
Abstract
A new method was developed for pre-concentration and determination of multiple drugs of abuse in human urine using dispersive liquid–liquid microextraction (DLLME) and capillary electrophoresis (CE) with photodiode array detection. The method was based on the formation of tiny droplets of an organic extractant in the prepared sample solution using water-immiscible organic solvent (chloroform) dissolved in water-miscible organic dispersive solvent (isopropyl alcohol). The organic phase, which extracted eight drugs of abuse from the prepared urine solution, was separated by centrifugation. The sedimented phase was transferred into a small volume CE auto-sampler vial with 10 µL of 1% HCl methanol solution and evaporated to dryness. The residue was reconstituted in lidocaine hydrochloride (internal standard) aqueous solution and introduced by electrokinetic injection into CE. Under the optimum conditions, acceptable linear relationship was observed in the range of 3.0–500 ng/mL with the correlation coefficient (r) of 0.9982–0.9994 for spiked urine samples. The limit of detection (LOD) (S/N = 3) was estimated to be 1.0 ng/mL. A recovery of 75.7%–90.6% was obtained for spiked samples. The mean relative error (MRE) was within ±7.0% and the relative standard deviation (RSD) was less than 6.9%. The proposed DLLME-CE procedure offers an alternative analytical approach for the sensitive detection of drugs of abuse in real urine samples.
The dispersive liquid-liquid microextraction (DLLME) was involved for the determination of drugs in urine with capillary electrophoresis with photodiode array detection (CE-PDA).
Good linearity, sensitivity, recovery and precision were achieved.
The proposed method was eco-friendly with microliter scale solvent consumption.
Key points
Introduction
Fighting against drugs of abuse and addiction is an ongoing struggle for society and public health systems. To control these drugs (such as amphetamines and heroin) effectively, it is necessary to develop selective and sensitive analytical methods suitable for their unambiguous identification and determination in illicit samples and biological matrices. This has traditionally been carried out using gas chromatography-mass spectrometry (GC-MS) [Citation1,Citation2], high-performance liquid chromatography-mass spectrometry-mass spectrometry (HPLC-MSMS) [Citation3,Citation4], capillary electrophoresis (CE) [Citation5,Citation6], immunosensor [Citation7] and immunoassay [Citation8], etc. In recent years, the mixing use of drugs is becoming one of the epidemiological characteristics of drug abuse patterns [Citation9]. The simultaneous screening and confirmation of multiple drugs in the body is of considerable importance for the investigation of intoxications, withdrawal and clinic treatment.
The structures of drugs and their metabolites are similar. They exist in complex matrices such as body fluids at different trace concentrations, so they are not easily determined both qualitatively and quantitatively. In this context, CE provides original characteristics in terms of separation mechanisms, rapid and efficient analysis with remarkably high resolution, small sample volume, high sample throughput, low operational cost and tolerance to biological matrices [Citation10]. All these characteristics ideally make it an attractive technique in comparison with liquid chromatography (LC) and gas chromatography (GC) for forensic toxicological analysis [Citation5,Citation6]. However, poor concentration sensitivity of traditional CE with on-column UV detection due to low sample injection volume and short optical path length limit the use of CE as an effective method to determine trace analytes in biological and environmental samples [Citation10].
To improve concentration detection limits in CE, several sample preparation techniques such as online sample concentration techniques [Citation11,Citation12], liquid–liquid extraction [Citation13,Citation14], and spectrometry (SPE) [Citation15,Citation16] have been developed, and these techniques have been applied to the forensic analysis [Citation5,Citation6,Citation17–19]. Compared with the other sample preparation techniques, dispersive liquid–liquid microextraction (DLLME) has the advantages of low cost, excellent sample clean-up effect, and high extraction efficiency [Citation20,Citation21], which has been applied to the determination of trace analytes in various samples [Citation22–25].
