
ABSTRACT
Ketamine is a phencyclidine derivative and a non-competitive antagonist of N-methyl-D-aspartate (NMDA) receptor for which glutamate is the full agonist. It produces a functional dissociation between the thalamocortical and limbic systems, a state that has been termed as dissociative anaesthesia. Considerable variability in the pharmacokinetics and pharmacodynamics between individuals that can affect dose-response and toxicological profile has been reported. This review aims to discuss pharmacokinetics of ketamine, namely focusing on all major and minor, active and inactive metabolites. Both ketamine optical isomers undergo hepatic biotransformation through the cytochrome P450, specially involving the isoenzymes 3A4 and 2B6. It is first N-demethylated to active metabolite norketamine. Different minor pathways have been described, namely hydroxylation of the cyclohexanone ring of ketamine and norketamine, and further conjugation with glucuronic acid to increase renal excretion. More recently, metabolomics data evidenced the alteration of several biological pathways after ketamine administration such as glycolysis, tricarboxylic acid cycle, amino acids metabolism and mitochondrial β-oxidation of fatty acids.
It is expected that knowing the metabolism and metabolomics of ketamine may provide further insights aiming to better characterize ketamine from a clinical and forensic perspective.
Introduction
Ketamine is a synthetic, non-barbiturate, injectable dissociative anaesthetic first synthesized by Calvin Stevens of the Parke-Davis Pharmaceutical Company in 1962 (Ann Arbor, Michigan) while searching for an alternative to the potent hallucinogenic agent phencyclidine [Citation1,Citation2]. Due to its quick onset and short duration of action with only slight cardio-respiratory depression in comparison with other general anaesthetics and the possibility of inhalation to maintain the anaesthetic state, ketamine is a preferred drug for short-term surgical procedures in veterinary and human medicine, especially in children [Citation3,Citation4]. Indeed, in adults, it induces severe psychomimetic reactions, namely hallucinations, delirium, nightmares, altered of short-term memory and cognition [Citation5–7]. It has also been proposed as analgesic and for the treatment of alcoholism [Citation8], heroin addiction [Citation9], anorexia [Citation10] and for the treatment of depression due to its long-lasting effects and rapid onset of action (within 4 h post-administration) [Citation11–14].
Ketamine produces dissociative anaesthesia (i.e. sense of dissociation from the body and the environment), a neologism first coined by Corssen and Domino [Citation15,Citation16]. This means that the patient remains conscious and appears to be awake (i.e. eyes may be open with presence of nystagmus) but exhibit no apparent response to surgical pain; “the lights are on, but no one's home” [Citation17]. Represents a “trancelike cataleptic state” characterized by profound and complete analgesia and total amnesia with preservation of protective airway reflexes (i.e. intubation is unnecessary), spontaneous respirations and cardiovascular stability (i.e. blood pressure and pulse rate do not decrease and may even increase slightly) [Citation6,Citation17]. The dissociative state seems to result from a functional dissociation: inhibition of thalamocortical pathways and stimulation of the limbic regions of the brain [Citation18]. These neuronal systems help to maintain neuronal connections required for consciousness.
Ketamine is primarily a stereoselective non-competitive antagonist of the ionotropic receptor of N-methyl-D-aspartate (NMDA) reducing calcium ion influx through this channel and therefore prevents neuronal activation required for conscious state [Citation19,Citation20]. This effect at NMDA receptors is able to reverse the enhanced pain sensitivity that is frequently present in major trauma or surgical injury and increases the antinociceptive effects of conventional opioid and nonsteroidal anti-inflammatory drugs (NSAIDs) [Citation21,Citation22]. In addition, ketamine also exerts non-NMDA related effects, interacting with several receptors, namely [Citation19,Citation23–27]: (1) μ, κ and δ opioid receptors, that contributes to its analgesic effects; (2) anticytokine effect; (3) inhibition of acetylcholine muscarinic and nicotinic receptors; (4) inhibition of L-type calcium and sodium channels current; (5) adrenergic receptors; (6) serotonin receptors; (7) dopaminergic D2 receptors; and (8) inhibition of neuronal sodium channels (producing a modest local anaesthetic action). In opposition to several other anaesthetics, it does not affect γ-aminobutyric acid receptors at clinically relevant concentrations [Citation28].
As shown in , ketamine [2-(O-chlorophenyl)-2-(methylamino)-cyclohexanone] has one stereogenic centre in the C2 position of the molecule and therefore can exist as two possible stereoisomers [R (l-ketamine) and S (d-ketamine; esketamine)]. Differences in pharmacological effects and pharmacokinetic properties between the two enantiomers have been described in vivo and in vitro [Citation29,Citation30]. The affinity of S-ketamine was demonstrated to be four times higher for the phencyclidine site of the NMDA receptor when compared with R-ketamine [Citation31,Citation32], resulting in a likewise increase in the hypnotic/anesthetic properties of the S-enantiomer [Citation29] and its analgesic potential was reported to be twice that of the racemic mixture and four times that of R-ketamine [Citation33]. Originally, it was used as racemic mixture, but nowadays in human medicine, S-ketamine is preferable due to its higher potency together with faster post-anaesthetic recovery times [Citation3,Citation4]. Unfortunately, the drug is still used as the racemic mixture.
Figure 1. Metabolism of ketamine.
