
Abstract
Definitive diagnosis to sudden cardiac death (SCD) is often challenging since the postmortem examination on SCD victims could hardly demonstrate an adequate cause of death. It is therefore important to uncover the inherited risk component to SCD. Signal transducer and activators of transcription 5 A (STAT5A) is a member of the STAT family and a transcription factor that is activated by many cell ligands and associated with various cardiovascular processes. In this study, we performed a systematic variant screening on the STAT5A to filter potential functional genetic variations. Based on the screening results, an insertion/deletion polymorphism (rs3833144) in 3’UTR of STAT5A was selected as the candidate variant. A total of 159 SCD cases and 668 SCD matched healthy controls was enrolled to perform a case-control study and evaluate the association between rs3833144 and SCD susceptibility in Chinese populations. Logistic regression analysis showed that the deletion allele of rs3833144 had significantly increased the SCD risk (odds ratio (OR) = 1.54; 95% confidence interval (CI) = 1.18–2.01; P = 0.000955). Further genotype-expression eQTL analysis showed that samples with deletion allele appeared to lower expression of STAT5A, and in silico prediction suggested the local 3 D structure changes of STAT5A mRNA caused by the variant. On the other hand, the bioinformatic analysis presented that promoters of RARA and PTGES3L-AARSD1 could interact with rs3833144, and eQTL analysis showed the higher expression of both genes in samples with deletion allele. Dual-luciferase activity assays also suggested the significant regulatory role of rs3833144 in gene transcription. Our current data thus suggested a possible involvement of rs3833144 to SCD predisposition in Chinese populations and rs3833144 with potential function roles may become a candidate marker for SCD diagnosis and prevention.
Introduction
Sudden cardiac death (SCD) refers to a sudden and unexpected death or arrest attributed to a cardiovascular cause, mostly occurring within 1 h from the onset of symptom or within 24 h of last been observed to be alive if not witnessed [Citation1]. As a leading cause of mortality, SCD accounts for half of all cardiovascular deaths with an annual incidence of 57.3–110.8 per 100 000 in USA and 40.7 per 100 000 in China [Citation2–4]. As work continues to pathological research, coronary heart diseases have been identified as the major etiology for SCD in older people [Citation5], and inheritable arrhythmogenic diseases were characterized as an important cause in young [Citation6, Citation7]. Nevertheless, a definitive diagnosis to SCD is still challenging since postmortem examination could hardly demonstrate an accurate cause of death [Citation8]. To uncover the molecular mechanisms of SCD and identify genetic markers, numerous efforts have been put into molecular genetics of SCD during the last few decades [Citation9, Citation10]. For instance, recent study has identified a causative variant within caveolin-3 (Cav-3) gene in a patient with SCD [Citation11]. Population studies also suggested a familial aggregation of SCD, characterizing the SCD family history as an important risk factor for sudden death [Citation12–14]. The casual rare variants with large effect size to SCD may show incomplete penetrance, while common pathogenic variants with small effect size may also generate the clinical picture of SCD. Therefore, it is noteworthy to aggregate common pathogenic variants to establish polygenic risk score analyses for SCD.
Signal transducer and activators of transcription (STATs) are a family of cytoplasmic transcription factors, activated by Janus kinases (JAKs) through tyrosine phosphorylation and translocated into nuclear after dimerization [Citation15, Citation16]. These activated STAT proteins regulate expression of target genes, implicated in renin-angiotensin system (RAS) [Citation17], hypertrophy [Citation18], angiogenesis [Citation19, Citation20], fibrosis [Citation21, Citation22] and other cellular processes in the cardiac myocytes. STAT5A is one member of the family and one of two highly homologous but non-redundant isoforms (STAT5A and STAT5B) [Citation23]. Previous studies [Citation24, Citation25] have identified that STAT5A of JAK/STAT pathway would be selectively activated in ischemia/reperfusion (I/R) injury and post-infarction remodeling. Moreover, a recent study determined that STAT5A was a target of miR-222, which could hamper neovascularization and suppress atherosclerotic disease progression [Citation20]. Considering the functional role of STAT5A in various cardiovascular processes, it merits deciphering the variants on STAT5A gene to explore the potential effect of STAT5A on cardiac lethal events. The 3’untranslated region (3’UTR) is the noncoding part of mRNA, harbour various functional elements and play an important role in gene regulation [Citation26]. We therefore screened polymorphisms within this region of STAT5A and performed a case-control study, finally identifying an insertion/deletion (indel) variation within STAT5A 3’UTR correlated with SCD susceptibility in Chinese populations. Further functional experiments were implemented to investigate underlying mechanisms.