The aim of this study was to develop and validate a method to simultaneous screening and confirmation of multiple drugs of abuse in human urine using the combination of DLLME sample pretreatment and CE with photo diode array (PDA) analyte detection. Different parameters affecting the extraction process were studied and optimized in detail. The performance of the proposed method can provide higher stability and extraction efficiency in comparison with other extraction methods and was successfully employed to determine the trace level target analytes in human urine samples.
Materials and methods
Reagents and materials
6-Monoacetylmorphine (6-MAM) hydrochloride, morphine hydrochloride, codeine phosphate, methamphetamine hydrochloride, amphetamine hydrochloride, 3,4-methylenedioxymethamphetamine (MDMA) hydrochloride, 3,4-methylenedioxyamphetamine (MDA) hydrochloride and ketamine hydrochloride were purchased from Cerilliant (Merck KGaA, Darmstadt, Germany). The HPLC-grade methanol and acetonitrile were obtained from TEDIA Company (Fairfield, OH, USA). Ethanol, isopropyl alcohol, dichloromethane (CH2Cl2), chloroform (CHCl3), carbon tetrachloride (CCl4) and 1,1,2,2-tetrachloroethane (C2H2Cl4) were purchased from Kermel (Tianjin, China). Lidocaine hydrochloride (≥98%) was purchased from Sigma-Aldrich (St. Louis, MO, USA) for internal standard (IS). All other analytical grade reagents used for experiments were purchased from Kermel. All electrolytes and standard solutions were prepared with doubly distilled water. They were stored in the refrigerator at 4 °C, filtered through 0.45 µm disposable membrane filter (Millipore, Milford, MA, USA) and degassed by ultrasonication before use.
The blank urine samples were obtained from drug-free students in Fujian Police College. The real samples from the drug abusers were obtained from the Institution of Forensic Science (Fujian Provincial Public Security Department, Fuzhou, China) anonymously. Written informed consent was signed by all individuals for sample collection and used in this study. Ethical approval was obtained from by Fujian Police College Internal Review Board.
Instrumentation
CE was performed with a CESI 8000 plus instrument equipped with a PDA detector (AB Sciex, Framingham, MA, USA). Separations were accomplished in a 75 µm I.D. fused silica capillary (Reafine, Handan, China) with an effective length of 50 cm (total length 60.3 cm). Detection was performed via on-capillary measurement of the UV absorptions at 214 nm. The measurements were performed at 25 °C.
Sample preparation
To make the possible impurity precipitation, 0.5 mL of methanol and 10 mg zinc acetate were added into 1 mL of urine sample. Then a 3-min centrifugation at 10 625 ×g and 4 °C was carried out and the top aqueous layer was removed and filtered through a 0.45 µm membrane filter.
DLLME
A 1.0 mL of prepared sample solution above was diluted with 1.0 mL of 30 mmol/L sodium tetraborate buffer (pH 9.2) and was placed in a 5 mL tube with conical bottom. The amount of 0.5 mL of isopropyl alcohol (disperser solvent) containing 41 µL of CHCl3 (extraction solvent) was injected rapidly into the sample solution by using a 1.00-mL syringe. A cloudy solution (water, isopropyl alcohol, and CHCl3) was formed in the test tube. In this step, the analytes were extracted into the fine droplets of CHCl3 in a few seconds. After centrifugation for 3 min at 10 625 ×g, the extraction solvent was sedimented in the bottom of the conical test tube (about (8.0 ± 0.2) µL). The sedimented phase (8 µL) was removed using a 10-µL GC microsyringe and placed into a small volume CE auto-sampler vial with 10 µL of 1% HCl methanol solution, then evaporated to dryness under nitrogen using an N-Evap evaporator (Organomation Assoc., Berlin, MA, USA) at room temperature. The residue was reconstituted in 20 µL of the 3 mg/L lidocaine hydrochloride aqueous solution and introduced by electrokinetic injection into CE.