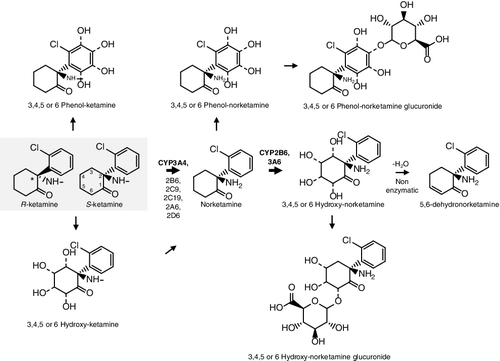
One of the objectives of metabolism and metabolomics is related to the qualitative and quantitative characterization of all pictures of biochemical and biological metabolic processes of an organism (i.e. the metabolome) and their changes over time [Citation34]. Several studies have demonstrated an unpredictable inter-individual variability of ketamine pharmacokinetics and pharmacodynamics [Citation35,Citation36]. Moreover, metabolic substrates and/or inhibitors or inducers of the same cytochrome P450 isoforms (CYP) implicated in ketamine metabolism are administered concurrently and thus important clinical and forensic consequences are expected. This work aims to perform a literature review of ketamine biotransformation and metabolomics, their pharmacological, toxicological effects, which have not been characterized sufficiently in most studies.
Methodology
An English exhaustive literature search was carried out to identify relevant articles. Ketamine metabolizing enzymes and metabolites, and metabolomics were searched in PubMed (U.S. National Library of Medicine) without a limiting period. Furthermore, electronic copies of the full papers were obtained from the retrieved journal articles as well as books on ketamine and then further reviewed to find possible additional publications related to human and non-human, in vivo and in vitro studies.
Absorption, distribution and excretion
Since it is water- and lipid-soluble xenobiotic, ketamine can be administered via almost any route depending on the intent. Oral, inhaled, rectal, smoked, intramuscular, subcutaneous, intravenous, epidural and intrathecal are the most frequently applied routes [Citation28,Citation32,Citation37]. Oral ketamine administration undergoes significant first-pass effects leading to the formation of norketamine and dehydronorketamine () [Citation38]. Rectal administration has a more rapid onset of action and has been applied specially in children [Citation39]. When used as a recreational drug, ketamine is usually inhaled (insufflated). Usually, the liquid commercial legal anaesthetic form is allowed to evaporate before administration [Citation32]. Because of ketamine crystalline appearance, the drug can be mistaken for or passed off as methamphetamine and cocaine. Bioavailabity largely depends on the route of administration (e.g. 20% oral; 90% intramuscular; 25% rectal; 50% intranasal; 77% epidural) [Citation32,Citation40,Citation41].
Since ketamine is very lipid-soluble and has a relatively low protein binding (about 20%–50%), a very large volume of distribution (3–5 L/kg) is attained [Citation32,Citation42]. Therefore, it rapidly crosses the blood–brain barrier to induce anaesthesia although the onset time is slower than thiopental. The affinity for α1-acid glycoprotein seems to be much higher than albumin [Citation43]. After intramuscular or subcutaneous injection, sedation or anaesthesia develops within 10–15 minutes and typically lasts for 30–120 minutes [Citation5]. When administered intravenously, the onset of action typically occurs within 1–2 minutes and anaesthesia lasts for approximately 20–60 minutes [Citation5]. After oral administration, the action onset typically occurs within 20–30 minutes and the duration of effect is between 60 and 90 minutes [Citation5]. Termination of anaesthesia is due to redistribution from the brain and plasma to other tissue. Ketamine is a week base with a pKa of 7.5 [Citation44]. At physiological pH of 7.4, it is 44.3% un-ionized [Citation45]. Since its pKa is close to physiological pH, small changes in pH result in a wide variation in the ionized and non-ionized fractions.
Analogous to other anaesthetics such as barbiturates and propofol, the elimination half-life is short; approximately 2–4 h by intravenous route [Citation46]. The pharmacokinetics in children is not very different from adults, although children do form more norketamine than adults [Citation47]. Indeed, children require higher infusion rates than adults to maintain the ketamine steady-state probably attributed to age-related pharmacokinetics [Citation48].
Ketamine and metabolites are excreted in urine; 2% is excreted unchanged, 2% in the form of norketamine, 16% as dehydronorketamine and 80% as conjugates of hydroxylated ketamine metabolites with glucuronic acid [Citation32,Citation49]. Other studies on human subjects given tritium-labelled ketamine intravenously have shown that while 91% of the administered radioactivity could be recovered in urine over a period of five days [Citation50], only 20% of the dose is presented as parent drug, norketamine and 5,6-dehydronorketamine [Citation51]. This means that a great proportion of an intravenous dose of ketamine is converted to other metabolites whose chemical structure and pharmacological activity are yet to be established. Hydroxylated metabolites of the parent drug and/or norketamine may be formed in vivo and subsequently eliminated in urine and bile as conjugates [Citation20,Citation52]. Several studies described that frequently repeated doses of ketamine prolonged its elimination time as long as 11 days [Citation49,Citation53]. In another study, norketamine was detected in urine samples up to 14 days after administration of a single intravenous dose of ketamine to children [Citation49].
Metabolism
The metabolism of ketamine has been described as extensive and stereoselective and occurs mainly in the liver [Citation52]. The major metabolic pathway is N-demethylation to an active metabolite norketamine by CYP3A4 () [Citation18]. Norketamine, besides some psychoactive properties, also retains anaesthetic effects; this fact explains the maintenance of the therapeutic efficacy at lower blood ketamine concentrations [Citation32]. In humans, at therapeutic concentrations, CYP2B6, CYP3A4 and CYP2C9 make only a minor contribution to ketamine N-demethylation [Citation54]. Nevertheless, there are some dissonant results regarding enzymes comparative contributions to clinical ketamine metabolism [Citation55]. Non-human data indicate that norketamine crosses the blood–brain barrier and has about one-fifth to one-third the potency of ketamine, contributing to the analgesic and psychomimetic side effects, especially in long infusions or chronic use [Citation56–58]. Paul and colleagues [Citation26] demonstrated that besides ketamine, also norketamine and hydroxynorketamine increase the levels of the activating phosphorylated form of mammalian target of rapamycine (mTOR; a protein kinase involved in protein synthesis, synaptic plasticity and neurotrophic signalling) and cognate signalling kinases in vitro and prefrontal cortex in the rat.