Methods and materials
Study populations
A total of 159 SCD cases and 668 healthy controls were enrolled in this case-control study. All the enrolled subjects were genetically unrelated Han Chinese. The 159 SCD patients were recruited from 2012 to 2020 at Sun Yat-sen University, Soochow University, Academy of Forensic Science, China and Xiangya Medical University. The recruitment criteria for both cases and controls were the same as those described previously [Citation27, Citation28]. Blood samples from SCD patients were assessed by exhaustive toxicological examinations to rule out toxic death. Comprehensive forensic pathological examinations were also conducted in all the cases and different extents of coronary atherosclerosis were detected in the corpses. Except these lesions, no other mortal pathological changes were observed during the autopsy. These deceased were thus assumed to suffer sudden death derived from coronary heart diseases. The 668 healthy controls were selected through community nutritional questionnaires performed in the same regions and during the same periods as the victims. They were all frequency matched to SCD victims based on age (±5 years) and gender and identified without any cardiovascular disease history or SCD family history. This study was approved by the Ethical Committee of Soochow University. All participants or their relatives provided written informed consents.
Genotyping of STAT5A Indel polymorphism
TIANamp Blood DNA Kit (TIANGEN, Beijing, China) was employed to extract genomic DNA from peripheral blood samples. The primers used for rs3833144 polymorphism were 5′-TGAAGCGGTCGTGTTGTGA-3′ (Forward), 5′-CACATCCCAGGACTGCACA-3′ (Reverse), which were generated by Genewiz Company (Suzhou, China). Genotyping analysis of PCR products amplified using above primers was completed with 7% non-denaturing polyacrylamide gel electrophoresis and silver staining [Citation29]. For quality control, 50 random DNA samples were used for validation of genotyping method by means of direct sequencing. Meanwhile, 10% masked random samples were analyzed twice by independent technicians to confirm 100% reproducibility.
Bioinformatic analysis
Mutations within STAT5A 3’UTR were collected from dbSNP database [Citation30]. The genotyping and mRNA expression data available online were employed for genotype-expression analysis in our study. The RNA sequencing data of 445 lymphoblastoid cell lines was available from 1000 Genomes Project (1KGP) database (https://www.ebi.ac.uk/arrayexpress/experiments/E-GEUV-3/). The genotypes of the variants observed in 2 504 samples in 1KGP database were directly obtained from Ensembl Genome Browser (http://asia.ensembl.org/Homo_sapiens/Info/Index). Based on above data, expression quality trait loci (eQTL) analysis was performed in R(https://www.datavis.ca/R/) to evaluate genotype-phenotype association. The eQTL and splicing quality trait loci (sQTL) analysis were also performed in Genotype-Tissue expression (GTEx) database. Linkage disequilibrium (LD) analysis was performed using LD heatmap package in R. The putative influence of insertion and deletion alleles on local 3 D structure of STAT5A mRNA was predicted using SimRNAweb (https://genesilico.pl/SimRNAweb/submit) based on a 170-base-pair region containing variant rs3833144. HaploReg [Citation31] and ENCODE [Citation32] database were used to perform functional annotations of the variant. The chromatin interaction partners for rs3833144 were extracted from high-throughput chromosome conformation capture (Hi-C) data in 3DIV database [Citation33].
Construction of reporter plasmids
A total of 210-bp or 206-bp DNA fragments including the variant rs3833144 were directly generated by Genewiz Company. These fragments were directionally subcloned into KpnI and SacI sites of pGL3-promoter vector, generating two wild type vectors containing insertion allele (pGL3-proWT-cis/trans) and two mutant type vectors containing deletion allele (pGL3-proMT-cis/trans). The sequence and direction of resultant constructs were testified by direct sequencing.
Cell culture and luciferase reporter assay
In vitro experiments were performed using 293 T cell lines purchased from Shanghai Cell Bank of Chinese Academy of Sciences (Shanghai, China). The cell lines were authenticated through short tandem repeat markers and maintained in Dulbecco’s modified eagle medium with 10% fetal bovine serum and 1% penicillin-streptomycin in a humidified atmosphere of 37 °C and 5% CO2.
Twenty-four hours after plated in the 24-well plates, 293 T cells were transfected with allele- and direction-different reporter plasmids (about 400 ng) with the corporation of jetPRIME® transfection reagent (Polyplus-transfection®, Illkirch, France). The empty pGL3-promoter vector transfected group was performed as negative control, and approximately 20 ng SV40 vector containing Renilla luciferase gene was co-transfected in each well for normalization of luciferase activity. Twenty-four hours after transfection, cells were harvested immediately by adding into 100 mL passive lysis buffer. Firefly luciferase activity was assessed by the Dual Luciferase® assay system (Promega, Madison, WI, USA) in FilterMaxF5 (Molecular Devices, San Jose, CA, USA). Each group had four duplicates and every transfection experiment was repeated at least three times.