CE-UV analysis
Carrier electrolytes were prepared by neutralization of 100 mmol/L NaH2PO4 solution containing 20% methanol (v/v) with H3PO4 to the pH 4.0. Each new fused silica capillary was rinsed at 20 p.s.i. with 0.1 mol/L NaOH for 30 min, double distilled water for 30 min and buffer for 30 min. All samples were introduced by electrokinetic injection for 5 s at 5 kV. Each sample was injected only once. During analysis the instrument was operated at 25 kV, generating a current level of approximately 115 µA. Between electrophoretic separations the capillary was rinsed at 20 p.s.i. with double distilled water, 0.1 mol/L NaOH, double distilled water and the running buffer for 3 min, respectively.
Method validation
Method validation was validated according to the published recommendations [Citation26,Citation27]. The linearity of the method was assessed in spiked samples. It was established from six calibration levels. For urine samples, the analytes at the concentration of 3.0, 10.0, 50.0, 100.0, 300.0 and 500.0 ng/mL were prepared, respectively. The spiked samples were treated in accordance with the above process (n = 5).
Based on the experimental data, the correlation of analytes concentration (x) with the peak area ratio of analytes to internal standard (y) was obtained. Linearity of the calibration curve was calculated using a line of best fit with correlation factor expected to be >0.99. Limit of detection (LOD) was calculated as 3 times of the signal to noise ratio (S/N). Lower limit of quantification (LLOQ) was calculated as 10 times of the S/N. The deviation of the measured concentration from the nominal concentration should be within ±15% except for the LLOQ level where a ± 20% deviation is acceptable.
Intra- and inter-day (over 5 days) precision and accuracy studies were investigated using quality control samples which were blank samples spiked with analytes at 10, 50 and 100 ng/mL (n = 5) for urine samples. Accuracy was expressed as a percentage mean relative error (MRE), which was deemed acceptable if the calculated concentrations fell within 20% of the concentration spiked. Precision was deemed acceptable if the percentage relative standard deviation (RSD) was <15%. Recovery percentage of the proposed DLLME procedure referred to comparing the peak area of the DLLME-treated analyte spiked sample with the pure analyte standard at the equivalent concentration without going through the DLLME process.
Results and discussion
Optimization of DLLME
Different parameters affecting the extraction efficiency such as the kind and volume of extraction and dispersive solvents, the pH of sample solution, and extraction time must be studied and optimized with the standard aqueous solution of the analytes. The enrichment factor (EF) and extraction recovery (ER) were used to assess the method optimized parameters as described by Rezaee et al. [Citation28] and Leong et al. [Citation29], as can be seen in the Supplementary Information.
CH2Cl2, CHCl3, CCl4 and C2H2Cl4 were pretested as extraction solvents to analyse the effect of the solvent on extraction efficiency. A series of sample solutions were studied by using 0.50 mL of methanol containing different volumes of extraction solvents to achieve 8.0 µL of settled phase [Citation28], accordingly, 83.5, 50.0, 25.3, 20.0 µL of CH2Cl2, CHCl3, CCl4, and C2H2Cl4 were selected, respectively. As shown in , CHCl3 possessed the highest ER as compared with other extraction solvents.
Table 1. Enrichment recoveries obtained with the different extraction solvents evaluated for the extraction of drugs by dispersive liquid–liquid microextraction (DLLME).
Methanol, acetonitrile, ethanol and isopropyl alcohol were tested as disperser solvent. To confirm the constant volume of the sedimented phase was constant (8.0 µL), the experiments were performed using 0.5 mL of methanol, acetonitrile, ethanol and isopropyl alcohol containing 50.0, 48.0, 20.8 and 41.0 µL of CHCl3, respectively. The results indicated that isopropyl alcohol exhibited the highest extraction efficiency (). Subsequently, various experiments were performed to optimize the volume of dispersive solvent by using different volumes of isopropyl alcohol (0.25, 0.50, 0.75, 1.00 and 1.50 mL) containing 17.0, 41.0, 54.4, 65.8 and 81.6 µL CHCl3, respectively. As shown in , 0.5 mL of isopropyl alcohol containing 41.0 µL CHCl3 gave a good extraction performance.
