30)). In this step, NO is released and hence a suitable amount of fuel has to be dosed in order to reduce the released NO.Citation60(30)
NO reduction is carried out by means of a reducing catalyst, such as an Rh-based system, incorporated in the catalyst formulation.Citation118 This reduction step is similar to that which occurs in a conventional three-way converter for the treatment of the exhausts from gasoline fueled engines. When the engine is switched to a fuel-rich condition, HC, CO, and H2 react with the NO species to form N2, CO2, and H2O (equations (24), (31) and (32)).Citation60(31)
(32)
The operating temperature of these NOx traps has a lower limit, which is determined by the Pt activity toward the NO oxidation, as well as the NOx release and reduction in the regeneration step; on the other hand, the upper limit is related to the stability of the NO3 species, which undergo thermal decomposition at high temperatures, even under lean conditions.Citation119
It is worth noting that several complex phenomena may appear on the surface of NOx traps, since they are multicomponent materials which have different functionalities. For instance, BaCO3 and Ba(OH)2 coexist with BaO on the catalyst surface.Citation120 Moreover, NOx release and reduction may not occur as consecutive steps, but, as a first step, it could appear via a direct nitrate reduction, without thermal decomposition of the adsorbed NOx species.Citation121 Therefore, it is very difficult to investigate the NOx storage/release mechanisms of these catalytic materials.
NOx adsorbents are sensitive to sulfur species: sulfur is a poison for Pt-sites and, in the form of SO3, it is competitive with NO2 in the formation of barium salts (e.g. BaSO4), thus causing a loss of activity toward the adsorption of NO2 (= competitive adsorption). BaSO4 is more stable than the corresponding nitrate, and hence its decomposition occurs at higher temperatures.Citation119 Finally, the possibility of having parasitic reactions that lead to undesired products (e.g. NH3, N2O, H2S) reduces the NOx trap efficacy, and a careful control of the secondary emissions is also necessary.
Multifunctional catalysts
NOx traps are multifunctional catalysts that display adsorbent, oxidation, and reduction functionalities. These materials consist of alkaline earth metals (Ba, Ca, Sr, Mg), alkali metals (K, Na, Li, Cs), and rare earth metals (La and Y).Citation121, 122 As a whole, they appear in the form of binary oxides, mixed oxides (perovskites), and metal-containing zeolites. In particular, alkali metals, such as potassium, show the highest NOx conversion efficacy among these metal groups: the inclusion of K-, Na-, and Cs- based oxides in the adsorbent catalyst formulation increases the NOx reduction to between 350 and 600 °C.Citation123 Another remarkable advantage of alkali metals is their good resistance to sulfur poisoning: their inclusion in Ba-containing materials leads to a better performance; however, this in turn leads to a lower NO conversion, due to the hydrocarbons, which makes the adsorbent catalyst more fuel demanding.Citation124 Finally, alkali-based materials exhibit low performances and give rise to leaching effects in the presence of water vapor.Citation125 It is therefore necessary to find a trade-off between the good NOx reduction activity at high temperatures of alkali metal oxides and their excessive mobility, in order to include them in Ba-based systems.
NOx traps for light-duty Diesel engines, which operate at lower temperatures, may eliminate the necessity of using alkali metals.
A washcoat is usually obtained using γ-Al2O3. Washcoats are often employed due to their high surface area (>100 m2 g−1); this allows high dispersion of the active sites. Ba and Al mixed oxides are designed to limit BaO sintering in the 700–800 °C temperature range.Citation123 Another washcoat component is TiO2, whose acidity gives a lower sulfur affinity, although it also reduces the stability of nitrates.Citation126 CeO2 can be considered a good washcoat component as it prevents Pt-sintering. However, its high oxygen storage capacity (OSC) can cause a higher fuel penalty during rich fuel conditions, since some of the hydrocarbons react with the released oxygen. The effects of BaO loading on the trapping capacity of NO2 and on the overall conversion performance of NOx are summarized in Fig. .Citation127 In this work, the catalyst consisted of Pt (2.20 wt.%) and BaO (16.3 wt.%) over Al2O3, and it showed a mean NO conversion of 85% over cycles lasting 60 s, with injection of C3H6 every 10 s. The complete cycle involves a fuel penalty which can be kept under 4% with an overall NO conversion above 80%. The regeneration strategy is closely related to the nature of the reductant species. The regeneration step is constituted by a short pulse of a reducing agent, such H2, CO, or HC, to convert the stored nitrates. The activity of a complex multicomponent catalyst toward NOx reduction is reported in the work by Takahashi et al.Citation128: the catalyst consisted of an Al2O3-based washcoat of CeO2–ZrO2 oxygen storage materials, with Ba and K oxides as the NOx storage compounds and with Pt and Rh as the supported noble metals. Figure reports a schematization of the adsorption capacity of the fresh/regenerated catalyst: the capacity of the reductant species to restore the BaO sites is considered. “Lean 1” is the inlet composition of the gas fed (namely at 250 °C), and represents a gas model for the exhausts; when the lean atmosphere is switched on, the outlet NOx concentration gradually increases with time, up to reach a constant level.
Figure 10 NOx conversion (Section A) and selectivity to N2 (Section B) on BaO loading. NO = 500 ppm, O2 = 5% (with C3H6 = 0.7% during regeneration), temperature = 375 °C and GHSV = 60,000 h−1
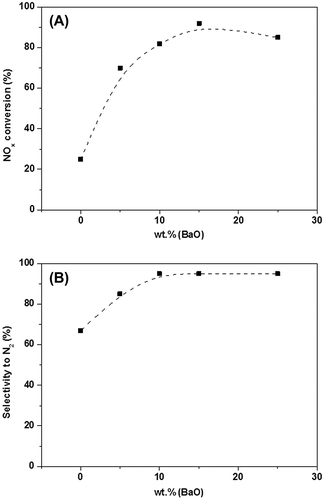
Figure 11 NOx concentration curves, during lean and rich periods, in the inlet and outlet using Lean1 and gases at 250 °C
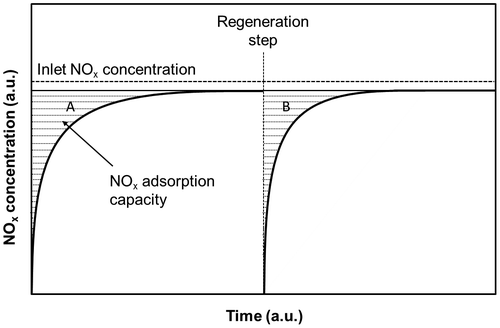
Shaded area A is related to the NOx amount stored in the catalyst, while shaded area B is related to the number of regenerated NOx storage sites on the catalyst. Takahashi et al.Citation128 then established that the effectiveness of the reducing agents follows the following order: C3H6 < CO < H2.
One extremely interesting attempt to improve the NOx absorbers activity is the one from Toyota,Citation129 which investigated the effect of an oscillating air: fuel ratio for the regeneration of the trap, in the so-called Di-Air system. The fundamental finding of this phenomenon is that a high-frequency injection of hydrocarbons might considerably improve the NOx regeneration phase, at high temperatures, with a very moderate fuel penalty (<2%).