Statistical analysis
The X2 test was performed to measure Hardy-Weinberg equilibrium for the representative of control samples. Unconditional logistic regression with adjustment for sex and age was used to evaluate the correlations between rs3833144 and SCD risk by estimating odds ratios (ORs) and their 95% confidence intervals (CIs). The mRNA expression levels between samples with different genotypes were compared by One-way ANOVA in eQTL analysis. Student’s t test was performed to evaluate the difference of the luciferase activities. The statistical analyses were implemented by Statistic Analysis System software (version 8.0; SAS Institute, Cary, NC, USA), a P-value of less than 0.05 was considered as the criterion of statistical significance. All statistical tests were two-sided in our study. The statistical power of the current sample size was calculated using the G*Power 3.1 software [Citation34].
Results
Bioinformatic screening of variants within 3’UTR of STAT5A gene
The workflow for this study was presented in . All mutations in STAT5A 3’UTR with frequency available online were summarized in . Among these mutations, only four mutations (rs3833144, rs3198502, rs115456777 and rs73983709) had a minimum allele frequency (MAF) of more than 0.01. Since rs115456777 was observed with only two genotypes, we excluded this variant to avoid potential deviation from Hardy-Weinberg equilibrium. Genotype-phenotype eQTL analysis was subsequently performed between remaining three variations and STAT5A mRNA expression levels by using genotyping and mRNA expression data from 1000 G database. As shown in , only rs3833144 polymorphism showed a significant genotype-phenotype association with its host gene STAT5A (P = 0.037), compared with rs3198502 (P = 0.273) and rs73983709 (P = 0.096). Also, the expression levels in samples with ins/del and del/del genotypes were lower than that with ins/ins genotype, which was shown in . Furthermore, we observed that rs3198502 was an sQTL significant variant for STAT5A and an eQTL significant variant for STAT3 based on GTEx database ( and ), while rs72983709 was not correlated with STAT5A transcription. Finally, our LD analysis suggested that rs3833144 and rs3198502 were in one LD block (). Unlike rs3198502, indel polymorphism rs3833144 contained the characteristic of length polymorphism, which was compatible with capillary electrophoresis (CE) platform commonly applied in forensic practice. We therefore chose the indel polymorphism rs3833144 to identify its association with risk to SCD based on case-control study.
Figure 1. The workflow for the study on regulatory variation within 3’UTR of STAT5A correlates with sudden cardiac death (SCD) in Chinese populations. 3’UTR: 3’untranslated region; MAF: minimum allele frequency; HWE: Hardy-Weinberg equilibrium; GTEx: Genotype-Tissue expression; eQTL: expression quality trait loci; sQTL: splicing quality trait loci; SCD: sudden cardiac death.
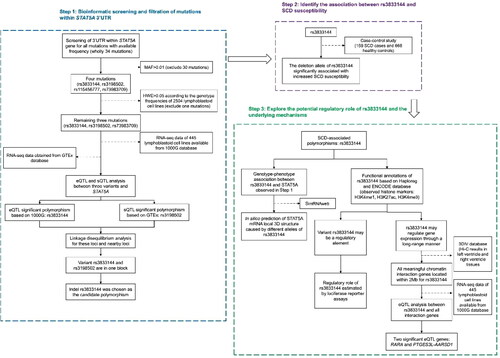
Figure 2. (A) Genotype-expression eQTL assay between rs3833144 and mRNA expression of STAT5A in 445 lymphoblastoid cell lines in 1000 Genomes Project database. (B) Genotype-phenotype sQTL assay between rs3198502 and STAT5A in GTex database. (C) Genotype-phenotype eQTL assay between rs3198502 and STAT3 in GTEx database. (D) Linkage disequilibrium (LD) analysis of rs3833144, rs3198502, rs73983709 and their nearby loci.
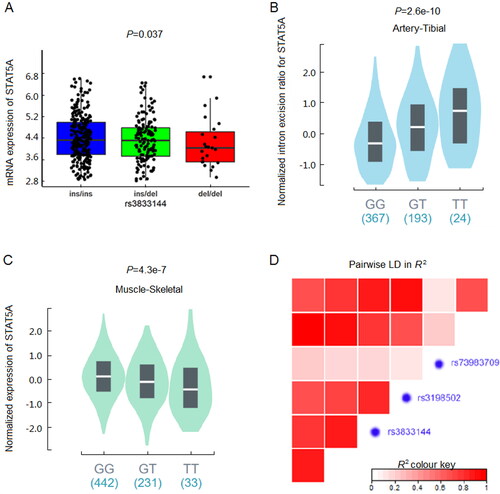
Table 1. Clinical characteristics of sudden cardiac death (SCD) cases and controls.
Table 2. Association between rs3833144 and sudden cardiac death (SCD) susceptibility.