Figure 1. Effect of the dispersive solvent (isopropyl alcohol) volume on extraction recovery.
Extraction conditions: aqueous sample volume, 1.0 mL; pH of sample, 9.0; dispersive solvent, isopropyl alcohol; extraction solvent, 41.0 μL CHCl3; room temperature; analytes concentration spiked, 50 ng/mL; internal standard, 3 µg/mL lidocaine. MA: methamphetamine; AM: amphetamine; MDMA: methylenedioxymethamphetamine; MDA: methylenedioxyamphetamine; MAM: monoacetylmorphine.
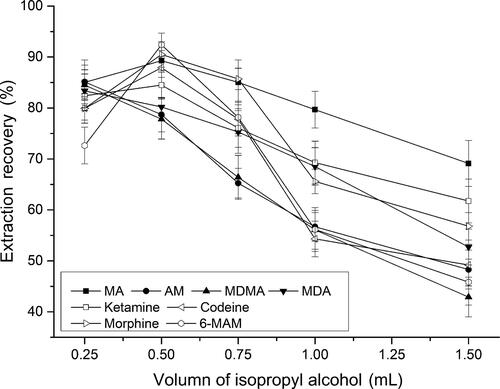
Table 2. Enrichment recoveries obtained with the different dispersive solvents evaluated for the extraction of drugs by dispersive liquid–liquid microextraction (DLLME).
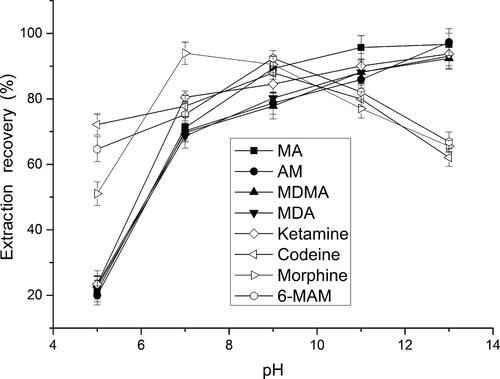
The effect of sample pH was tested in the pH range from 5 and 13. As shown in , the ERs of amphetamines and ketamine were maximized at pH 11 and then slightly increased. However, a gradual increase of the ERs of heroin metabolites was observed with increasing sample pH, up to 9.0, then decreased, which was attribute to the amphoteric characteristic of morphine. Then, the pH 9.0 was selected for the sample solution. The desired pH was achieved by diluting with sodium tetraborate buffer (pH 9.2) to the prepared sample solution.
Figure 2. Effect of the pH of sample solution on extraction recovery.
Extraction conditions: aqueous sample volume, 1.0 mL; dispersive solvent, 0.5 mL isopropyl alcohol; extraction solvent, 41.0 µL CHCl3; room temperature; analytes concentration spiked, 50 ng/mL; internal standard, 3 µg/mL lidocaine. MA: methamphetamine; AM: amphetamine; MDMA: methylenedioxymethamphetamine; MDA: methylenedioxyamphetamine; MAM: monoacetylmorphine.
Previous studies [Citation26–Citation29] indicated that the extraction quickly reached equilibrium and the DLLME method is time-independent. However, the good reproducibility cannot be ensured in an extraction time as short as a few seconds. Consequently, 1 min of extraction time was chosen in the following experiments.
Method validation
In order to proceed with the current evaluation of the proposed DLLME technique, linearity, LOD, and repeatability were investigated under optimized conditions with the spiked samples. The performance of the developed procedure is summarized in . The calibration curve was linear for concentrations of all analytes in the range of 3.0–500 ng/mL with the correlation coefficient ranging from 0.9982 to 0.9994 for spiked urine samples. The LOD (S/N = 3) was estimated to be 1.0 ng/mL. The LLOQ (S/N = 10) was estimated to be 3.0 ng/mL. The recoveries vary from 75.7% to 90.6% with RSD ≤6.9%. Precision and accuracy at the three concentration levels were determined to be satisfactory. The repeatability was acceptable and comparable with other methods reported in the literature (). The results suggested that the methods had high sensitivity and reproducibility, which could be applied to monitoring multiple drugs of abuse in real samples by DLLME-CE.
