Graphical abstract
 Five novel nonenolides, phomolides D–H (1–5), and phomonol (6), phomotone (7) and phomopene (8), plus previously know compounds 4-formyl-7-methoxyl-6-methyl-phthalide (9), dihydrogladiolic acid methyl lactal (10), cytochalasin H (11) and cytochalasin J (12) were isolated from the endophytic fungal strain Phomopsis sp. A123. This strain was isolated from leaves of the mangrove species, Kandelia candel, collected in the Fugong Mangrove Conservation Area, Fujian, China. The chemical structures were elucidated by spectroscopic analyses, including 1D- and 2D-NMR, and on the basis of HR‐Q‐TOF mass spectrometry. Antibacterial assays with the novel compounds 1–8 were carried out; they had no effect on the growth of the tested bacteria or yeasts.
Five novel nonenolides, phomolides D–H (1–5), and phomonol (6), phomotone (7) and phomopene (8), plus previously know compounds 4-formyl-7-methoxyl-6-methyl-phthalide (9), dihydrogladiolic acid methyl lactal (10), cytochalasin H (11) and cytochalasin J (12) were isolated from the endophytic fungal strain Phomopsis sp. A123. This strain was isolated from leaves of the mangrove species, Kandelia candel, collected in the Fugong Mangrove Conservation Area, Fujian, China. The chemical structures were elucidated by spectroscopic analyses, including 1D- and 2D-NMR, and on the basis of HR‐Q‐TOF mass spectrometry. Antibacterial assays with the novel compounds 1–8 were carried out; they had no effect on the growth of the tested bacteria or yeasts.
1. Introduction
Endophytes are microorganisms that live in the internal tissues of living plants (Hyde and Soytong 2008) and are a relatively unstudied potential source of novel and biologically active natural products (Brady et al. Citation2001; Tan and Zou Citation2001; Lu and Shen Citation2003; Strobel and Daisy Citation2003; Zhang et al. Citation2006; Huang et al. 2009). In investigating endophytic fungi of the mangrove species, Kandelia candel, collected from the Fugong Mangrove Conservation Area in Fujian, China, we isolated a fungal strain named A123, which, based on its complete ITS1–5.8S–ITS2 gene sequence, was identified as Phomopsis sp. Here, we report the isolation and structure elucidation of five novel nonenolides, i.e. phomolides D–H (1–5), phomonol (6), phomotone (7) and phomopene (8), and the previously known metabolites 4–formyl–7–methoxyl–6–methyl–phthalide (9) (Grove Citation1953), dihydrogladiolic acid methyl lactal (10) (Ichihara et al. Citation1985), cytochalasin H (11) (Beno et al. Citation1977) and cytochalasin J (12) (Cole et al. Citation1981), together with an antibacterial assay of the new compounds (1–8) ().
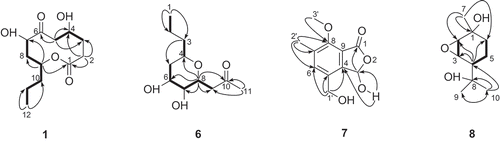
Figure 1. Structure of compounds 1 and 6–8, and the selected HMBC correlations (H→C) and 1H–1H-COSY (bold line).
1. Methods
2.1. General
Column chromatography (CC): silica gel (SiO2, 200–300 and 80–100 mesh; Qingdao Marine Chemical Factory, Qingdao, China), SiO2 GF254 (Merck), RP-18 (Merck), and Sephadex LH-20 (Amersham Biosciences) were used. TLC: precoated SiO2 GF254 plates (0.20–0.25 mm; Qingdao Marine Chemical Factory). Optical rotations: Perkin-Elmer 341 polarimeter with CHCl3 or MeOH as solvent. UV spectra: Amersham Biosciences Ultrospec 1100 pro spectrophotometer; λmax (log ε) in nm. IR spectra: Nicolet FT-IR 380 in KBr. HR-Q-TOF-MS: Bruker Daltonios BioTOF-Q mass spectrometers; in m/z (rel. %). 1H- and 13C-NMR spectra: Bruker DRX-600 spectrometer, at 600 (1H), and 150 (13C) MHz; in CDCl3 or (CD3)2CO; d in ppm relative to Me4Si, J in Hz.