Correlations between STAT5A rs3833144 polymorphism and SCD susceptibility
The demographic characteristics of both SCD subjects and their matching controls in present study were summarized in . The characteristics of the SCD cases are listed in Supplementary Table S3. The median age of the SCD patients was 50 years old. Among the case group, there was a remarkable discrepancy in the amount of two genders, at a male/female ratio of 9.6:1, suggesting male as a significant risk factor for SCD. More than half (58%) of the death occurred without previous symptom, and 70 cases (44%) took place without heavy physical or emotional stress, indicating that SCD was an unexpected and unpredictable disease. Examples of genotyping and sequencing output were displayed in . The genotype frequencies of STAT5A rs3833144 observed in the controls were in correspondence with Hardy-Weinberg equilibrium. The G*Power 3.1 software was employed, examining a statistical power of 0.712 with α set at 0.05 under the dominant model.
Figure 3. Examples of sequencing and genotyping output of rs3833144.
(A) The sequencing output of insertion and deletion allele of rs3833144. The underlined bases are the “GTGT” insertion in coding strands. (B) The examples of the genotyping results. The amplified DNA fragments were analyzed by 7% non-denaturing polyacrylamide gel electrophoresis and silver staining (lane 3, 6, 7, 8 and 13, ins/del genotype; lane 5 and 14, del/del genotype; remaining lanes, ins/ins genotype
Genotypic frequencies of rs3833144 along with OR and its 95%CI in both cases and controls were presented in . Carriers of del/del genotype significantly increased SCD risk in comparison with ins/ins genotype under a codominant model (adjusted OR: 2.53, 95%CI: 1.41–4.51, P = 6.83 × 10−4). Similar trends were also pronounced in both dominant and recessive model (adjusted OR: 1.57, 95%CI: 1.08–2.27, P = 0.0124; adjusted OR: 2.17, 95%CI: 1.26–3.72, P = 2.42 × 10−3). The 4-bp deletion allele had 1.54-fold elevated risk of SCD (adjusted OR: 1.54, 95%CI: 1.18–2.01, P = 9.55 × 10−4). As a result, these findings indicated that deletion allele of rs3833144 would contribute to a higher susceptibility to SCD.
rs3833144 alters local 3D structure of STAT5A mRNA
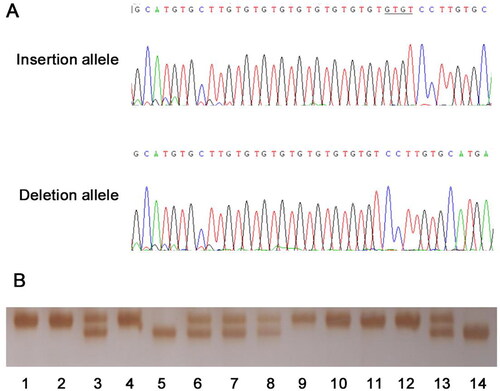
Although rs3833144 locates in 3’UTR of STAT5A gene harbouring abundant miRNA binding sites, it was difficult to predict a matched miRNA whose binding could be interrupted by this “GTGT” indel variant, since “GT” repeats significantly abound in this locus.
RNA structure plays important roles in gene expression regulation and biological function. To explore mechanisms underlying the genotype-expression association between rs3833144 and STAT5A presented by our eQTL analysis, we used SimRNAweb to predict local 3 D structure of STAT5A mRNA. As presented in , the insertion of “GTGT” or not contributed to a visualized discrepancy in mRNA local structures. The altered local structure within 3’UTR might influence mRNA expression of STAT5A at a post-transcriptional level.
Figure 4. the altered local 3 D structure of STAT5A mRNA caused by insertion and deletion allele.
Functional annotation and bioinformatic analysis of rs3833144
To further interrogate the potential regulatory roles of variants rs3833144, we next conducted in silico functional annotation based on Haploreg and ENCODE database. As summarized in Supplementary Table S4, among most of cell lines and tissues available in Haploreg database, the rs3833144 polymorphism located in a region with enrichment of H3K4me1 and H3K27ac and depletion of H3K4me3. Similar modification profiles can also be found in ENCODE database, which were shown in . These results all indicated that rs3833144 locus may reside in a long-range regulatory element. We therefore speculated that some nearby genes may be interacting with and regulated by this regulatory element, and examined all genes located within 2 Mb for interactions based on Hi-C data from 3DIV database. Two tissues, left and right ventricle, were chosen as model systems and distance normalized interaction frequency >2.00 was considered as the criterion of biologically significant interactions. The significant chromatin interaction partners (genes) for rs3833144 locus were listed in Supplementary Table S5 and S6. A total of 47 (left ventricle) and 43 (right ventricle) genes were identified to possibly interact with rs3833144 locus, and 26 genes were found in both two tissues.
Figure 5. (A) Functional annotation of the rs3833144 loci using ENCODE database. Coloured histograms for ChIP-sequencing data containing histone modification markers (H3K4me1, H3K4me3 and H3K27ac) in seven human cell types (GM12878, H1-hESC, HSMM, HUVEC, K562, NHEK and NHLF). (B) Genotype-expression eQTL analysis between rs3833144 and mRNA expression of RARA and (C) PTGES3L-AARSD1 in 445 lymphoblastoid cell lines in 1000 Genomes project database. ins/ins, N = 274; ins/del, N = 147; del/del, N = 24.