The combination of different catalytic functionalities could also be implemented to achieve synergies that increase the overall NOx abatement efficiency. A relevant example is the embodiment of an SCR catalytic functionality in a NOx trap. This system was presented by Ford in 2004, and widely studied by the group at the University of Houston (see the general concept inCitation130). The concept is based on the fact that, in the presence of H2 during the rich pulses of the NOx trap regeneration, NH3 can be formed by direct reaction with the previously stored NOxCitation131; CO instead leads to NH3 formation through intermediate isocyanate species, in water-assisted reaction. The water gas shift reaction makes both pathways to co-exist, due to the presence of CO2 and H2O in the exhausts.Citation131 The suitable proportion of the SCR and NOx trap catalysts in the overall formulation is determined by the amount of NH3 generated in the NOx trap during its regeneration, which should be utilized by the SCR adsorption sites. Since it was found that only the neighboring SCR catalyst concurs in adsorbing the NH3 produced in the NOx trap in the reduction stage, a dual-layer configuration (two layers with different composition) outperforms the dual-brick (consecutive washcoat formulations along the axial length), largely because the NH3 generated in the NOx trap layer is better utilized in an adjacent SCR layer.Citation130 The optimal configuration in terms of relative thickness of the layers depends on the operating temperatures and SCR site density.
Plasma-assisted catalytic NOx reduction
Non-thermal, atmospheric pressure plasma has been widely studied for the removal of VOC from waste gas streams for almost 20 years.Citation132
Non-thermal plasma (NTP) discharges in exhaust gas have been investigated as a potential technology to reduce NOx and PM emissions in Diesel exhaust as well as NOx and cold start hydrocarbons in lean gasoline exhaust.
NTP is generated, by means of an electrical excitation, to induce thermodynamically unstable NOx species; the latter should decompose into N2 and O2.
In automotive applications, the reduction of NOx is preferable. On the other hand, oxidative NOx reactions dominate the subsequent kinetics, converting most flue gas NO to NO2 and HNO3Citation133–135 through reactions with plasma-produced oxygen and hydroxyl (equations (33) and (34)).(33)
(34)
Several other byproducts can be produced, including N2O, N2O3, N2O4, HONO, and HONO2, and the selectivity of the plasma aftertreatment therefore remains an important drawback, as does the large power requirement. The electric power supply has similar characteristics for each experimental setup: the ΔV range is from 5 to 35 kV and the frequency is about 1 kHz. Park et al. investigated NOx abatement for Diesel exhaust, for which pulsed voltages (namely 24 kV and 600 Hz) were shown to be more effective than a direct current (about 80% NOx conversion).Citation134 HNO3 (and other acidic byproducts) can be removed easily at a stationary combustion facility in the form of solid nitrates using a downstream scrubber. However, neither chemical scrubbing nor the conversion of NO to HNO3 constitutes a practical approach for mobile engine NOx control, due to the requirements of onboard scrubbing chemicals, the periodic disposal of accumulated solid waste, and the requirement of an acid scrubber. Yamamoto et al.Citation135 have proposed a plasma reactor followed by a Na2SO3 solution scrubber to reduce volumes, even though this technology can only be used for Diesel engine trucks.
Combinations of plasma with solid catalysts, referred to as “plasma-assisted catalysts” or simply “plasma catalysts”, have been suggested for NOx reduction.Citation135
As a whole, plasma reactors are pulsed corona-type reactors and the catalyst is generally placed after the plasma device (Fig. ).Citation139 Although plasma aftertreatment favors oxidation over reduction, a useful synergism can be observed when it is combined with a catalyst.Citation134 The reducing capability of some catalysts is enhanced considerably when NOx is presented to catalytic surfaces as NO2 (peroxyl radicals favor the conversion of NO to NO2) rather than NO.Citation137–140 This means that the NTP technology may significantly improve catalyst selectivity and removal efficiency. Thus, lower temperatures can be used in NOx reduction with plasma-catalyst technology than with NTP. An increase in temperature improves catalytic activity but the action of plasma is only retained from room temperature to 300 °C. Moreover, the presence of water vapor and oxygen promotes the NO removal rate, as a consequence of synergistic phenomena on the catalyst surface.Citation137
Figure 12 Scheme of the plasma-assisted catalytic reduction system developed at Lawrence Livermore National laboratory
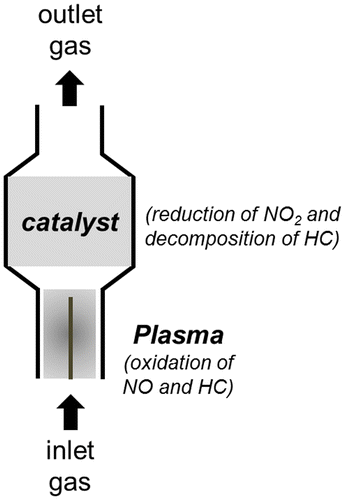
Several catalysts, including γ-Al2O3, TiO2, MnO2/TiO2, Ag/TiO2, V2O5–WO3/TiO2, HZSM-5, NaX, Cu-ZSM and Na-Y and Ba-Y, have been proposed to be active for this technology.Citation135, 138, 139 The best performance for NOx removal at low temperatures (150–270 °C) has been achieved using Na- and Ba-doped zeolite Y. However, promising results (90% NOx removal) have also been obtained with In/γ-Al2O3 catalysts in the presence of sulfur oxides.Citation140
Over the years, several strategies have been proposed to improve NOx abatement via plasma-assisted catalytic technologies. Among these, the multiple-step treatment strategy, whereby two or more plasma-catalyst reactors are utilized in series, has been shown to increase the maximum NOx conversion.Citation141 Moreover, this technology should reduce the energy and/or hydrocarbon supplies for fixed conversion efficiency. Similarly, the HC-SCR process occurs via oxidation of NO, followed by the reduction of NO2 with hydrocarbons (vide supra); hence, NTP can oxidize NO without depleting the number of hydrocarbons available for the reduction of NO2 to N2.Citation142 This means that the function of SCR catalysts could be greatly simplified by focusing on the reduction of NO2 by hydrocarbons.
A different approach has been studied by Bittenson et al.,Citation143 who proposed generating atomic nitrogen in an electric discharge external to the exhaust stream, followed by rapid injection and mixing into exhaust gas to achieve an NOx chemical reduction.(35)
This approach leads to the elimination of the generation of very reactive oxygen and hydroxyl species by the discharge, thus preventing contact between soot and wet particles and the electrode of the plasma reactor; in this case, the electrical power supply has a larger frequency: 40–60 kHz.
Simultaneous NOx and soot removal systems
The stringent regulation limits on both NOx and PM are forcing the automotive industry, one hand, to maximize engine control to reduce pollutant emissions, and on the other hand to pile up a number of costly catalytic converters; this results in rather high pressure drops, complex control features, inefficiency linked to considerable weight and space consumption and elevated costs. In this context, Diesel particulate traps appear to be necessary. Our research group has been involved in various European R&D programs (e.g. CATATRAP, DEXA-cluster, TOP-Expert, ATLANTIS) for several years, with the aim of developing novel multifunctional catalytic traps.Citation144 The simultaneous removal of soot and NOx in a single catalyzed filter (single brick solution) represents the most ambitious strategy in this field, in view of the considerable advantages in terms of both investment costs and pressure drop reduction.