2.2. Isolation and fermentation of the fungal strain
The endophytic fungus was isolated from the leaves of Kandelia candel, collected during December of 2003 in the Fugong Mangrove Conservation Area of Fujian, China. Both traditional morphological assessment and internal transcribed spaces (ITS) sequence analysis were performed to characterize it as Phomopsis sp. and named as A123. A nucleotide-to-nucleotide BLAST query of the NCBI database yielded Phomopsis liquidambari AY601919 as the closest match to the ITS rDNA of A123 (98%). The strain A123 was cultivated on potato-dextrose agar (PDA) plates for 15 days at 28°C in darkness. Every plate contained 20 ml PDA media and, in total, 10 l were cultivated under the above fermentation conditions.
2.3. Extraction and isolation
The cultured agar was chopped, diced and extracted with 10 l EtOAc/MeOH/AcOH (80:15:5, v/v) at room temperature overnight. The organic solution was collected through filtration and the remaining agar residue was extracted successively with the same solvent until the filtrate was colorless. The combined filtrate, upon evaporation, yielded a crude brown syrupy extract. This was partitioned between water and EtOAc (1:1, v/v) until the EtOAc layer was colorless. The combined organic layer was concentrated in vacuo to obtain EtOAc extract (10 g). The extract was subjected to MPLC (RP-18, 145 g; gradient aq. MeOH 0, 30, 50, 70 and 100% resp., 1 l each) to obtain five fractions, i.e. Fr.a–Fr.e.
Fr.b (2.1 g) was separated by CC (Sephadex LH-20; 140 g; MeOH) to obtain six fractions Fr.b.1–Fr.b.6. Fr.b.2 (230 mg) and was purified using MPLC (RP-18; 30 g; gradient aq. MeOH 25, 30 and 40% resp., 100 ml each) twice, and subjected to CC (SiO2; petroleum ether (PE)/ethyl acetate (EA) 3:1, 2:1, and 3:2, v/v) to obtain Fr.b.2.a and Fr.b.2.b. Fr.b.2.a (10 mg) was purified by CC (SiO2; PE/EA, 5:1, v/v), and purified on prepared TLC (EA/Me2CO, 30:1, v/v) twice to obtain 6 (4 mg). Fr.b.2.b (14 mg) was purified by CC (SiO2; PE/EA, 4:1, v/v), and purified on prepared TLC (EA/Me2CO, 30:1, v/v) twice to obtain 5 (2 mg). Fr.b.3 (140 mg) was purified on MPLC (RP-18, 30 g; gradient aq. MeOH 0, 30, 40 and 100% resp.) to obtain four fractions Fr.b.3.a–Fr.b.3.d. Fr.b.3.a (45 mg) was purified on MPLC (RP-18, 30 g; 30% MeOH), and then subjected to CC (SiO2; PE/Me2CO, 6:1 and 1:1, v/v) to obtain three fractions Fr.b.3.a.1–Fr.b.3.a.3. Fr.b.3.a.1 (8 mg) was purified on CC (SiO2; PE/Me2CO, 10:1, v/v) to obtain 8 (3 mg). Fr.b.3.a.2 (12 mg) was purified by CC (SiO2; PE/EA, 5:1 and 3:1, v/v, resp.) twice, and further purified on prepared TLC (EA/Me2CO, 20:1, v/v) twice to obtain 2 (2 mg). Fr.b.3.a.3 (4 mg) was purified by CC (SiO2; PE/EA 3:1 and 2:1, v/v, resp.) twice, and then purified on prepared TLC (EA/Me2CO, 30:1, v/v) twice to obtain 3 (3 mg). Fr.b.3.c (35 mg) was purified by CC (SiO2; PE/ Me2CO, 12:1, 10:1, 8:1 and 5:1, v/v) to obtain two fractions Fr.b.3.c.1 and