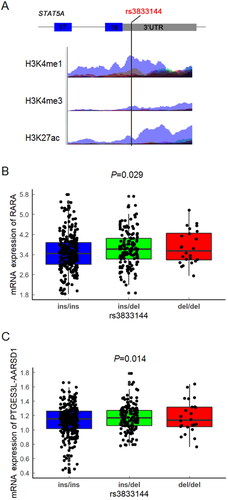
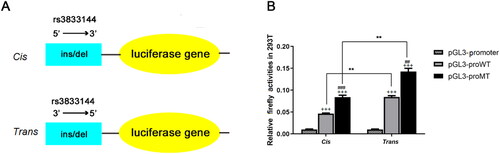
Figure 6. The effect of rs3833144 on gene transcriptional activity as determined by luciferase reporter assay.
(A) plasmids used for luciferase reporter assays. DNA fragments containing variant rs3833144 (210-bp for insertion allele and 206-bp for deletion allele) were directionally subcloned into pGL3-promoter vector. (B) The luciferase activities were compared between construct groups containing different alleles in 293T lines, regardless of 5’ or 3’ direction of the fragments (###P < 0.001, compared with pGL3-proWT in cis group; ##P < 0.01, compared with pGL3-proWT in trans group). The luciferase activities of both insertion or deletion construct group were compared between construct groups including different directions of the fragments (**P < 0.01); Cells transfected with pGL3-proWT-cis/trans or pGL3-proMT-cis/trans showed a significantly higher luciferase activity as compared with cells transfected with pGL3-promoter (+++P < 0.001).
To further investigate whether the transcription of interaction genes would be influenced by rs3833144 through an allele-dependent manner, we performed genotype-expression eQTL analysis between rs3833144 and the interaction genes. Only two genes, retinoic acid receptor alpha (RARA) and PTGES3L-AARSD1 readthrough (PTGES3L-AARSD1) were identified to possess genotype-phenotype correlations with rs3833144. Intriguingly, the chromatin interactions of the two genes with rs3833144 were not found in either of the two tissues (). As shown in , the mRNA level of RARA (P = 0.029) and PTGES3L-AARSD1 (P = 0.014) both appeared to an apparent difference between samples with different genotypes, manifesting an increasing trend in samples with ins/del and del/del genotypes compared with ins/ins genotype. These findings revealed the possibility that the rs3833144 locus might interact with the promoters of RARA and PTGES3L-AARSD1 and thereby regulate the gene transcription through a long-range regulation mechanism.
The regulatory role of rs3833144 on gene transcription activity
Based on Hi-C data and eQTL analysis, we have observed the chromatin interactions and genotype-phenotype correlations between rs3833144 and two interaction genes. To validate the effects of the indel variant on transcription activity, we next performed luciferase reporter assays using pGL3-promoter vector with allele- and direction-different fragments harbouring rs3833144 (). As shown in , the luciferase activities appeared to be significantly different between the groups transfected with vectors harbouring insertion allele (pGL3-proWT) and the groups harbouring deletion allele (pGL3-proMT), regardless of 5′ or 3′ direction of the fragments (P<0.01). Moreover, the constructs harbouring trans (3′→5′) fragments had significant higher luciferase expression than constructs harboring cis (5′→3′) fragments (P<0.01, P<0.001). They both had significant higher luciferase expression than fragments transfected with pGL3-prometer (P<0.001). These results all demonstrated the regulatory properties of rs3833144 on gene transcription.
Discussion
In this study, a novel indel polymorphism has been identified to associate with risk of SCD through bioinformatic screening and subsequent case-control study. On the basis of our bioinformatic and functional analysis, we discovered that rs3833144 played a functional role probably through the following two manners: firstly, rs3833144 might affect 3 D structure of STAT5A mRNA and thereby influence its expression through a structure-dependent manner; secondly, rs3833144 could interact with promoters of nearby genes and regulate gene transcription activities via a long-range mechanism. These results uncovered the possible biological mechanisms underlying the correlation between rs3833144 and SCD risk. Thus, the indel polymorphism may become a potential marker for forensic molecular diagnosis and genetic counseling of SCD.