The possibility of obtaining a contemporary reduction in NOx and soot from Diesel engine exhausts has clearly been pointed out in several kinetic studies (Fig. ).Citation14, 15, 145–149. Fino et al.Citation145 studied the kinetics of a soot-NO-O2 reacting system over perovskite-type catalysts and formulated two reaction mechanisms (Fig. ). In mechanism 1, soot combustion leads to the formation of two oxygen vacancies on the catalytic surface, which become active centers for the chemisorption of two NO molecules. Hence, the adsorbed NO dissociates into N (ads) and O (ads) with subsequent formation of either N2 or N2O. However, this mechanism does not account for the positive effect of molecular oxygen, which should conversely lower the reaction rate by filling up the oxygen vacancies over the oxidation catalysts.Citation145–147 On the other hand, according to mechanism 2, carbon plays a role in the reduction of NO through the formation of C(N,O) adducts. The latter are formed through the combination of reactive free carbon (Cf) and the NO molecule adsorbed on the catalyst surface (NOad). Finally, N2 can be formed through the reaction of C(N,O) with further NOad (LH-type kinetics) or directly with a gaseous NO molecule (ER-type kinetics). However, it is also possible for N2 to be formed through the decomposition of C(N,N) adducts. According to this mechanism, the promoting effect of oxygen can be ascribed to the formation of active centers for NO chemisorption on Cf as a consequence of carbon combustion. The other beneficial effect is the easy formation of NO2, which is much more active than NO for the reduction of soot. Thus, the presence of soot has a beneficial effect in a combined SCR + CSF at low temperature, since it may promote the reduction of NO2 (via the “fast SCR reaction”). On the other hand, the NO2/NO ratio has to be kept close to one for effective SCR + SCF systems.
Figure 13 Reaction mechanisms proposed for the soot/NO2/O2 reacting system over perovskite-type catalysts
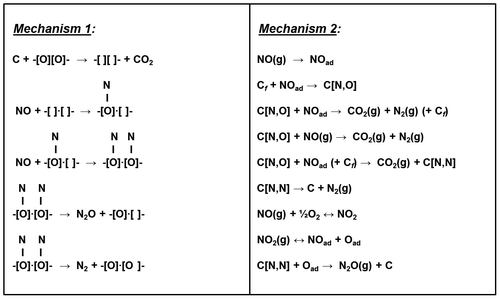
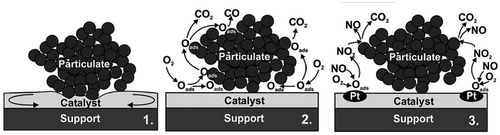
Figure 14 Simultaneous abatement of NOx and soot over different solid catalysts: 1 = mobile catalyst; 2 = catalyst promoting oxygen spillover; 3 = catalyst coupling a NO → NO2 functionalityCitation148
DeSoot-DeNOx Catalysts
La-K-Cu-V-based perovskitesCitation145 have shown their potential application as active catalysts for the simultaneous removal of NOx and soot. The role of vanadium species in the perovskite lattice has been found to be prevalent in obtaining outstanding NOx abatement efficiencies. Nevertheless, several other complex reaction pathways involving reaction intermediates, either present on the catalyst surface or on that of the carbon particulate itself, could take place.Citation145 On the other hand, nanostructured spinel-type oxide catalysts, that is, AB2O4 (where A = Co and Mn, B = Cr and Fe) have proved to be particularly active in the simultaneous removal of soot and NOx in the 350–450 °C temperature range.Citation146, 147 The best compromise between soot and NO abatement below 400 °C has been shown by the CoCr2O4 catalyst. The relevant activity of chromite catalysts could be explained by their higher amount of suprafacial, weakly chemisorbed oxygen (α-species), which contributes actively to soot combustion through spillover in the 300–500 °C temperature range. Similarly, nanostructured perovskite-type lanthanum ferrites, that is, La1−xAxFe1−yByO3 (where A = Na, K, Rb and B = Cu) have displayed high catalytic activity toward carbon combustion and NO conversion in the same temperature range.Citation144 Other multicomponent catalysts, characterized by the highest possible α-oxygen type concentration, have been prepared by our group over the last few years, with careful attention being paid to their compatibility with the substrate material or to the poisoning components present in the Diesel exhaust gas (e.g. sulfur oxides). Using a standard protocol on an engine bench, promising results have been obtained with the La0.9K0.1Cr0.9O3−δ + 1 wt% Pt catalyst over a wall-flow SiC trap.Citation150 The presence of Pt in fact aids the oxidization of NO and therefore allows more NO-NO2-NO cycles before it leaves the catalyst (the efficiency of NOx utilization can be as high as 140% on average). Li et al.Citation151, 152 and Kotarba et al.Citation153 have reported that K-promoted CoMgAlO hydrotalcite can favor soot combustion, but can also lead to a 30% conversion of NOx under “real” conditions. Lin et al.Citation154 have shown high catalytic activity of BaAl2O4 systems for the simultaneous removal of soot and NOx under “loose” contact conditions.
It is worth noting that ceria-based catalysts have received a great deal of attention because CeO2 alone, or in combination with other metals/metal oxides, may exhibit promising oxidation activity under either O2 or in a NOx/O2 atmosphere.Citation155 Thus, zirconium and many rare earth elements (e.g. La, Pr, Sm, Y, Gd, Tb, Lu, Hf and Nd) have been introduced into the ceria framework to improve the oxidation activity (OSC and redox properties) of CeO2-based materials and their structural stability. The redox behavior and the availability of chemisorbed oxygen (α-species) are important features for/of these materials.Citation156–158 Atribak et al.Citation149 have reported interesting results with Ce–Zr mixed oxides for soot combustion under NOx/O2. Among the different mixed oxides, Ce0.76Zr0.24O2 provided the best performance.Citation159 The same authors observed that the most active mixed oxides are those which combine high surface areas with a homogeneous distribution of cerium and zirconium throughout the particles (Ce/Zr surface ratio about 3.2).Citation149
The number of soot-catalyst contact points also plays a significant role in complex solid–solid–gas systems, just as the overall activity for NOx and soot removal depend on the interaction between the two solids and the gas mixture.Citation160, 161 Nanostructured CeO2-based materials, or other mixed oxides, are particularly interesting because of their small-featured size, which endows them with size- and shape-dependent properties due to the high surface-to-volume ratio (= higher number of coordinated unsaturated sites), and their unique electronic features (quantum size effects).Citation162–165 Moreover, the effects of cooperation between active sites (or between different phases) are favored, thus leading to a simultaneous NOx and soot abatement.