Fr.b.3.c.2. Fr.b.3.c.1 (4 mg) was purified on prepared TLC (chloroform/MeOH, 15:1, v/v) twice to obtain 1 (3 mg). Fr.b.3.c.2 (25 mg) was subjected to MPLC (RP-18, 30 g; 35 and 40% MeOH), then purified on prepared TLC (PE/EA, 1:3, v/v) four times to obtain 4 (3 mg). Fr.b.4 precipitated a white powder in MeOH, which was washed with Me2CO several times to obtain 7 (6 mg). Fr.b.5 (120 mg) was purified on MPLC (RP-18, 30 g; 25 and 100% MeOH), and then the chloroform soluble fraction (21 mg) was further purified by CC (SiO2; PE/chloroform, 4:1 and 3:1, v/v) twice to obtain dihydrogladiolic acid methyl lactal (6 mg). Fr.b.6 (160 mg) was subjected to CC (SiO2; PE/EA, 10:1 and 3:1, v/v) to obtain 4-formyl-7-methoxyl-6-methyl-phthalide (6 mg).
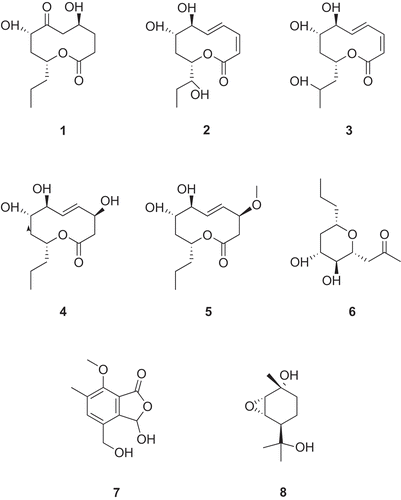
Scheme 1. Chemical structures of novel compounds from Phomopsis sp. A123.
Fr. c (1.3 g) were separated by CC (Sephadex LH-20, 140 g; MeOH) to afford four fractions Fr.c.1–Fr.c.4. Fr.c.1 (12 mg) was purification by CC (SiO2; PE/Me2CO, 12:1 and 10:1, v/v) to obtain cytochalasin H (3 mg). Fr.c.3 (170 mg) was purified by MPLC (RP-18, 30 g; 55 and 60% MeOH), and then subjected to CC (SiO2; PE/Me2CO, 8:1 and 6:1, v/v) to obtain cytochalasin J (3 mg).
3. Results and discussion
3.1. Structure elucidation
The morphological properties of isolate A123 were examined after incubation for 40 days at room temperature in PDA medium. This organism was identified to be Phomopsis sp. according to the ITS rDNA sequence (ITS1–5.8S–ITS2). The fermentation culture was extracted successively with EtOAc/MeOH/AcOH (80:15:5, v/v), and partitioned between water and EtOAc (1:1, v/v). The EtOAc extract was purified by repeated column chromatography (RP-18, Sephadex LH-20 and SiO2) to afford compounds 1–8.
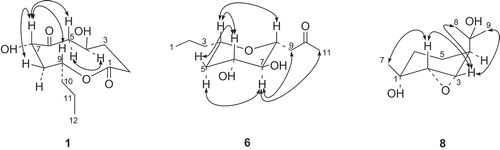
Compound 1 was obtained as a colorless oil. The molecular formula of 1 was determined to be C12H20O5 according to the HR-Q-TOF-MS and NMR data. The IR absorptions at 3444 and 1738 cm−1 indicated the presence of OH and C=O groups, respectively. The 13C-NMR and DEPT spectra of 1 () exhibited 12 signals corresponding to one Me, six CH2, three oxygenated CH groups, as well as two C=O groups. Inspection of the 1H–1H COSY, HMQC and HMBC revealed a nonenolide core for 1 (Rivero-Cruz et al. Citation2003). The HMBC correlations from H2–C(2) to C(1), C(3) and C(4), from H2–C(5) to C(3), C(4) and C(6), from H–C(9) to C(1), C(7) and C(8), from Me(12) to C(10) and C(11), along with 1H–1H COSY correlations H2–(3)↔H-C(4)↔H2-C(5), and H–C(7)↔H2–C(8)↔H–C(9)↔H2–C(10)↔H2–C(11)↔Me(12) established the structure of 1 (). The relative configurations of 1 were determined by analysis of the ROESY spectrum. The presence of NOE correlations H–C(7)↔H–C(9) indicated that H–C(7) and H–C(9) were β-oriented, while H–C(4) was in α-orientation (). Thus, the structure of 1 was established to be 5,8-dihydroxy-10-propyloxecane-2,7-dione, named as phomolide D.