Ketamine and norketamine are also hydroxylated at 3–6 carbons of the cyclohexanone ring leading to the formation of both inactive free and glucuronidated hydroxylated derivatives, which are more water-soluble compounds in order to facilitate urinary excretion [Citation55,Citation59–62].
The hydroxylation of the alicyclic ring led to the introduction of a second chiral centre and thereby diastereomers with two chiral centres are formed. Although previous studies did not show formation of phenolic metabolites of ketamine [Citation63], more recent studies evidence their existence in urine of subjects after oral ketamine administration [Citation62].
Cyclohexanone ring also undergoes oxidative metabolism by dehydrogenation to an active metabolite 5,6-dehydronorketamine [Citation64]. Only the 5-hydroxynorketamine stereoisomers proved to be precursors of 5,6-dehydronorketamine [Citation60,Citation65,Citation66]. 5,6-dehydronorketamine has been posted as the most useful metabolite to target in a forensic context since it has a longer plasma half-life (i.e. detected for 6–10 days in volunteers) [Citation3,Citation67]. Hydroxylated dehydronorketamine or dihydroxynorketamine metabolites were not obtained from microsomal incubates of ketamine or norketamine [Citation62].
Drug–drug interactions and pharmacogenomics in metabolism
Since ketamine undergoes extensive metabolism, drug–drug interactions and inter-individual variability due to polymorphisms in genes coding for drug-metabolizing enzymes are expected to result in clinically important consequences. However, there are few reports about drug interactions and pharmacogenomics of ketamine in humans. The treatment with diazepam, a substrate of CYP2C19 and CYP3A4, or secobarbital, an inhibitor of CYP2B, increased plasma half-lives of ketamine in humans and therefore their sedative effects [Citation68,Citation69]. Since ketamine decreases CYP3A enzyme activity, diazepam metabolism may be also slowed [Citation70]. When ketamine was associated with bupivacaine, local anaesthetic effect was significantly enhanced as well as its elimination half-life [Citation71]. In rats, ketamine also decreases the clearance of the CYP2D1 substrate flecainide and the CYP3A substrate ethosuximide, by 13% and 18%, respectively [Citation72]. Pre-treatment of rat and rabbit with phenobarbital, an inducer of the CYP2B subfamily, causes a marked increase in the rate of ketamine metabolism by hepatic tissue in vitro [Citation65]. Rifampicin, a potent inducer of many CYP enzymes, particularly of CYP3A, significantly reduces the plasma concentrations of ketamine and norketamine after oral administration of S-ketamine; this effect was only moderately registered after its intravenous administration [Citation73,Citation74]. Clarithromycin, a potent inhibitor of CYP3A and P-glycoprotein transporter, also increased serum S-ketamine concentrations (263%) [Citation75]. Unexpectedly, itraconazole, a potent inhibitor of CYP3A and P-glycoprotein transporter, had no effect on the pharmacokinetics of S-ketamine, whereas inhibition of CYP2B6 by ticlopidine significantly increased its serum concentrations, emphasizing the role of CYP2B6 in the elimination of ketamine [Citation74]. Further studies are needed to evaluate the influence of polymorphisms of CYP3A4, but especially for CYP2B6 and/or CYP2C9, on ketamine pharmacokinetics. Until now, a significant impact of the CYP2B6*6 allele (i.e. the most prevalent and clinically important variant) on N-demethylation of ketamine was registered in vitro [Citation76]. Corroborating these results, CYP2B6*6 allele was associated with reduced steady-state ketamine plasma clearance in chronic pain patients [Citation36]. Authors hypostatized that the higher plasma concentrations obtained may predispose to higher incidence of ketamine adverse effects.
Ketamine also causes “self-induction” of multiple hepatic P450 isoforms in rat liver microsomes (i.e. 1A, 2B, 2E1 and 3A proteins by 2-, 13-, 2- and 2-fold, respectively), meaning that after chronic administration of ketamine, higher doses may be required to obtain the therapeutic effect [Citation77,Citation78]. There is some evidence that cocaine may interact with ketamine to cause hepatic injury, at least in experimental animals [Citation77]. The effect strongly suggests that due to ketamine P450 induction, liver toxicity may increase after exposure to cocaine and/or increase formation of norcocaine (i.e. the main hepatotoxic oxidative metabolite) [Citation79]. Nevertheless, results are not conclusive since other authors have shown that gene expression of CYP3A4 was suppressed by ketamine by disturbing cytoskeleton remodelling due to reduction of calcium mobilization and adenosine triphosphate (ATP) synthesis [Citation80]. Perhaps, differences in experimental designs may explain such discrepancies since ketamine need time to achieve its inductive effect [Citation81]. Indeed, while single-dose administration of ketamine inhibited CYP3A4-involved N-demethylation of erythromycin [Citation80], repetitive exposure can induce CYP activities, similarly to ethanol [Citation82,Citation83].