As a signal transducer and transcriptional factor (TF), the role of STAT5A in the progression of cardiovascular diseases remains contradictory. On one hand, STAT5A would be activated by JAK2 during ischemia/reperfusion (I/R) injury and bind to the specific domain located in promoter of angiotensinogen gene, contributing to activation of cardiac renin angiotensin system (RAS) [Citation24]. Moreover, depression of STAT5A attributed to miR-222 in endothelial cells would protect against advanced neovascularized atherosclerotic lesions [Citation20]. These results suggested that STAT5A would play a pathological role in atherosclerosis and myocardial ischemia injury. On the other hand, there was also some evidence indicating the cardioprotective role of STAT5A. For instance, STAT5A was reported to be indispensable for the cardiacprotection of ischemia preconditioning (PC) against myocardial I/R injury [Citation35, Citation36]. When treated with berberine, relaxin, a protein known for its anti-fibrotic effects would be endogenously upregulated because of reduced STAT3 and increased STAT5A binding to the relaxin promoter [Citation22]. The discrepancy might be attributed to different upstream stimulations and downstream targets for STAT5A. Despite the contradictory role of STAT5A in the heart function, there is no doubt that either upregulation or repression of STAT5A would trigger some transient factors and cause malignant effects on cardiovascular system. In our present study, we have identified that the deletion allele of rs3833144 linked to higher SCD susceptibility, together with repression of STAT5A mRNA expression. Previous study have reported that genetic variants could change RNA local structure, affect the mRNA stability and regulate gene expression [Citation37, Citation38], and our in silico analysis exhibited the altered 3 D structures of STAT5A mRNA caused by different alleles of the variant indeed. We therefore proposed a hypothesis that the deletion allele of rs3833144 may impair the stability of STAT5A mRNA, which thereby contributes to aberrant suppression of STAT5A expression, interfere cardioprotective role of STAT5A and finally facilitate the occurrence of SCD.
SCD is a complex disease caused by various factors, such as cardiac diseases, genetic factors, drastic emotional change or exercises. Consistently, STAT5A regulation may also be interfered by various regulatory factors or genetic variants. Our eQTL analysis based on 1000 G database have identified that rs3833144 is associated with STAT5A mRNA expression. Apart from this variant, the sQTL analysis performed based on GTEx database also uncovered that rs3198502 may influence intron splicing of STAT5A, which may interfere STAT5A function. These bioinformatic results have suggested two possible regulatory factors for STAT5A regulation. Further, our LD analysis also shows that polymorphism rs3833144 and rs3198502 are in one LD block. Considering the contribution of rs3833144 to SCD risk, we therefore speculated that rs3833144 and rs3198502 may play a synergetic role in STAT5A regulation as well as SCD occurrence.
Cis-regulatory elements (CREs), such as promoters, enhancers, silencers, and insulators, are non-coding DNA regions which regulate gene expression at a transcriptional level. Genome-wide association studies have suggested a series of disease-associating non-coding mutants could disrupt the CREs and influence expression of nearby genes via a long-range mechanism [Citation39–41]. Our functional annotations found that the region containing rs3833144 showed enrichment of H3K4me1 and H3K27ac and depletion of H3K4me3, which were all indicative markers of enhancers [Citation42]. Further bioinformatic analysis identified that RARA and PTGES3L-AARSD1 interacted with rs3833144, and eQTL analysis manifested significant genotype-expression correlations between rs3833144 and the two genes with a higher expression linked to deletion allele. The luciferase assays finally demonstrated the regulatory role of the variant on gene expression, suggesting the possibility that rs3833144 may reside in a potential CRE and control the transcription of RARA and PTGES3L-AARSD1 through a long-range mechanism.
RARA, a member of nuclear retinoic acid (RA) receptor family, acts as a transcription factor and control gene transcription through a ligand-dependent manner. It has been reported that cardiac impairment of RARA would lead to the development of diastolic dysfunction [Citation43], while activated RARA would protect artery from atherosclerosis by derepressing miR-10a in vascular endothelial cells in oscillatory shear regions [Citation44]. Despite these cardioprotective role, overexpression of RARA could also stimulate differentiation of aortic endothelial cells and enhance RA-induced angiogenesis [Citation45]. Considering that increased neovascularization within atherosclerotic plaques would enhance the risk of plaque rupture [Citation46], RARA overexpression may play a pathological role in plaque rupture and finally increase SCD risk, especially to the victims with high extents of atherosclerosis. PTGES3L-AARSD1 represents naturally readthrough transcription and encodes a fusion protein of two nearby genes: prostaglandin E synthase 3 like (PTGES3L) and alanyl-tRNA synthetase domain containing 1 (AARSD1). Although few studies have been reported about PTGES3L-AARSD1, this kind of transcription-induced chimera may harbour the properties of both genes [Citation47]. PTGES3L encodes a protein possessing similar region with prostaglandin E synthase 3 (PTGES3), a synthase of prostaglandin E2 (PGE2). It is previously reported that PGE2 could contribute to cardiac hypertrophy [Citation48], atherogenesis [Citation49] and inflammatory cardiomyopathies [Citation50]. Meanwhile, both PTGES3 (also known as p23) and AARSD1 functions as co-chaperones of heat shock protein 90 (HSP90), which participates in cardiac hypertrophy and heart failure [Citation51]. Based on the evidence exemplified above, we could certainly speculate that the PTGES3L-AARSD1 play a prominent role in cardiovascular diseases and dysregulation of PTGES3L-AARSD1 would be harmful to human heart. Taken together, it is plausible that the overexpression of RARA and PTGES3L-AARSD1 derived from deletion allele of rs3833144 might cause malignant changes to our heart and participate in the pathophysiology of SCD.