Figure 2. Selected NOE correlations for compounds 1, 6 and 8 (H↔H).
Compound 2 was obtained as a colorless oil. The molecular formula of 2 was deduced to be C12H18O5 from the HR-Q-TOF-MS and NMR data. The structure of 2 was established by comparison of its NMR data with those of 1. The spectroscopic data of both compounds were similar ( and ), except the presence of C(2)=C(3) and C(4)=C(5) due to the dehydrogenation and dehydration between each of them, respectively, and the addition of a OH group at C(6) along with the absence of a C=O group, and a OH substitution at C(10). The relative configurations of 2 were determined based on the same ROSEY correlations as in 1. The configurations of C(2)=C(3) and C(4)=C(5) were identified to be cis and trans, respectively, according to the coupling constants between them. The absence of NOE correlations H–C(6)↔H–(7) or H–C(6)↔H–(9) indicated that H–C(6) was in α–orientation. Thus, the structure of 2 was established to be (3Z*, 5E*,7S*,8S*,10S*)-7,8-dihydroxy-10-((S*)-1-hydroxypropyl)-7,8,9,10-tetrahydro-2H-oxecin-2-one, named as phomolide E.
Table 1. 1H-NMR data of 1–3 (CDCl3) and 4, 5 ((CD3)2CO) (600 MHz, δ in ppm, J in Hz)
Table 2. 13C-NMR data of 1–3 (CDCl3) and 4, 5 ((CD3)2CO) (150 MHz, δ in ppm, J in Hz)
Compound 3 was obtained as a colorless oil. The molecular formula of 3 was established to be C12H18O5 by its HR-Q-TOF-MS and NMR data. The 1H- and 13C-NMR spectral data of 3 ( and ) were similar to those of 2, except δ 45.7 (C(10)) and d 65.4 (C(11)) due to a OH substitution at C(11) along with the absence of a OH group at C(10). The relative configurations of 3 were determined based on the same ROSEY correlations as in 2. Thus, the structure of 3 was established to be (3Z*,5E*,7S*,8S*,10R*)-7,8-dihydroxy-10-(2-hydroxy propyl)-7,8,9,10-tetrahydro-2H-oxecin-2-one, named as phomolide F.
Compound 4 was obtained as white powder. The molecular formula of 4 was deduced to be C12H20O5 based on the HR-Q-TOF-MS and NMR data. The 1H- and 13C-NMR spectra ( and ) showed that 4 had a similar structure as 2, except δ 45.2 (C(2)) and d 71.6 (C(3)) due to a OH substitution at C(3) along with the absence of C(2)=C(3), and d 39.0(C(10)) due to the absence of a OH group at C(10). The relative configurations of 4 were determined based on the same ROSEY correlations as in 2. The configuration of H–C(3) was determined to be α–oriented according to the NOE correlations H–C(3)↔H–(4)↔H–C(6). Thus, the structure of 4 was established to be (4S*,7S*,8S*,10R*,E*)-4,7,8-trihydroxy-10-propyl-3,4,7,8,9,10-hexahydro-2H-oxecin-2-one, named as phomolide G.