Table 3. The performance of the proposed methods in spiked urine samples.
Table 4. Comparison of DLLME with SPME and LPME for determination of MA and morphine in aqueous solution.
Analysis of real biological samples
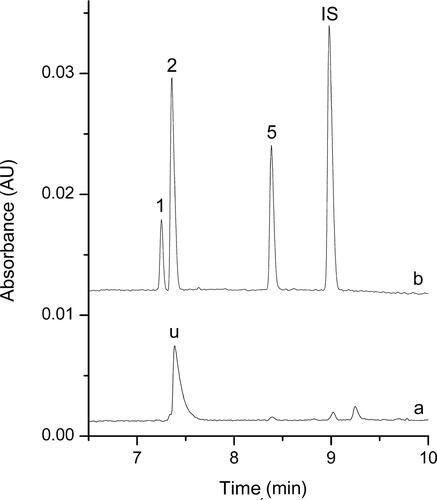
The proposed DLLME-CE procedure described above was applied to determine the multiple drugs of abuse in spiked and real samples. In addition to the high preconcentration, electropherograms obtained for the spiked biological samples and the real biological samples after DLLME extraction demonstrated a substantial sample clean-up effect. Electropherograms of these samples before and after DLLME extraction were shown in and , no other peaks were present in the electropherograms, and high preconcentration was obtained after DLLME extraction.
Figure 3. Electropherograms obtained for the spiked biological samples before (a) and after (b) dispersive liquid–liquid microextraction (DLLME) extraction under the optimum conditions.
Extraction conditions: dispersive solvent, 0.5 mL isopropyl alcohol; extraction solvent, 41.0 µL CHCl3; room temperature; pH of sample, 9.0; analytes concentration spiked, urine: 50 ng/mL; internal standard: 3 µg/mL lidocaine.
Peak identification: (IS) lidocaine, (1) AM: amphetamine, (2) MA: methamphetamine, (3) MDA: methylenedioxyamphetamine, (4) MDMA: methylenedioxymethamphetamine, (5) ketamine, (6) codeine, (7) morphine, (8) 6-MAM: 6-monoacetylmorphine.
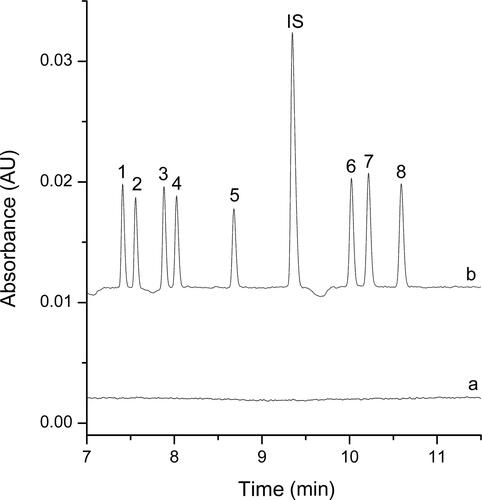
Figure 4. Electropherograms obtained for the real biological samples from the drug abuser before (a) and after (b) dispersive liquid–liquid microextraction (DLLME) extraction under the optimum conditions. Extraction conditions: dispersive solvent, 0.5 mL isopropyl alcohol; extraction solvent, 41.0 μL CHCl3; room temperature; pH of sample, 9.0; analytes concentration spiked, urine: 50 ng/mL; internal standard: 3 μg/mL lidocaine. Peak identification: (IS) lidocaine, (1) AM: amphetamine, 42.1 ng/mL, (2) MA: methamphetamine, 102.6 ng/mL, (5) ketamine, 84.7 ng/mL.