Some issues pertaining to Diesel NOx aftertreatment catalysts
One of the main current issues for NOx aftertreatment catalysts is that no material seems to ensure a high NOx abatement throughout the whole Diesel exhaust temperature range (i.e. 200–500 °C).Citation2–4, 8, 14, 30 As reported above, many multicomponent catalysts have been designed over the years to increase their NOx abatement activity and to cover a broader temperature range. This is often possible since synergistic (beneficial) effects may arise through phase-cooperation (namely, the structural relations among atoms/ions/electrons) and spillover phenomena.Citation166, 167 Moreover, solid catalysts must be active and stable during operating conditions. As far as the former aspect is concerned, catalytic materials are usually tested under laboratory conditions, which can be sometimes be different from real operating conditions. For instance, researchers may perform the activity tests with relatively low GHSV or O2 concentrations; however, high values of these parameters are known to adversely affect NOx abatement. Similarly, two features of Diesel exhausts can be critical for catalyst stability, that is, the sulfur content and the presence of water vapor associated with high temperatures. Sensitivity to sulfur poisoning can also be a major problem for metal-exchanged zeolites.Citation95 Feeley et al.Citation102, 168 have found that long-term exposure of Cu-ZSM-5 to SO2 leads to its permanent deactivation. Conversely, Pt-based catalysts may exhibit good resistance to poisoning by SO2. Nevertheless, these catalysts favor the oxidation of SO2 and thus cause an increase in the total mass of the particulate emitted. Moreover, phosphorous from lubricating oil can cause a decline in catalyst performance.
In the case of low-temperature DeNOx catalysts, a further problem still has to be solved, that is, the formation of N2O instead of N2, with a subsequent negative environmental impact, due to the fact that N2O is a greenhouse gas.Citation169
Sensitivity to water vapor is a very critical limitation for the automotive application of solid catalysts. In fact, since combustion exhaust streams are rich in water vapor (10–16%), catalysts should be able to operate in the presence of high levels of water vapor. Given the fact that NOx levels are often <2% (typically at ca. 50 ppm level), it is very difficult to find a catalyst with active sites, which are necessary for NO sorption, that will not be flooded by water. Because of the high exhaust velocities (GHSV > 30,000 h−1), it is impractical to try to separate such large quantities of water from such a huge volume of gas. Therefore, the thermal and hydrothermal stability of catalysts is pivotal for any successful NOx decomposition technology. The effect of water on the stability of Cu-ZSM-5 has been widely investigated (vide supra).Citation47–50 Zeolite activity decreases to various extents in the presence of water, according to the amount of water and the reaction temperature. This effect is probably due to the fact that water, like NOx, is a good Lewis base and competes for the same active sites on which NOx reduction must occur.Citation95 Conversely, Corma et al.Citation54 observed that the deactivation of Cu-ZSM-5 occurs through the partial dealumination of the zeolite, the reversible migration of copper species, and the irreversible formation of catalytically inactive and stable Cu–Al clusters, which have some resemblance to CuAl2O4, but without the symmetry of Cu in the spinel structure. In order to improve the hydrothermal resistance of zeolites, several methods have been proposed over the years, but none of them seems so far to have completely solved the problem. On the other hand, as previously mentioned, Pt-ZSM-5 (Pt nanoparticles dispersed on the zeolite surface) is more resistant to water vapor.Citation170 Nevertheless, the low selectivity, and hence the relatively high amounts of N2O formed in the presence of this catalyst are important issues that still need to be solved. Nanostructured mixed metal oxides, on the other hand, can be considered suitable candidates for NOx abatement, due to their unique electronic features and (usually) high hydrothermal stability.
This analysis is consistent also in the case of the use of biofuels: as far as emissions are concerned, biodiesel produces substantially smaller amounts of CO, hydrocarbons (HC), and soot, which has been reported to dramatically decrease by 50, 70, and 50%, respectively, for pure biodiesel and to moderately decrease by 12, 20, and 12% for a diesel with a 20% biodiesel content.Citation171 These are average values, but a more than 80% particulate matter emission reduction was recorded in tests with pure biodieselCitation172 with respect to fossil one. These emission reductions are reached at the price of a modest 10% emission increase in nitrogen oxides (NOx) for pure biodiesel, which is close to zero for a 20% biodiesel content,Citation171 therefore not entailing radically different catalytic formulations.
Moreover, as in the case of innovative biofuels like farnasane, which often require a re-calibration of the engine map to achieve combustions efficiencies comparable to the ones of conventional diesel, beneficial results in terms of emissions can be achieved at the same time, thus eliminating the problem of increased pollutant emissions.Citation173
Conclusions
The progressive requirements for fuel-efficient diesel cars highlight the problem of the necessity of removing NOx under lean-burn conditions in order to satisfy current legislation. Since direct NO decomposition cannot be applied successfully to control Diesel engine emissions, researchers have begun to investigate alternative approaches, such as the selective catalytic reduction of NOx by means of ammonia/urea or hydrocarbons. The SCR of NOx by ammonia/urea has become a key technology for the aftertreatment of exhaust emitted from Diesel and other lean-burn engines. In fact, a high efficiency of NOx removal can be obtained with NH3-SCR (namely 70–98%). The uniqueness of this technology is that the reaction can occur in the presence of excess O2. Thus, the NH3-SCR approach has received a great deal of interest for Diesel-engine vehicles. On the other hand, the HC-SCR of NOx has been investigated extensively since the early 1990s, and many catalysts have been tested. Although promising results have been obtained with this technology (more than 80% NOx conversion), most of the research was conducted in the absence, or low presence, of sulfur. Moreover, it has been revealed that the narrow activity temperature range and the poor activity below 300 °C are relevant drawbacks for the application of HC-SCR catalysts.
Recently, growing interest has been shown in the adsorption of NOx (NOx traps) from lean exhaust, followed by release and catalytic reduction under rich conditions. This strategy has been shown to achieve ca. 70–90% NOx reduction, and seems the most promising approach for NOx abatement in diesel engines, since it does not require a reducing agent. NTP has been proposed as a promising technology to reduce NOx and PM emissions in Diesel exhaust. At present, however, this approach appears more complicated than other advanced technologies for NOx abatement.
The stringent regulation limits for both NOx and PM emissions are forcing the automotive industry to pile up a number of costly catalytic converters, which results in rather high pressure drops, sophisticated control systems, inefficiency linked to considerable weight and space consumption and hence elevated costs. Therefore, the simultaneous removal of soot and NOx in a single filter catalyzed represents the most ambitious strategy, in view of the great advantages that can be obtained in terms of both investment costs and pressure drop reduction. In this scenario, synergistic effects play a key role in catalytic converters. This means that it is necessary to obtain more detailed knowledge of the surface phenomena (reaction mechanisms, cooperative effects, etc.) in order to be able to develop effective catalytic systems.
References
- J. M. Pardiwala, F. Patel and S. Patel: Proc. International Conference on Current Trends in Techology, Nuicone, 2011.
- R. M. Heck, R. J. Farrauto and S. T. Gulati: ‘Catalytic air pollution control: commercial technology’, 3rd edn; 2009, New Jersey, NJ, (Wiley-VCH), Hooken.
- G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp: ‘Handbook of heterogeneous catalysis’, 2nd edn; ; 2008, Weinheim: (Wiley-VCH).
- P. Eastwood: ‘Critical topics in exhaust gas aftertreatment’; 2000, Baldock, Research Studies Press Ltd.
- I. Fechete, Y. Wang and J. C. Védrine: Catal Today, 2012, 189, 2–27.10.1016/j.cattod.2012.04.003
- O. Deutschmann: ‘Modeling and Simulation of Heterogeneous Catalytic Reactions’, 330–331; 2012, Weinheim, Wiley-VCH.