Moreover, some studies demonstrated that ketamine noncompetitively inhibits the glucuronidation of morphine to morphine-3-glucuronide in rats [Citation38]. This result may explain the prolonged duration of morphine-induced analgesia, less administrations and reduced adverse effects of morphine in humans [Citation15,Citation16]. Since ketamine is often used in combination with other drugs, especially cocaine and heroin (for which morphine is the active metabolite), these possible interactions should not be disregarded [Citation79,Citation84,Citation85]. Insights are needed to explain mechanisms of alterations of CYP activities since these may compromise drug metabolism, drug–drug interactions and the toxicity and carcinogenicity of foreign chemicals [Citation81].
Endogenous metabolome
Besides exposure metabolites, several other endogenous metabolic changes are expected after ketamine administration, but were not yet well explored in detail. Zhang and colleagues [Citation86] provide data regarding serum metabolites changes due to ketamine abuse in rats. Authors shown that ketamine induced alterations in the levels of phosphate, propanoic acid, butanoic acid, ribitol, propanetricarboxylic acid, hexadecanoic acid, d-fructose, l-leucine, l-threonine, alanine, glycine, butanoic acid, valine, l-serine, l-proline, mannonic acid, octadecanoic acid and cholesterol. Particularly important are the phosphate, propanoic acid, ribitol and d-fructose alterations, which are intermediate substances in energy metabolism. Propanoic acid is a product of glycolysis that can be metabolized into propionyl-CoA and then enters the tricarboxylic acid cycle (TCA cycle). Moreover, since phosphate is used to ATP synthesis, ketamine abuse in rats may result in changes in the energetic status of cells. Regarding alanine, glycine, butanoic acid, valine, l-serine, l-proline, malfunctioned protein synthesis and degradation are expected. Ketamine may be also implicated in liver damage since lipid metabolism was also compromised. In other study, Wen et al. [Citation87] observed that compared with the control group, the levels of glycerol, uridine and cholesterol in rat brain of the ketamine group decreased, while the urea levels increased. Glycerol and fatty acids are released into blood when the body uses stored fat as a source of energy. Uridine plays a role in the glycolysis pathway of galactose, which is converted to glucose and metabolized in the common glucose pathway. Urea is an end product of the metabolism of nitrogen containing compounds by animals that is excreted in urine. Authors also found biomarkers differently expressed in plasma and brain: compared with the control group, the levels of butanoic acid, phosphate, aminomalonic acid, gluconic acid, hexadecanoic acid, oleic acid and arachidonic acid increased in the plasma of the ketamine group, while the levels of glycine, l-lysine and cholesterol decreased [Citation87]. Several hippocampal pathways including glycolysis/gluconeogenesis, pentose phosphate pathway and citrate cycle are also affected after ketamine administration [Citation88].
An interesting study was performed by Villaseñor et al. [Citation89]. Authors performed a metabolomic analysis of plasma samples from patients receiving ketamine for the treatment of bipolar depression. Results evidenced that lysophosphatidylethanolamines and lysophosphatidylcholines increased in responder's patients suffering from resistant bipolar depression. These findings suggest that alterations in the mitochondrial β-oxidation of fatty acids are not registered in ketamine-administered group. Authors concluded that metabolomics may be useful to predict response aiming a ketamine personalized therapy. More recently, Rotroff and colleagues [Citation90] used metabolomics to provide new insights aiming to map global blood metabolic effects of ketamine racemic mixture and S-ketamine in treatment-refractory major depressive disorder patients. Tryptophan, the amino-acid precursor of serotonin, and related metabolites such as indole-3-acetate, indole-3-lactate and tyrosine, the amino-acid precursor of dopamine, were all decreased after ketamine administration. However, neither dopamine nor serotonin levels were significantly changed, meaning that further studies are required. Authors also observed that glutamate and circulating phospholipids (e.g. phosphatidylcholine and phosphoethanolamine) levels were significantly increased and were associated with decreases in depression severity. Since ketamine blocks the glutamatergic NMDA receptor, thus the possible effect of increased glutamate levels could shift glutamatergic signalling from NMDA to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor receptors to enhance the 5HT1B receptor activity that is claimed to be required for antidepressant effects [Citation91]. Moreover, since phosphatidylcholine is a major component of cell membranes, authors hypothesized that their increased synthesis is greater in the patients whose depression severity is decreasing. Other structurally non-identified metabolites also proved to be potentially interesting.
Pan and colleagues [Citation92] demonstrated that ketamine influenced the major energy and amino acid metabolic pathways in macaques. Indeed, serum levels of α-glucose, myoinositol, lactate and succinate, and urine levels of pyruvate and lactate were all decreased. In contrast, the levels of leucine in serum and arginine in urine were significantly higher in the ketamine group.
Conclusion and future perspectives
For more than 50 years, ketamine has been widely used to induce anaesthesia and to produce analgesia [Citation6]. Nevertheless, its use is limited due to its unfavourable psychoactive effects. In contrast to classic hallucinogens (e.g. lysergic acid diethylamide, psilocybin, mescaline, etc.), ketamine presents a higher risk of dependence and addiction (i.e. to develop a compulsive drug-seeking behaviour) [Citation1]. Indeed, hallucinogens (i.e. compounds able to change in lower doses the perception of reality regarding thought, time and space and alterations of humour and consciousness, without causing marked psychomotor stimulation or depression) differ from most other psychoactive drugs since they induce neither dependence nor addiction nor are used for prolonged periods; in other words, these drugs do not interfere with mesolimbic rewarding system [Citation66]. However, repetitive exposure causes rapid tolerance (also called tachyphylaxis) leading abusers to escalate doses to achieve the full hallucinogenic experience [Citation66,Citation93]. In recent years, ketamine has been used as a recreational and “club drug” due to its hallucinogenic, dream-like state, sensation of floating outside the body, “cosmic” experiences, body distortions and stimulant effects at dance parties and raves [Citation94–96]. After being illegally diverted from legal suppliers (e.g. veterinary), ketamine has also been misused as a “date-rape” drug since it induces amnesia as γ-hydroxybutyric acid and flunitrazepam [Citation97]. The narcotic effects of ketamine are comparable to those achieved by phencyclidine, and the hallucinogen action is similar to that of lysergic acid diethylamide [Citation59].