Conclusion
This study has demonstrated the successful application of DLLME-CE-UV method in the determination of multiple drugs of abuse in human urine. The optimum conditions of extraction performance have been obtained. The experimental results reveal that this method provides high extraction efficiency within a short time, good selectivity, low LODs and good linearity over the investigated concentration range. The performance of this procedure in the extraction of 6-MAM, morphine, codeine, MA, AM, MDMA, MDA and ketamine from human urine was satisfactory. Compared with SPME and LPME, besides the short extraction time, low cost, and feasibility, the best advantages of the presented method are the high sensitivity and good linearity. Therefore, it has the potential to be a powerful tool for the analysis of drugs of abuse in forensic investigations.
Authors’ contributions
Liang Meng designed the experiments, studied the sample pretreatment method, and drafted the manuscript; Shuhai Ye conducted the CE method; Yilin Wu, Linda You conceived of the study, and participated in the method developing. All authors contributed to the final text and approved it.
Compliance with ethical standards
Written informed consent was signed by all the healthy individuals and drug users for the sample collection and used in this study. Ethical approval was granted by Fujian Police College Internal Review Board.
Supplemental Material
Download MS Word (20.7 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Kranenburg RF, Lukken CK, Schoenmakers PJ, et al. Spotting isomer mixtures in forensic illicit drug casework with GC-VUV using automated coelution detection and spectral deconvolution. J Chromatogr B Analyt Technol Biomed Life Sci. 2021;1173:122675.
- Orfanidis A, Mastrogianni O, Koukou A, et al. A GC-MS method for the detection and quantitation of ten major drugs of abuse in human hair samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1047:141–150.
- Orfanidis A, Gika H, Theodoridis G, et al. Development of a UHPLC-MS/MS method for the determination of 84 pharmaceuticals and drugs of abuse in human liver. J Chromatogr B Analyt Technol Biomed Life Sci. 2020;1151:122192.
- Bassotti E, Merone GM, D’Urso A, et al. A new LC-MS/MS confirmation method for the determination of 17 drugs of abuse in oral fluid and its application to real samples. Forensic Sci Int. 2020;312:110330.
- Cui X, Ni C, Liang C, et al. Screening and quantitation of forty-six drugs of abuse and toxic compounds in human whole blood by capillary electrophoresis: application to forensic cases. Microchem J. 2019;144:403–410.
- Saar-Reismaa P, Brilla C-A, Leiman K, et al. Use of a newly-developed portable capillary electrophoresis analyser to detect drugs of abuse in oral fluid: a case study. Talanta. 2020;211:120662.
- Yang Y, Pan J, Hua W, et al. An approach for the preparation of highly sensitive electrochemical impedimetric immunosensors for the detection of illicit drugs. J Electroanal Chem. 2014;726:1–6.
- Pelletti G, Rossi F, Garagnani M, et al. Optimization of cloned enzyme donor immunoassay cut-offs for drugs of abuse in post-mortem whole blood. Forensic Sci Int. 2020;312:110291.
- Meng L, Dai Y, Chen C, et al. Determination of amphetamines, ketamine and their metabolites in hair with high-speed grinding and solid-phase microextraction followed by LC-MS. Forensic Sci Res. 2021;6:273–280.
- Shah M, Patel N, Tripathi N, et al. Capillary electrophoresis methods for impurity profiling of drugs: a review of the past decade. J Pharm Anal. 2022;12:14.
- Liu Z, Wang C, Wang X, et al. On-line screening of indoleamine 2,3-dioxygenase 1 inhibitors by partial filling capillary electrophoresis combined with rapid polarity switching. J Chromatogr A. 2021;1651:462305.
- John AS, Sidek MM, Thang LY, et al. Online sample preconcentration techniques in nonaqueous capillary and microchip electrophoresis. J Chromatogr A. 2021;1638:461868.
- Mbaye AR, Foulon C, Lecoeur M. Capillary electrophoresis as a versatile tool for quality control and epidermis permeation studies of transdermal formulations. J Pharm Biomed Anal. 2021;193:113732.
- Pérez-Alcaraz A, Borrull F, Aguilar C, et al. An electrokinetic supercharging approach for the enantiodetermination of cathinones in urine samples by capillary electrophoresis. Microchem J. 2020;158:105300.