- A. G. Konstandopoulos, M. Kostoglou and N. Vlachos: Int. J. Vehicle Des., 2006, 41, 256–284.10.1504/IJVD.2006.009676
- D. Duprez and F. Cavani: ‘Handbook of advanced methods and processes in oxidation catalysis’, 25–50; 2014, London, Imperial College Press.
- V. Vestreng, L. Ntziachristos, S. Semb, I. S. A. Isaksen and L. Tarrason. Atmos. Chem. Phys., 2009, 9, 1503–1520.10.5194/acp-9-1503-2009
- P. Barbaro and C. Bianchini: ‘Catalysis for sustainable energy production’; 2009, 393–438, Weinheim, Wiley-VCH.10.1002/9783527625413
- I. Nova and E. Tronconi: ‘Urea-SCR technology for deNOx after treatment of diesel exhausts’; 2014, Verlag, Springer.
- S. Yashnik and Z. Ismagilov: Appl. Catal. B Environ., 2015, 170–171, 241–254.10.1016/j.apcatb.2015.01.021
- M. Rutkowska, U. Díaz, A. E. Palomares and L. Chmielarz: Appl. Catal. B: Environ., 2015, 168–169, 531–539.10.1016/j.apcatb.2015.01.016
- P. Granger and V. L. Parvulescu: Chem. Rev., 2011, 111, 3155–3207.10.1021/cr100168g
- Z. Liu and S. I. Woo: Catal. Rev., 2006, 48, 43–89.10.1080/01614940500439891
- J. Hagen: ‘Industrial catalysis: a practical approach’, 2nd edn, 317–328; 2006, Weinheim, Wiley-VCH .
- R. A. van Santen and M. Neurock: ‘Molecular heterogeneous catalysis’; 2006, Weinheim, Wiley-VCH.10.1002/9783527610846
- H. Falsig, T. Bligaard, C. H. Christensen and J. K. Nørskov: Pure Appl. Chem., 2007, 79, 1895–1903.
- C. T. Goralski and W. F. Schneider: Appl. Catal. B, 2002, 37, 263–277.10.1016/S0926-3373(01)00317-4
- V. Johánek, S. Schauermann, M. Laurin, C. S. Chinnakonda, S. Gopinath, J. Libuda and H.-J. Freund: J. Phys. Chem. B, 2004, 108, 14244–14254.10.1021/jp031371f
- Q. Ge and M. Neurock: J. Am. Chem. Soc., 2004, 126, 1551–1559.10.1021/ja036575o
- J. K. Nørskov, F. Studt, F. Abild-Pedersen and T. Bligaard: ‘Fundamental concepts in heterogeneous catalysis’; 2014, Veinheim, Wiley.
- H. Hamada, Y. Kintaichi, M. Sasaki and T. Ito. Chem. Lett., 1990, 7, 1069–1070.10.1246/cl.1990.1069
- A. Ogata, K. Obuchi, K. Mizuno, A. Ohi, H. Aoyama and H. Ohuki. Appl. Catal., 1990, 65, L11–L15.10.1016/S0166-9834(00)81582-1
- H. Huang, Y. Xu, Q. Feng and D. Y. C. Leung: Catal. Sci. Technol., 2015, 5, 2649–2669.10.1039/C4CY01733A
- R. J. Wu, T. Y. Chou and C. T. Yen: Appl. Catal. B Environ., 1995, 6, 105–116.10.1016/0926-3373(95)00005-4
- B. Frank, G. Emig and A. Renken: Appl. Catal. B Environ., 1998, 19, 45–57.10.1016/S0926-3373(98)00057-5
- B. Frank, R. Lubke, G. Emig and A. Renken: Chem. Eng. Technol., 1998, 21, 494–502.
- M. Iwamoto and H. Hamada: Catal. Today, 1991, 10, 57–71.10.1016/0920-5861(91)80074-J
- S. Roy, M. S. Hegde and G. Madras: Appl. Energy, 2009, 86, 2283–2297.10.1016/j.apenergy.2009.03.022
- M. Iwamoto, H. Yahiro, Y. Torikai, T. Yoshioka and N. Mizuno: Catal. Lett., 1990, 11, 1967–1970.
- Z. Say, E. I. Vovk, V. I. Bukhtiyarov and E. Ozensoy: Appl. Catal. B Environ., 2013, 142–143, 89–100.10.1016/j.apcatb.2013.04.075
- Sharma V, P. A. Crozier, R. Sharmaa and J. B. Adams: Catal. Today, 2012, 180, 2–8.10.1016/j.cattod.2011.09.009
- X. Zhang, A. B. Walters and M. A. Vannice: Appl. Catal. B, 1996, 7, 321–336.10.1016/0926-3373(95)00042-9
- P. Esteves, Y. Wu, C. Dujardin, M. K. Dongare and P. Granger: Catal. Today, 2011, 176, 453–457.10.1016/j.cattod.2010.10.068
- D. Marrocchelli, N. H. Perry and S. R. Bishop: Phys. Chem. Chem. Phys., 2015, 17, 10028–10039.10.1039/C4CP05885B
- L. F. Liotta, M. Ousmane, G. Di Carlo, G. Pantaleo, G. Deganello, G. Marcì, L. Retailleaud and A. Giroir-Fendler: Appl. Catal. A, 2008, 347, 81–88.10.1016/j.apcata.2008.05.038
- C. Tofan, D. Klvana and J. Kirchnerova: Appl. Catal. A, 2002, 223, 275–286.10.1016/S0926-860X(01)00764-5
- Y. Shimada, S. Miyama and H. Kuroda: Chem. Lett., 1998, 10, 1797–1800.