Although ketamine therapy is generally considered safe, several toxicological consequences can be highlighted [Citation49]: (1) at lower doses it causes mild intoxication, dreamy thinking, alterations of speech, hearing and seeing, muscular discoordination, disorientation, anxiety, disinhibition, euphoria, seeing the world differently and irrational behaviour; (2) higher doses cause great difficulty in moving, respiratory disturbances, seizures and nausea; (3) extreme doses produce complete dissociation from reality and loss of consciousness, hallucinations, out-of-body experiences and so-called “near-death experiences” or the “K-hole” [Citation93]; (4) in absence of other drugs, deaths by overdose (e.g. due to respiratory depression) are rare since doses used by addicts are typically lower than necessary for therapeutic anaesthesia. Accidental deaths have been reported as consequence of falling, hypothermia, traffic accident or drowning [Citation75]; and (5) the frequent use of ketamine can lead to addiction and dependence [Citation98]. Sometimes ketamine abused is mixed with other drugs such as cocaine, methamphetamine, 3,4-methylenedioxymethamphetamine and benzodiazepines [Citation99].
Despite some studies, the metabolism and metabolomics of ketamine remain poorly understood. In this work, exogenous and endogenous metabolome of ketamine was fully reviewed. Ketamine undergoes hepatic N-demethylation by several isoforms of cytochrome P450 to norketamine, which is the main metabolite excreted in urine. In order to better comprehend clinical effects, pharmacokinetic studies should focus on both active and inactive metabolites and on polymorphisms in genes encoding enzymes (e.g. CYP2B6) involved in ketamine metabolism [Citation100,Citation101]. Moreover, due to its extensive metabolism, potentially dangerous interactions are expected when other drugs are taken together. The identification of additional metabolites will be particularly useful to confirm xenobiotic exposure [Citation102] and further sensitive analytical methods are needed to prove consumption in a wider detection window [Citation49]. Since ketamine has become a popular club and date-rape drug, research on biotransformation acquires particular importance in forensic toxicology. Moreover, the mechanisms underlying the emergence psychedelic alterations are not fully understood. Based on the time course of their appearance and due to the low levels of ketamine registered, active metabolites are expected to be involved. Scarce data exist in the literature regarding the binding of ketamine and metabolites such as norketamine and dehydronorketamine to serum proteins namely to albumin and α1-acid glycoprotein. The clinical relevance for patients with serum protein levels severely altered also needs to be further explored. Finally, particularly interesting will be the metabolomics elucidation of the mechanisms by which ketamine rapidly treats depressive symptoms. To be accomplished, the identification of unknown metabolites may provide novel insights into the biological pathways involved.
Acknowledgments
Ricardo Dinis-Oliveira acknowledges Fundação para a Ciência e a Tecnologia (FCT) for his Investigator Grant [grant number IF/01147/2013].
Disclosure statement
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending or royalties. No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
References
- Liu Y, Lin D, Wu B, et al. Ketamine abuse potential and use disorder. Brain Res Bull. 2016;126:68–73.
- Domino EF, Chodoff P, Corssen G. Pharmacologic effects of ci-581, a new dissociative anesthetic, in man. Clin Pharmacol Ther. 1965;6:279–291.
- Hijazi Y, Bolon M, Boulieu R. Stability of ketamine and its metabolites norketamine and dehydronorketamine in human biological samples. Clin Chem. 2001;47:1713–1715.
- Fischer MM. Ketamine hydrochloride in severe bronchospasm. Anaesthesia. 1977;32:771–772.
- Barash PG, Cullen BF, Stoelting RK, et al. Clinical anesthesia. 7th ed. Philadelphia (PA): Lippincott Williams & Wilkins; 2013.
- James K, Briggs S, Lewis R, et al. Introduction to specialist therapeutics. In: Tomlin M, editor. Pharmacology & pharmacokinetics: a basic reader. London: Springer London; 2010. p. 53–66.
- Niesters M, Martini C, Dahan A. Ketamine for chronic pain: risks and benefits. Br J Clin Pharmacol. 2014;77:357–367.
- Petrakis IL, Limoncelli D, Gueorguieva R, et al. Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry. 2004;161:1776–1782.
- Krupitsky EM, Burakov AM, Dunaevsky IV, et al. Single versus repeated sessions of ketamine-assisted psychotherapy for people with heroin dependence. J Psychoactive Drugs. 2007;39:13–19.
- Mills IH, Park GR, Manara AR, et al. Treatment of compulsive behaviour in eating disorders with intermittent ketamine infusions. QJM. 1998;91:493–503.
- Murrough JW. Ketamine as a novel antidepressant: from synapse to behavior. Clin Pharmacol Therap. 2012;91:303–309.
- Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338:68–72.
- Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964.
- Coyle CM, Laws KR. The use of ketamine as an antidepressant: a systematic review and meta-analysis. Hum Psychopharmacol. 2015;30:152–163.