Some limitations should be emphasized in our study. Since the RNA-seq and genotyping data of cardiomyocytes are not available at present, our eQTL analysis mainly focused on lymphoblastoid cell lines in 1KGP database. Therefore, based on limited bioinformatic results we can only propose possible hypotheses about how the variant rs3833144 contribute to SCD risk, further functional assays are needed to verify our results. Additionally, the significance of the finding that the deletion allele of rs3833144 contributed to the SCD risk are still limited by small sample size in our study. Further replicated case-control studies with different or expanded populations are needed to guarantee our observations. Finally, since our SCD cases mostly suffer sudden death originated from coronary atherosclerosis, our results raised the possibility that rs3833144 may be associated with coronary atherosclerosis susceptibility in natural populations. However, this implication still needs to be further investigated by coronary atherosclerosis related studies, which may have potential interests for cardiovascular community.
Conclusion
Here, we have provided the initial evidence that the indel polymorphism (rs3833144) within the 3’UTR of STAT5A gene was significantly associated with SCD risk. Our current data thus suggested a possible involvement of rs3833144 to SCD predisposition in Chinese populations and rs3833144 with potential function roles may become a candidate marker for SCD diagnosis and prevention.
Authors’ contributions
Yuzhen Gao and Bin Luo conceived and designed the experiments; Huan Yu, Zhenzhen Yang, Qing Zhang, Qi Yang, Yiling Qu, and Rui Tan performed the experiments; Yadong Guo, Jiabin Xu, Lijuan Li and Yan He analyzed the data; Chengtao Li and Suhua Zhang contributed reagents, materials and analysis tools; Huan Yu and Yuzhen Gao contributed to the writing and editing of the manuscript. All authors contributed to the final text and approved it.
Compliance with ethical standards
The study was approved by the Ethical Committee of Soochow University. Written informed consent was obtained from relatives of each sudden death case before the investigation.
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–2351.
- Sara JD, Eleid MF, Gulati R, et al. Sudden cardiac death from the perspective of coronary artery disease. Mayo Clin Proc. 2014;89:1685–1698.
- Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492.
- Feng XF, Hai JJ, Ma Y, et al. Sudden cardiac death in Mainland China: a systematic analysis. Circ Arrhythm Electrophysiol. 2018;11:e006684.
- Myerburg RJ, Junttila MJ. Sudden cardiac death caused by coronary Heart Disease. Circulation. 2012;125:1043–1052.
- Goff ZD, Calkins H. Sudden death related cardiomyopathies: hypertrophic cardiomyopathy. Prog Cardiovasc Dis. 2019;62:212–216.
- Emery MS, Kovacs RJ. Sudden cardiac death in athletes. JACC Heart Fail. 2018;6:30–40.
- Basso C, Carturan E, Pilichou K, et al. Sudden cardiac death with normal heart: molecular autopsy. Cardiovasc Pathol. 2010;19:321–325.
- Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res. 2015;116:1919–1936.
- Magi S, Lariccia V, Maiolino M, et al. Sudden cardiac death: focus on the genetics of channelopathies and cardiomyopathies. J Biomed Sci. 2017;24:56.
- Lariccia V, Nasti AA, Alessandrini F, et al. Identification and functional analysis of a new putative caveolin-3 variant found in a patient with sudden unexplained death. J Biomed Sci. 2014;21:58.
- Friedlander Y, Siscovick DS, Arbogast P, et al. Sudden death and myocardial infarction in first degree relatives as predictors of primary cardiac arrest. Atherosclerosis. 2002;162:211–216.
- Jouven X, Desnos M, Guerot C, et al. Predicting sudden death in the population: the Paris Prospective Study I. Circulation. 1999;99:1978–1983.
- Kaikkonen KS, Kortelainen ML, Linna E, et al. Family history and the risk of sudden cardiac death as a manifestation of an acute coronary event. Circulation. 2006;114:1462–1467.
- Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–217.
- Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci. 2000;25:496–502.
- Satou R, Gonzalez-Villalobos RA. JAK-STAT and the renin-angiotensin system: the role of the JAK-STAT pathway in blood pressure and intrarenal renin-angiotensin system regulation. JAKSTAT. 2012;1:250–256.
- Haghikia A, Stapel B, Hoch M, et al. STAT3 and cardiac remodeling. Heart Fail Rev. 2011;16:35–47.
- Osugi T, Oshima Y, Fujio Y, et al. Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J Biol Chem. 2002;277:6676–6681.
- Dentelli P, Rosso A, Orso F, et al. microRNA-222 controls neovascularization by regulating signal transducer and activator of transcription 5A expression. Arterioscler Thromb Vasc Biol. 2010;30:1562–1568.