- H. Iwakuni, Y. Shinmyou, H. Yano, H. Matsumoto and T. Ishihara: Appl. Catal. B, 2007, 74, 299–306.10.1016/j.apcatb.2007.02.020
- Y. Teraoka, H. Ogawa, H. Furukawa and S. Kagawa: Catal. Lett., 1992, 12, 361–366.10.1007/BF00765065
- C. H. Kim, G. Qi, K. Dahlberg and W. Li: Science, 2010, 327, (5973), 1624–1627.10.1126/science.1184087
- V. Blasin-Aubé, J. Belkouch and L. Monceaux: Appl. Catal. B Environ., 2003, 43, (2), 175–186.10.1016/S0926-3373(02)00302-8
- S. Banerjee and V. R. Choudhary: J. Chem. Sci., 2000, 112, 535–542.10.1007/BF02709286
- A. Corma: J. Catal., 2003, 216, 298–312.10.1016/S0021-9517(02)00132-X
- J. Čejka, A. Corma and S. Zones: ‘Zeolites and catalysis’; 2010, Weinheim, Wiley-VCH.10.1002/9783527630295
- H. Sjövall, L. Olsson, E. Fridell and R. J. Blint: Appl. Catal. B Environ., 2006, 64, 180–188.10.1016/j.apcatb.2005.12.003
- J. Park, H. J. Park, J. H. Baik, I. Nam, C. Shin, J. Lee, B. K. Cho and S. H. Oh: J. Catal., 2006, 240, 47–57.10.1016/j.jcat.2006.03.001
- P. N. Panahi, D. Salari, A. Niaei and S. M. Mousavi: J. Ind. Eng. Chem. 2013, 19, 1793–1799.10.1016/j.jiec.2013.02.022
- C. Seo, B. Choi, H. Kim, C. H. Lee and C. B. Lee: Chem. Eng. J., 2012, 191, 331–340.10.1016/j.cej.2012.03.027
- L. Pang, C. Fan, L. Shao, K..Song, J. Yi, X. Cai, J..Wang, M. Kang.and T. Li: Chem. Eng. J., 2014, 253, 394–401.10.1016/j.cej.2014.05.090
- M. Yu. Kustova, S. B. Rasmussen, A. L. Kustov and C. H. Christensen: Appl. Catal. B, 2006, 67, 60–67.10.1016/j.apcatb.2006.04.014
- W. Weisweiler and R. Wunsch: Chem. Eng. Process, 1998, 37, 229–232.10.1016/S0255-2701(98)00027-0
- P. N. R. Vennestrøm, T. V. W. Janssens, A. Kustov, M. Grill, A. Puig-Molina, L. F. Lundegaard, R. R. Tiruvalam, P. Concepción and A. Corma: J. Catal., 2014, 309, 477–490.10.1016/j.jcat.2013.10.017
- Q. Ye, L. Wang and R. T. Yang: Appl. Catal. A Gen., 2012, 427–428, 24–34.10.1016/j.apcata.2012.03.026
- G. Busca: ‘Heterogeneous catalytic materials’, 478; 2014, Amsterdam, Elsevier.
- R. D. Reitz: Combust. Flame, 2013, 160, 1–8.10.1016/j.combustflame.2012.11.002
- M. V. Twigg: Appl. Catal. B Environ., 2007, 70, 2–15.10.1016/j.apcatb.2006.02.029
- B. Guan, R. Zhan, H. Lin and Z. Huang: J. Environ. Manag., 2015, 154, 225–258.10.1016/j.jenvman.2015.02.027
- J. Yan: ‘Handbook of clean energy systems’, Vol. 2, 1083–1109; 2015, New York, NY, Wiley.
- I. Lezcano-Gonzalez, U. Deka, B. Arstad and A. Van Yperen-De: Phys. Chem. Chem. Phys., 2014, 16, 1639–1650.10.1039/C3CP54132K
- M. Koebel, M. Elsener and G. Madia: SAE Tech. Pap. 2001-01-3625, 2001.
- I. Gekas, L. Nyengaard.and T. Lund: SAE Tech. Pap. 2002-01-0289, 2002.
- W. Mathes, F. Witzel and S. Schnapp: International patent application WO 99/05402, 1999.
- J. T. Tarabulski and J. D. Peter-Hoblyn, US Patent 5809775, 1998.
- www.dieselnet.com
- B. K. Yun and M. Y. Kim: Appl. Therm. Eng., 2013, 50, (1), 152–158.10.1016/j.applthermaleng.2012.05.039
- Y. Shu, H. Sun, X. Quan and S. Chen: J. Phys. Chem. C, 2012, 116, (48), 25319–25327.10.1021/jp307038q
- J. A. Dumesic, N. Y. Topsøe, H. Topsøe, Y. Chen and T. Slabiak: J. Catal., 1996, 163, 409–417.10.1006/jcat.1996.0342
- ‘Advanced Clean-Energy Vehicles (ACEVs), Project Summary’; 2004, Tokyo, Japan, Japan Automobile Research Institute JARI.
- ‘Chemicals known to the state to cause cancer or reproductive toxicity’; 2005, Oakland, CA, California Environmental Protection Agency, OEHHA.
- O. Kröcher, M. Devadas, M. Elsener, A. Wokaun, N. Söger, M. Pfeifer, Y. Demel and L. Mussmann: Appl. Catal. B, 2006, 66, 208–216.10.1016/j.apcatb.2006.03.012
- J. Gieshoff, M. Pfeifer, A. Schafer-Sindlinger, P. Spurk, G. Garr and T. Leprince: SAE Tech. Pap. 2001-01-0514, 2001.
- S. J. Schmieg, S. H. Oh, C. H. Kim, D. B. Brown, J. H. Lee, C. H. F. Peden and D. H. Kim: Catal. Today, 2012, 184, 252–261.10.1016/j.cattod.2011.10.034
- V. Bacher, C. Perbandt, M. Schwefer, R. Siefert, S. Pinnow and T. Turek: Appl. Catal. B, 2015, 162, 158–166.10.1016/j.apcatb.2014.06.039
- M. A. Zamudio, N. Russo and D. Fino: Ind. Eng. Chem. Res., 2011, 50, 6668–6672.10.1021/ie200227u
- P. Zelenka, W. Cartellieri and P. Herzog: Appl. Catal. B, 1996, 10, 3–28.10.1016/0926-3373(96)00021-5
- A. Frobert, S. Raux, S. Rousseau and G. Blanchard: Top. Catal., 2013, 56, (1–8), 125–129.10.1007/s11244-013-9940-5
- J. M. Herreros, P. George, M. Umar and A. Tsolakis: Chem. Eng. J., 2014, 252, 47–54.10.1016/j.cej.2014.04.095
- V. Houel, P. Millington, R. Rajaram and A. Tsolakis: Appl. Catal. B Environ., 2007, 73, (1–2), 203–207.10.1016/j.apcatb.2006.12.005
- A. Frobert, S. Raux, A. Lahougue, C. Hamon, K. Pajot and G. Blanchard: SAE Int. J. Fuels Lubr., 2012, 5, (1), 389–398.
- H. Gu, K. M. Chun and S. Song: Int. J. Hydrogen Energy, 2015, 40, 9602–9610.10.1016/j.ijhydene.2015.05.070
- B. Guan, R. Zhan, H. Lin and Z. Huang: Appl. Therm. Eng., 2014, 66, 395–414.10.1016/j.applthermaleng.2014.02.021
- R. Mrad, A. Aissat, R. Cousin, D. Courcot and S. Siffert: Appl. Catal. A, 2014, available at http://dx.doi.org/10.1016/j.apcata.2014.10.02.
- A. Obuchi, I. Kaneko, J. Oi, A. Ohi, A. Ogata, G. R. Bawenda and S. Kushiyama: Appl. Catal. B, 1998, 15, 37–47.10.1016/S0926-3373(97)00035-0
- A. Obuchi, I. Kaneko, J. Oi, A. Ohi, A. Ogata, G. R. Bawenda and S. Kushiyama: Appl. Catal. B Environ., 1998, 15, 37–47.10.1016/S0926-3373(97)00035-0
- J. P. Breen and R. Burch: Top. Catal., 2006, 39, 53–58.10.1007/s11244-006-0037-2
- P. S. Kim, M. K. Kim, B. K. Cho, I. S. Nam and S. H. Oh: J. Catal., 2013, 301, 65–76.10.1016/j.jcat.2013.01.026
- G. G. De Soete: Comb. Sci. Technol., 1996, 121, 103–121.