- Corssen G, Domino EF. Dissociative anesthesia: further pharmacologic studies and first clinical experience with the phencyclidine derivative CI-581. Anesth Analg. 1966;45:29–40.
- Domino EF, Chodoff P, Corssen G. Pharmacologic effects of CI-581, a new dissociative anesthetic, in man. Clin Pharmacol Ther. 1965;6:279–291.
- Roberts JR, Hedges JR. Roberts and Hedges’: clinical procedures in emergency medicine. 6th ed. Philadelphia (PA): Elsevier Saunders; 2014.
- White PF, Way WL, Trevor AJ. Ketamine–its pharmacology and therapeutic uses. Anesthesiology. 1982;56:119–136.
- Smith KM, Larive LL, Romanelli F. Club drugs: methylenedioxymethamphetamine, flunitrazepam, ketamine hydrochloride, and gamma-hydroxybutyrate. Am J Health Syst Pharm. 2002;59:1067–1076.
- Mion G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci Therap. 2013;19:370–380.
- Baker AK, Hoffmann VL, Meert TF. Dextromethorphan and ketamine potentiate the antinociceptive effects of mu- but not delta- or kappa-opioid agonists in a mouse model of acute pain. Pharmacol, Biochem, Behav. 2002;74:73–86.
- Pawson P, Forsyth S. Chapter 5, Anesthetic agents. In: Maddison JE, Page SW, Church DB, editors. Small animal clinical pharmacology. 2nd ed. Edinburgh: W.B. Saunders; 2008. p. 83–112.
- Welters ID, Hafer G, Menzebach A, et al. Ketamine inhibits transcription factors activator protein 1 and nuclear factor-kappaB, interleukin-8 production, as well as CD11b and CD16 expression: studies in human leukocytes and leukocytic cell lines. Anesth Analg. 2010;110:934–941.
- Durieux ME. Inhibition by ketamine of muscarinic acetylcholine receptor function. Anesth Analg. 1995;81:57–62.
- Udesky JO, Spence NZ, Achiel R, et al. The role of nicotinic inhibition in ketamine-induced behavior. Anesth Analg. 2005;101:407–411.
- Paul RK, Singh NS, Khadeer M, et al. (R,S)-Ketamine metabolites (R,S)-norketamine and (2S,6S)-hydroxynorketamine increase the mammalian target of rapamycin function. Anesthesiology. 2014;121:149–159.
- Yamakage M, Hirshman CA, Croxton TL. Inhibitory effects of thiopental, ketamine, and propofol on voltage-dependent Ca2+ channels in porcine tracheal smooth muscle cells. Anesthesiology. 1995;83:1274–1282.
- Peltoniemi MA, Hagelberg NM, Olkkola KT, et al. Ketamine: a review of clinical pharmacokinetics and pharmacodynamics in anesthesia and pain therapy. Clin Pharmacokinetics. 2016;55:1059–1077.
- White PF, Schuttler J, Shafer A, et al. Comparative pharmacology of the ketamine isomers. Studies in volunteers. Br J Anaesth. 1985;57:197–203.
- Himmelseher S, Pfenninger E, Georgieff M. The effects of ketamine-isomers on neuronal injury and regeneration in rat hippocampal neurons. Anesth Analg. 1996;83:505–512.
- Klepstad P, Maurset A, Moberg ER, et al. Evidence of a role for NMDA receptors in pain perception. Euro J Pharmacol. 1990;187:513–518.
- Karch SB, Drummer OH. Karch's pathology of drug abuse. 5th ed. Boca Raton (FL): CRC Press, Taylor & Francis Group; 2016.
- Schmid RL, Sandler AN, Katz J. Use and efficacy of low-dose ketamine in the management of acute postoperative pain: a review of current techniques and outcomes. Pain. 1999;82:111–125.
- Dinis-Oliveira RJ. Metabolomics of drugs of abuse: a more realistic view of the toxicological complexity. Bioanalysis. 2014;6:3155–3159.
- Aroke EN, Dungan JR. Pharmacogenetics of anesthesia: an integrative review. Nurs Res. 2016;65:318–330.
- Li Y, Jackson KA, Slon B, et al. CYP2B6*6 allele and age substantially reduce steady-state ketamine clearance in chronic pain patients: impact on adverse effects. British J Clin Pharmacol. 2015;80:276–284.
- Johnson BA. Addiction medicine: science and practice. London: Springer; 2011.
- Qi X, Evans AM, Wang J, et al. Inhibition of morphine metabolism by ketamine. Drug Metabolism Disposition: Biol Fate Chem. 2010;38:728–731.
- Idvall J, Aronsen KF, Stenberg P, et al. Pharmacodynamic and pharmacokinetic interactions between ketamine and diazepam. Euro J Clin Pharmacol. 1983;24:337–343.
- Fanta S, Kinnunen M, Backman JT, et al. Population pharmacokinetics of S-ketamine and norketamine in healthy volunteers after intravenous and oral dosing. Eur J Clin Pharmacol. 2015;71:441–447.
- Malinovsky JM, Servin F, Cozian A, et al. Ketamine and norketamine plasma concentrations after i.v., nasal and rectal administration in children. Br J Anaesth. 1996;77:203–207.
- Little B, Chang T, Chucot L, et al. Study of ketamine as an obstetric anesthetic agent. Am J Obstetrics Gynecol. 1972;113:247–260.
- Dayton PG, Stiller RL, Cook DR, et al. The binding of ketamine to plasma proteins: emphasis on human plasma. Euro J Clin Pharmacol. 1983;24:825–831.