- Martínez-Pérez-Cejuela H, Benavente F, Simó-Alfonso EF, et al. A hybrid nano-MOF/polymer material for trace analysis of fluoroquinolones in complex matrices at microscale by on-line solid-phase extraction capillary electrophoresis. Talanta. 2021;233:122529.
- Pero-Gascon R, Benavente F, Neusüß C, et al. Evaluation of on-line solid-phase extraction capillary electrophoresis-mass spectrometry with a nanoliter valve for the analysis of peptide biomarkers. Anal Chim Acta. 2020;1140:1–9.
- Baciu T, Botello I, Borrull F, et al. Capillary electrophoresis and related techniques in the determination of drugs of abuse and their metabolites. TrAC Trends Anal Chem. 2015;74:89–108.
- Cruces-Blanco C, García-Campaña AM. Capillary electrophoresis for the analysis of drugs of abuse in biological specimens of forensic interest. TrAC Trends Anal Chem. 2012;31:85–95.
- Costa JL, Morrone AR, Resende RR, et al. Development of a method for the analysis of drugs of abuse in vitreous humor by capillary electrophoresis with diode array detection (CE-DAD). J Chromatogr B Analyt Technol Biomed Life Sci. 2014;945–946:84–91.
- Rykowska I, Ziemblińska J, Nowak I. Modern approaches in dispersive liquid-liquid microextraction (DLLME) based on ionic liquids: a review. J Mol Liq. 2018;259:319–339.
- Mousavi L, Tamiji Z, Khoshayand MR. Applications and opportunities of experimental design for the dispersive liquid-liquid microextraction method—a review. Talanta. 2018;190:335–356.
- Farajzadeh MA, Kiavar L, Pezhhanfar S. Development of a method based on dispersive liquid-liquid microextraction followed by partial vaporization of the extract for ultra-preconcentration of some pesticide residues in fruit juices. J Chromatogr A. 2021;1653:462427.
- Meng L, Zhang W, Meng P, et al. Comparison of hollow fiber liquid-phase microextraction and ultrasound-assisted low-density solvent dispersive liquid-liquid microextraction for the determination of drugs of abuse in biological samples by gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;989:46–53.
- Kahl JMM, da Cunha KF, Rodrigues LC, et al. Quantification of amphetamine and derivatives in oral fluid by dispersive liquid-liquid microextraction and liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2021;196:113928.
- Qiao L, Sun R, Yu C, et al. Novel hydrophobic deep eutectic solvents for ultrasound-assisted dispersive liquid-liquid microextraction of trace non-steroidal anti-inflammatory drugs in water and milk samples. Microchem J. 2021;170:106686.
- Peters FT, Drummer OH, Musshoff F. Validation of new methods. Forensic Sci Int. 2007;165:216–224.
- Scientific Working Group for Forensic Toxicology (SWGTOX). Standard practices for method validation in forensic toxicology. J Anal Toxicol. 2013;37:452–474.
- Rezaee M, Assadi Y, Milani Hosseini M-R, et al. Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A. 2006;1116:1–9.
- Leong M-I, Fuh M-R, Huang S-D. Beyond dispersive liquid-liquid microextraction. J Chromatogr A. 2014;1335:2–14.
- Cháfer-Pericás C, Campíns-Falcó P, Herráez-Hernández R. Application of solid-phase microextraction combined with derivatization to the determination of amphetamines by liquid chromatography. Anal Biochem. 2004;333:328–335.
- Halvorsen TG, Pedersen-Bjergaard S, Reubsaet JL, et al. Liquid-phase microextraction combined with flow-injection tandem mass spectrometry Rapid screening of amphetamines from biological matrices. J Sep Sci. 2001;24:615–622.
- Yazdi AS, Eshaghi Z. Surfactant enhanced liquid-phase microextraction of basic drugs of abuse in hair combined with high performance liquid chromatography. J Chromatogr A. 2005;1094:1–8.