- L. J. Lobree, A. W. Aylor, J. A. Reimer and A. T. Bell: J. Catal., 1997, 169, 188–193.10.1006/jcat.1997.1699
- A. W. Aylor, L. J. Lobree, J. A. Reimer and A. T. Bell: J. Catal., 1997, 170, 390–401.10.1006/jcat.1997.1776
- L. J. Lobree, I. Hwang, J. A. Reimer and A. T. Bell: Catal. Lett., 1999, 63, 233–240.10.1023/A:1019033508479
- L. J. Lobree, A. W. Aylor, J. A. Reimer and A. T. Bell: J. Catal. 1998, 181, 189–204.
- M. Iwamoto and H. Yahiro: Catal. Today, 1994, 22, 5–18.10.1016/0920-5861(94)80089-8
- T. Maunula, J. Ahola and H. Hamada: Appl. Catal. B Environ., 2006, 64, 13–24.10.1016/j.apcatb.2005.10.016
- F. Bin, C. Song, G. Lv, J. Song, X. Cao, H. Pang and K. Wang: J. Phys. Chem. C, 2012, 116, (50), 26262–26274.10.1021/jp303830x
- S. Erkfeldt, A. Palmqvist and M. Petersson: Appl. Catal. B, 2011, 102, 457–554.
- A. Sultana, M. Sasaki, K. Suzuki and H. Hamada: Catal. Commun., 2013, 41, 21–25.10.1016/j.catcom.2013.06.028
- G. Landi, L. Lisi, R. Pirone, G. Russo and M. Tortorelli: Catal. Today, 2012, 191, 138–141.10.1016/j.cattod.2012.01.018
- H. K. Shin, H. Hirabayashi, H. Yahiro, M. Watanabe and M. Iwamoto: Catal Today, 1995, 26, 13–21.10.1016/0920-5861(95)00125-Y
- J. S. Feeley, M. Deeba, R. J. Farrauto, D. Dang: US Patent 5776423 A, 1998.
- M. Deeba, J. Feeley, R. Farrauto, N. Steinbock and A. Punke: SAE Tech. Pap. 952491, 1995.
- T. N. Burdeinaya, V. A. Matyshak, V. F. Tretyakov, L. S. Glebov, A. G. Zakirova and M. A. Carvajal: Appl. Catal. B, 2007, 70, 128–137.10.1016/j.apcatb.2005.11.027
- M. K. Neylon, M. J. Castagnola, N. B. Castagnola and C. L. Marshall: Catal. Today, 2004, 96, 53–60.10.1016/j.cattod.2004.05.007
- T. Miyadera: Appl. Catal. B, 1997, 13, 157–165.10.1016/S0926-3373(96)00100-2
- K. Arve, K. Svennerberg, F. Klingstedt, K. Eränen, L. R. Wallenberg, J.-O. Bovin, L. Čapek and D Yu Murzin: J. Phys. Chem. B, 2006, 110, 420–427.10.1021/jp055147x
- K. Masuda, K. Shinoda, T. Kato and K. Tsujimura: Appl. Catal. B, 1998, 15, 63–73.
- H. Kannisto, H. H. Ingelten and M. Skoglundh: Top. Catal., 2009, 52, 1817–1820.10.1007/s11244-009-9359-1
- G. R. Bamwenda, A. Ogata, A. Obuchi, J. Oi, K. Mizuno and J. Skrzypek: Appl. Catal. B, 1995, 6, 311–323.10.1016/0926-3373(95)00020-8
- Y. Miyahara, M. Takahashi, T. Masuda, S. Imamura, H. Kanai, S. Iwamoto, T. Watanabe and M. Inoue: Appl. Catal. B, 2008, 25, 289–296.10.1016/j.apcatb.2008.04.005
- Y. Zhang, R. W. Cattrall, I. D. McKelvie and S. D. Kolev: Gold Bull., 2011, 41, 145–153.10.1007/s13404-011-0025-6
- A. Ueda, T. Oshima and M. Haruta:. Appl Catal. B, 1997, 12, 81–93.10.1016/S0926-3373(96)00069-0
- A. Ueda and M. Haruta: Appl. Catal. B, 1998, 18, 115–121.10.1016/S0926-3373(98)00026-5
- V. G. Milt, C. A. Querini, E. E. Mirò and M. A. Ulla: J. Catal., 2003, 220, 424–432.10.1016/S0021-9517(03)00285-9
- N. Rankovic, C. Chizallet, A. Nicolle, D. Berthout and P. Da Costa: Oil Gas Sci. Technol. 2013, 68, (6), 951–1113.
- W. S. Epling, L. E. Campbell, A. Yezerets, N. W. Currier and J. E. Parks II: Catal. Rev., 2004, 46, 163–245.10.1081/CR-200031932
- A. Lindholm, N. W. Currier, E. Fridell, A. Yezerets and L. Olsson: Appl. Catal. B, 2007, 75, 78–87.10.1016/j.apcatb.2007.03.008
- L. J. Gill, P. G. Blakeman, M. V. Twigg and A. P. Walker: Top. Catal., 2004, 28, 157–164.10.1023/B:TOCA.0000024345.85369.73
- V. G. Milt, A. Querini, E. E. Mirò and M. A. Ulla: J. Catal., 2003, 220, 424–432.10.1016/S0021-9517(03)00285-9
- L. Lietti, P. Forzatti, I. Nova and E. Tronconi: J. Catal., 2001, 204, 175–191.10.1006/jcat.2001.3370
- I. Nova, L. Lietti, L. Castoldi, E. Tronconi and P. Forzatti: J. Catal., 2006, 239, 244–254.10.1016/j.jcat.2006.01.013
- K. Krutzsch, D. Webster, E. Chaize, S. Hodjati, C. Petit, V. Pitchon, A. Kiennemann, R. Loenders, O. Monticelli, P. A. Jacobs, J. A. Martens, B. Kasemo, M. Weibel and G. Wenninger: SAE Tech. Pap. 982593, 1998.
- J. S. Hepburn and W. Watkins, European Patent Application, EP 0 857 510 A1, 1998.
- D. Dou and J. Balland: SAE Tech. Pap. 2002-01-0734, 2002.
- W. A. Cutler and J. P. Day: SAE Tech. Pap. 1999-01-3500, 1999.
- I. Hachisuka, T. Yoshida, H. Ueno, N. Takahashi, A. Suda and M. Sugiura: SAE Tech. Pap. 2002-01-0732, 2002.
- K. S. Kabin, R. L. Muncrief, M. P. Harold and Y. Li: Chem. Eng. Sci., 2004, 59, 5319–5327.10.1016/j.ces.2004.07.114
- N. Takahashi, K. Yamazaki, H. Sobukawa and H. Shinjoh: Appl. Catal. B, 2007, 70, 198–204.10.1016/j.apcatb.2005.10.029
- Y. Bisaiji, K. Yoshida, M. Inoue, K. Umemoto and T. Fukuma, JSAE 20119272, SAE Tech. Pap. 2011-01-2089.