- Moffat AC, Osselton MD, Widdop B, et al. Clarke's analysis of drugs and poisons. 4th ed. London: Pharmaceutical Press; 2011.
- Taylor E. Chapter 24, Ketamine. In: Bissonnette B, editor. Pediatric anesthesia: basic principles-state of the art-future. Shelton (CT): People's Medical Publishing House; 2011. p. 376–386.
- Domino EF, Zsigmond EK, Domino LE, et al. Plasma levels of ketamine and two of its metabolites in surgical patients using a gas chromatographic mass fragmentographic assay. Anesth Analg. 1982;61:87–92.
- Grant IS, Nimmo WS, McNicol LR, et al. Ketamine disposition in children and adults. Br J Anaesth. 1983;55:1107–1111.
- Dallimore D, Anderson BJ, Short TG, et al. Ketamine anesthesia in children–exploring infusion regimens. Paediatr Anaesth. 2008;18:708–714.
- Adamowicz P, Kala M. Urinary excretion rates of ketamine and norketamine following therapeutic ketamine administration: method and detection window considerations. J Anal Toxicol. 2005;29:376–382.
- Chang T, Savory A, Albin M, et al. Metabolic disposition of tritium-labelled ketamine. Clin Res. 1970;18:597–601.
- Wieber J, Gugler R, Hengstmann JH, et al. Pharmacokinetics of ketamine in man. Anaesthesist. 1975;24:260–263.
- Chang T, Glazko AJ. Biotransformation and disposition of ketamine. Int Anesthesiol Clin. 1974;12:157–177.
- Jansen KL. A review of the nonmedical use of ketamine: use, users and consequences. J Psychoactive Drugs. 2000;32:419–433.
- Hijazi Y, Boulieu R. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metabolism Disposition: Biol Fate Chem. 2002;30:853–858.
- Yanagihara Y, Kariya S, Ohtani M, et al. Involvement of CYP2B6 in n-demethylation of ketamine in human liver microsomes. Drug Metabolism Disposition: Biol Fate Chem. 2001;29:887–890.
- Leung LY, Baillie TA. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J Med Chem. 1986;29:2396–2399.
- Shimoyama M, Shimoyama N, Gorman AL, et al. Oral ketamine is antinociceptive in the rat formalin test: role of the metabolite, norketamine. Pain. 1999;81:85–93.
- Ebert B, Mikkelsen S, Thorkildsen C, et al. Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. Euro J Pharmacol. 1997;333:99–104.
- Moore KA, Kilbane EM, Jones R, et al. Tissue distribution of ketamine in a mixed drug fatality. J Forensic Sci. 1997;42:1183–1185.
- Schmitz A, Theurillat R, Lassahn PG, et al. CE provides evidence of the stereoselective hydroxylation of norketamine in equines. Electrophoresis. 2009;30:2912–2921.
- Adams JD, Jr., Baillie TA, Trevor AJ, et al. Studies on the biotransformation of ketamine. 1-Identification of metabolites produced in vitro from rat liver microsomal preparations. Biomed Mass Spectrom. 1981;8:527–538.
- Turfus SC, Parkin MC, Cowan DA, et al. Use of human microsomes and deuterated substrates: an alternative approach for the identification of novel metabolites of ketamine by mass spectrometry. Drug Metabolism Disposition: Biol Fate Chem. 2009;37:1769–1778.
- Leung LY, Baillie TA. Studies on the biotransformation of ketamine. II–Quantitative significance of the N-demethylation pathway in rats in vivo determined by a novel stable isotope technique. Biomed Environ Mass Spectrom. 1989;18:401–404.
- Curran HV, Monaghan L. In and out of the K-hole: a comparison of the acute and residual effects of ketamine in frequent and infrequent ketamine users. Addiction. 2001;96:749–760.
- Woolf TF, Adams JD. Biotransformation of ketamine, (Z)-6-hydroxyketamine, and (E)-6-hydroxyketamine by rat, rabbit, and human liver microsomal preparations. Xenobiotica; Fate Foreign Compd Biol Syst. 1987;17:839–847.
- Katzung BG, Masters SB, Trevor AJ. Basic & clinical pharmacology. 12th ed. New York (NY): McGraw-Hill; 2012.
- Parkin MC, Turfus SC, Smith NW, et al. Detection of ketamine and its metabolites in urine by ultra high pressure liquid chromatography-tandem mass spectrometry. J Chromatography B, Anal Technol Biomed Life Sci. 2008;876:137–142.
- Lo JN, Cumming JF. Interaction between sedative premedicants and ketamine in man in isolated perfused rat livers. Anesthesiology. 1975;43:307–312.
- Domino EF, Domino SE, Smith RE, et al. Ketamine kinetics in unmedicated and diazepam-premedicated subjects. Clin Pharmacol Therap. 1984;36:645–653.
- Sweeney BP, Bromilow J. Liver enzyme induction and inhibition: implications for anaesthesia. Anaesthesia. 2006;61:159–177.
- Gantenbein M, Abat C, Attolini L, et al. Ketamine effects on bupivacaine local anaesthetic activity and pharmacokinetics of bupivacaine in mice. Life Sci. 1997;61:2027–2033.
- Loch JM, Potter J, Bachmann KA. The influence of anesthetic agents on rat hepatic cytochromes P450 in vivo. Pharmacology. 1995;50:146–153.
- Noppers I, Olofsen E, Niesters M, et al. Effect of rifampicin on S-ketamine and S-norketamine plasma concentrations in healthy volunteers after intravenous S-ketamine administration. Anesthesiology. 2011;114:1435–1445.