- Chen Y, Surinkaew S, Naud P, et al. JAK-STAT signalling and the atrial fibrillation promoting fibrotic substrate. Cardiovasc Res. 2017;113:310–320.
- Gu HP, Lin S, Xu M, et al. Up-regulating relaxin expression by G-quadruplex interactive ligand to achieve antifibrotic action. Endocrinology. 2012;153:3692–3700.
- Buitenhuis M, Coffer PJ, Koenderman L. Signal transducer and activator of transcription 5 (STAT5). Int J Biochem Cell Biol. 2004;36:2120–2124.
- Mascareno E, El-Shafei M, Maulik N, et al. JAK/STAT signaling is associated with cardiac dysfunction during ischemia and reperfusion. Circulation. 2001;104:325–329.
- El-Adawi H, Deng L, Tramontano A, et al. The functional role of the JAK-STAT pathway in post-infarction remodeling. Cardiovasc Res. 2003;57:129–138.
- Mayr C. Regulation by 3’-untranslated regions. Annu Rev Genet. 2017;51:171–194.
- Wang S, Zhang Z, Yang Y, et al. An insertion/deletion polymorphism within 3’UTR of RYR2 modulates sudden unexplained death risk in Chinese populations. Forens Sci Int. 2017;270:165–172.
- Yin Z, Zhang Q, Zhou W, et al. Influence of functional polymorphism in MIF promoter on sudden cardiac death in Chinese populations. Forens Sci Res. 2017;2:152–157.
- Allen RC, Graves G, Budowle B. Polymerase chain reaction amplification products separated on rehydratable polyacrylamide gels and stained with silver. Biotechniques. 1989;7:736–744.
- Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311.
- Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012:D930–D934.
- Davis CA, Hitz BC, Sloan CA, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801.
- Yang D, Jang I, Choi J, et al. 3DIV: a 3D-genome interaction viewer and database. Nucleic Acids Res. 2018;46:D52–D57.
- Faul F, Erdfelder E, Buchner A, et al. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods. 2009;41:1149–1160.
- Yamaura G, Turoczi T, Yamamoto F, et al. STAT signaling in ischemic heart: a role of STAT5A in ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H476–82.
- Chen H, Jing XY, Shen YJ, et al. Stat5-dependent cardioprotection in late remote ischaemia preconditioning. Cardiovasc Res. 2018;114:679–689.
- Duan J, Wainwright MS, Comeron JM, et al. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. 2003;12:205–216.
- Mooers BH, Logue JS, Berglund JA. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc Natl Acad Sci USA. 2005;102:16626–16631.
- Pomerantz MM, Ahmadiyeh N, Jia L, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884.
- Harismendy O, Notani D, Song X, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268.
- Smemo S, Campos LC, Moskowitz IP, et al. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet. 2012;21:3255–3263.
- Mora A, Sandve GK, Gabrielsen OS, et al. In the loop: promoter-enhancer interactions and bioinformatics. Brief Bioinform. 2016;17:980–995.
- Zhu S, Guleria RS, Thomas CM, et al. Loss of myocardial retinoic acid receptor α induces diastolic dysfunction by promoting intracellular oxidative stress and calcium mishandling in adult mice. J Mol Cell Cardiol. 2016;99:100–112.
- Lee DY, Yang TL, Huang YH, et al. Induction of microRNA-10a using retinoic acid receptor-α and retinoid x receptor-α agonists inhibits atherosclerotic lesion formation. Atherosclerosis. 2018;271:36–44.
- Gaetano C, Catalano A, Illi B, et al. Retinoids induce fibroblast growth factor-2 production in endothelial cells via retinoic acid receptor α activation and stimulate angiogenesis in vitro and in vivo. Circ Res. 2001;88:E38–47.
- Roth L, Rombouts M, Schrijvers DM, et al. Chronic intermittent mental stress promotes atherosclerotic plaque vulnerability, myocardial infarction and sudden death in mice. Atherosclerosis. 2015;242:288–294.
- Akiva P, Toporik A, Edelheit S, et al. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16:30–36.
- Mendez M, LaPointe MC. PGE2-induced hypertrophy of cardiac myocytes involves EP4 receptor-dependent activation of p42/44 MAPK and EGFR transactivation. Am J Physiol Heart Circ Physiol. 2005;288:H2111–H2117.
- Gross S, Tilly P, Hentsch D, et al. Vascular wall-produced prostaglandin E2 exacerbates arterial thrombosis and atherothrombosis through platelet EP3 receptors. J Exp Med. 2007;204:311–320.
- Toth AD, Schell R, Levay M, et al. Inflammation leads through PGE/EP3 signaling to HDAC5/MEF2-dependent transcription in cardiac myocytes. EMBO Mol Med. 2018;10:e8536.
- Ranek MJ, Stachowski MJ, Kirk JA, et al. The role of heat shock proteins and co-chaperones in heart failure. Phil Trans R Soc B. 2018;373:20160530.