- B. M. Shakya, M. P. Harold and V. Balakotaiah: Chem. Eng. J., 2014, 237, 109–122.10.1016/j.cej.2013.10.008
- F. Can, X. Courtois, S. Royer, G. Blanchard, S. Rousseau and D. Duprez: Catal. Today, 2012, 197, 144–154.10.1016/j.cattod.2012.07.032
- D. Duprez and F. Cavani: ‘Handbook of advanced methods and processes in oxidation catalysis’, 155–172; 2014, London, Imperial College Press.
- T. Hammer and S. Broer: SAE Tech. Pap. 982428, 1998.
- M. C. Park, D. R. Chang, M. H. Woo, G. J. Nam and S. P. Lee: SAE Tech. Pap. 982514, 1998.
- T. Yamamoto and C. L. Yang: SAE Tech. Pap. 982432, 1998.
- B. M. Penetrante and R. C. Brusasco: SERDP Project Cp-1077, 2001.
- G. Yu, Q. Yu, K. Zeng and X. Zhai: J. Environ. Sci., 2005, 17, 846–848.
- J. Van Durme, J. Dewulf, C. Leys and H. Van Langenhove: Appl. Catal. B, 2008, 78, 324–333.10.1016/j.apcatb.2007.09.035
- J. H. Kwak, J. Szanyi and C. H. F. Peden: J. Catal., 2003, 10, 291–298.10.1016/j.jcat.2003.08.007
- D. N. Tran, C. L. Aardahl, K. G. Rappe, P. W. Park and C. L. Boyer: Appl. Catal. B, 2004, 48, 155–164.10.1016/j.apcatb.2003.10.008
- R. G. Tonkyn, S. E. Barlow and J. W. Hoard: Appl. Catal. B, 2003, 40, 207–217.10.1016/S0926-3373(02)00150-9
- P. Talebizadeh, M. Babaie, R. Brown, H. Rahimzadeh, Z. Ristovski and M. Arai: Renewable Sustainable Energy Rev., 2014, 40, 886–901.10.1016/j.rser.2014.07.194
- S. N. Bittenson and F. E. Becker: SAE Tech. Pap. 982515, 1998.
- D. Mescia, J. C. Caroca, N. Russo, N. Labhsetwar, D. Fino, G. Saracco and V. Specchia: Catal. Today, 2008, 137, 300–305.10.1016/j.cattod.2007.11.010
- D. Fino, P. Fino, G. Saracco and V. Specchia: Appl. Catal. B: 2003, 43, 243–259.10.1016/S0926-3373(02)00311-9
- D. Fino, N. Russo, G. Saracco and V. Specchia: J. Catal., 2006, 242, 38–47.10.1016/j.jcat.2006.05.023
- D. Fino, N. Russo, G. Saracco and V. Specchia: Power Technol., 2008, 180, 74–78.10.1016/j.powtec.2007.03.003
- D. Fino and V. Specchia: Power Technol., 2008, 180, 64–73.10.1016/j.powtec.2007.03.021
- I. Atribak, A. Bueno-Lopez and A. Garcia-Garcia: Top. Catal., 2009, 52, 2088–2091.10.1007/s11244-009-9388-9
- E. Cauda, D. Fino, G. Saracco and V. Specchia: Top. Catal. 2004, 30, (31), 299–303.10.1023/B:TOCA.0000029766.91816.d2
- Q. Li, M. Meng, N. Tsubaki, X. Li, Z. Li, Y. Xie, T. Hu and J. Zhang: Appl. Catal. B, 2009, 91, 406–415.10.1016/j.apcatb.2009.06.007
- Q. Li, M. Meng, Z. Q. Zou, X. G. Li and Y. Q. Zha: J. Hazard. Mater., 2009, 161, 366–372.10.1016/j.jhazmat.2008.03.103
- B. Ura, J. Trawczynski, A. Kotarba, W. Bieniasz, M. J. Illá-Gόmez, A. Bueno-López and F. E. López-Suárez: Appl. Catal. B, 2011, 101, 169–175.10.1016/j.apcatb.2010.09.018
- H. Lin, Y. Li, W. Shangguan and Z. Huang: Combust. Flame, 2009, 156, 2063–2070.10.1016/j.combustflame.2009.08.006
- A. Trovarelli and P. Fornasiero: ‘Catalysis by ceria and related materials’, 2nd edn, 565–621; 2013, London, Imperial College Press
- E. Aneggi, M. Boaro, C. de Leitenburg, G. Dolcetti and A. Trovarelli: J. Alloy Compd. 2006, 408–412, 1096–1102.10.1016/j.jallcom.2004.12.113
- S. Bernal, G. Blanco, J. M. Pintado, J. M. Rodrìguez-Izquierdo and M. P. Yeste: Catal. Commun., 2005, 6, 582–585.10.1016/j.catcom.2005.05.011
- S. Carrettin, P. Concepciόn, A. Corma, J. M. Lόpez-Nieto and V. F. Puntes: Angew Chem. Int. Ed., 2004, 43, 2538–2540.10.1002/(ISSN)1521-3773
- I. Atribak, A. Bueno-López and A. García-García: J. Catal., 2008, 259, 123–132.10.1016/j.jcat.2008.07.016
- S. Bensaid, N. Russo and D. Fino: Catal. Today, 2013, 216, 57–63.10.1016/j.cattod.2013.05.006
- M. Piumetti and S. Bensaid, N. Russo and D. Fino: Appl. Catal. B, 2015, 165, 742–751.10.1016/j.apcatb.2014.10.062
- S. J. Tans, A. R. M. Verschueren and C. Dekker: Nature, 1998, 393, 49–52.
- A. K. Geim and K. S. Novoselov: Nat. Mater., 2007, 3, 183–191.10.1038/nmat1849
- D. L. Feldheim and C. A. Foss: ‘Metal nanoparticles’, 352; 2002, New York, NY, Marcel Dakker.
- J. M. Thomas and R. Raja: Top. Catal., 2010, 53, 848–858.10.1007/s11244-010-9517-5
- J. C. Védrine: Appl. Catal. A, 2014, 474, 40–50.10.1016/j.apcata.2013.05.029
- L. T. Weng and B. Delmon: Appl. Catal. A, 1992, 81, 141–213.10.1016/0926-860X(92)80093-R
- J. S. Feeley, M. Deeba and R. J. Farrauto: SAE Tech. Pap. 950747, 1995.
- C. Lambert, D. Dobson, C. Gierczak, G. Guo, J. Ura and J. Warner: Int. J. Powertrains, 2014, 3, (1), 4–25.10.1504/IJPT.2014.059410
- C. Chen, F. Chen, L. Zhang, S. Pan, C. Bian, X. Zheng, X. Meng and F. S. Xiao: Chem. Commun., 2015, 51, 5936–5938.10.1039/C4CC09383F
- ‘A comprehensive analysis of biodiesel impacts on exhaust emissions’. U.S. Environmental Protection Agency, EPA420-P-02-001, October 2002.
- J. C. Caroca, F. Millo, D. Vezza, T. Vlachos, A. De Filippo, S. Bensaid, N. Russo and D. Fino: Ind. Eng. Chem. Res., 2011, 50, 2650–2658.10.1021/ie1006799
- F. Millo, S. Bensaid, D. Fino, S. J. Castillo Marcano, T. Vlachos and B. K. Debnath: Fuel, 2014, 138, 134–142.10.1016/j.fuel.2014.07.060