- Peltoniemi MA, Saari TI, Hagelberg NM, et al. Exposure to oral S-ketamine is unaffected by itraconazole but greatly increased by ticlopidine. Clin Pharmacol Ther. 2011;90:296–302.
- Hagelberg NM, Peltoniemi MA, Saari TI, et al. Clarithromycin, a potent inhibitor of CYP3A, greatly increases exposure to oral S-ketamine. Eur J Pain. 2010;14:625–629.
- Li Y, Coller JK, Hutchinson MR, et al. The CYP2B6*6 allele significantly alters the N-demethylation of ketamine enantiomers in vitro. Drug Metabolism Disposition: Biol Fate Chem. 2013;41:1264–1272.
- Chan WH, Sun WZ, Ueng TH. Induction of rat hepatic cytochrome P-450 by ketamine and its toxicological implications. J Toxicol Environ Health Part A. 2005;68:1581–1597.
- Marietta MP, White PF, Pudwill CR, et al. Biodisposition of ketamine in the rat: self-induction of metabolism. J Pharmacol Exp Therap. 1976;196:536–544.
- Rofael HZ. Effect of ketamine pretreatment on cocaine-mediated hepatotoxicity in rats. Toxicol Lett. 2004;152:213–222.
- Chang HC, Chen TL, Chen RM. Cytoskeleton interruption in human hepatoma HepG2 cells induced by ketamine occurs possibly through suppression of calcium mobilization and mitochondrial function. Drug Metabolism Disposition: Biol Fate Chem. 2009;37:24–31.
- Chen JT, Chen RM. Mechanisms of ketamine-involved regulation of cytochrome P450 gene expression. Expert Opin Drug Metabol Toxicol. 2010;6:273–281.
- Dinis-Oliveira RJ. Oxidative and non-oxidative metabolomics of ethanol. Curr Drug Metabolism. 2016;17:327–335.
- Ueng TH, Ueng YF, Tsai JN, et al. Induction and inhibition of cytochrome P-450-dependent monooxygenases in hamster tissues by ethanol. Toxicology. 1993;81:145–154.
- Rofael HZ, Abdel-Rahman MS. The role of ketamine on plasma cocaine pharmacokinetics in rat. Toxicol Lett. 2002;129:167–176.
- Wu LT, Schlenger WE, Galvin DM. Concurrent use of methamphetamine, MDMA, LSD, ketamine, GHB, and flunitrazepam among American youths. Drug Alcohol Depend. 2006;84:102–113.
- Zhang M, Wen C, Zhang Y, et al. Serum metabolomics in rats models of ketamine abuse by gas chromatography-mass spectrometry. J Chromatography B, Anal Technol Biomed Life Sci. 2015;1006:99–103.
- Wen C, Zhang M, Zhang Y, et al. Brain metabolomics in rats after administration of ketamine. Biomed Chromatography: BMC. 2016;30:81–84.
- Weckmann K, Labermaier C, Asara JM, et al. Time-dependent metabolomic profiling of Ketamine drug action reveals hippocampal pathway alterations and biomarker candidates. Translational Psychiatry. 2014;4:e481.
- Villaseñor A, Ramamoorthy A, Silva dos Santos M, et al. A pilot study of plasma metabolomic patterns from patients treated with ketamine for bipolar depression: evidence for a response-related difference in mitochondrial networks. Br J Pharmacol. 2014;171:2230–2242.
- Rotroff DM, Corum DG, Motsinger-Reif A, et al. Metabolomic signatures of drug response phenotypes for ketamine and esketamine in subjects with refractory major depressive disorder: new mechanistic insights for rapid acting antidepressants. Translational Psychiatry. 2016;6:e894.
- Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224:107–111.
- Pan X, Zeng X, Hong J, et al. Effects of ketamine on metabolomics of serum and urine in Cynomolgus Macaques (Macaca fascicularis). J Am Assoc Lab Animal Sci. 2016;55:558–564.
- Muetzelfeldt L, Kamboj SK, Rees H, et al. Journey through the K-hole: phenomenological aspects of ketamine use. Drug Alcohol Depend. 2008;95:219–229.
- Jansen KL. Non-medical use of ketamine. BMJ. 1993;306:601–602.
- Wolff K, Winstock AR. Ketamine : from medicine to misuse. CNS Drugs. 2006;20:199–218.
- Gahlinger PM. Club drugs: MDMA, gamma-hydroxybutyrate (GHB), Rohypnol, and ketamine. Am Family Phys. 2004;69:2619–2626.
- Dinis-Oliveira RJ, Magalhaes T. Forensic toxicology in drug-facilitated sexual assault. Toxicol Mechanisms Methods. 2013;23:471–478.
- Jansen KL, Darracot-Cankovic R. The nonmedical use of ketamine, part two: A review of problem use and dependence. J Psychoactive Drugs. 2001;33:151–158.
- Lora-Tamayo C, Tena T, Rodriguez A, et al. The designer drug situation in Ibiza. Forensic Sci Int. 2004;140:195–206.
- van Velzen M, Dahan A. Ketamine metabolomics in the treatment of major depression. Anesthesiology. 2014;121:4–5.
- Dinis-Oliveira RJ. Metabolomics of methadone: clinical and forensic toxicological implications and variability of dose response. Drug Metabolism Rev. 2016;48:568–576.
- Dinis-Oliveira RJ. Metabolomics of Delta(9)-tetrahydrocannabinol: implications in toxicity. Drug Metabolism Rev. 2016;48:80–87.