Compound 5 was obtained as white powder. The molecular formula of 5 was established to be C13H22O5 from the HR-Q-TOF-MS and NMR data. The structure of 5 was established by comparison of its NMR data with those of 4. The spectroscopic data of both compounds were similar ( and ), except the addition of a MeO group at C(3). The relative configurations of 5 were determined based on the same ROSEY correlations as in 4. Thus, the structure of 5 was established to be (7S*,8S*,10R*,E*)-4,7-dihydroxy-8-methoxy-10-propyl-3,4,7,8,9,10-hexahydro-2H-oxecin-2-one, named as phomolide H.
Compound 6 was isolated as colorless oil. The molecular formula of 6 was determined to be C11H16O3 according to the HR-Q-TOF-MS and NMR data. The IR absorptions at 3417 and 1743 cm−1 indicated the presence of OH and C=O groups, respectively. The 13C-NMR and DEPT spectra () of 6 exhibited 11 signals corresponding to two Me, four CH2, four oxygenated CH groups, and one C=O group. The HMBC correlations from Me(1) to C(2) and C(3), from H2–C(3) to C(4) and C(5), from H–C(8) to C(4), C(6), C(7), C(9) and C(10), from Me(11) to C(9) and C(10), along with the 1H–1H COSY correlations Me(1)↔H2–C(2), H2–C(3)↔H–C(4), and H2–C(5)↔H–C(6)↔H–C(7)↔H–C(8)↔H2–C(9), led to the establishment of the structure of 6 (). The relative configurations of 6 were established from the NOE spectra. The presence of NOE correlations H–C(6)↔H–C(4)↔H–C(8) indicated that H–C(4), H–C(6), and H–C(8) were in β-orientation, while Hα–C(5)↔H–C(7)↔H2–C(9), and H–C(7)↔H2–C(11) indicated that H–C(7) was α-oriented (). Thus, the structure of 6 was elucidated as 1-(3,4-dihydroxy -6-propyltetrahydro-2H-pyran-2-yl)propan-2-one, named as phomonol.
Table 3. 1H- and 13C-NMR data of 6 (600 and 150 MHz, resp. in CDCl3; δ in ppm, J in Hz)
Compound 7 was obtained as white powder. The molecular formula of 7 was determined to be C11H12O5 according to the HR-Q-TOF-MS and NMR data. The IR spectrum indicated the presence of HO groups (3417 cm−1) and C=O groups (1743 cm−1). The 13C-NMR spectra of 7 () displayed two Me (one being oxygenated), one oxygenated CH2 group, one CH and seven quaternary C‐atoms (six olefinic and one carboxyl). Inspection of the HMQC and HMBC revealed an isobenzofuranone-type structure for 7 (Grove Citation1953; Ichihara and Sawamura Citation1985). The HMBC correlations from H2–C(1') to C(4), C(5) and C(6), from Me(2') to C(6), C(7) and C(8), from Me(3') to C(8), from HO–C(3) to C(3) and C(4), from H–C(3) to C(1), led to the establishment of the structure of 7 (). Thus, the structure of 7 was elucidated as 3‐ hydroxy-4-(hydroxymethyl)-7-methoxy-6-methylisobenzo furan-1(3H)-one, named as phomotone.
Table 4. 1H- and 13C-NMR data of 7 (600 and 150 MHz, resp. in (CD3)2CO; δ in ppm, J in Hz)
Compound 8 was obtained as white powder. The molecular formula of 8 was determined to be C10H18O3 according to the HR-Q-TOF-MS and NMR data. The IR absorption at 3441 cm−1 indicated the presence of OH groups. The 13C-NMR and DEPT spectra () of 8 exhibited 10 signals corresponding to three Me, two CH2, three CH (two being oxygenated) and two oxygenated quaternary C-atoms. The HMBC correlations from H–C(4) to C(2), C(3) and C(6), from Me(7) to C(1), C(2) and C(6), from Me(10) to C(4), C(8) and C(9), along with the 1H–1H COSY correlations H–C(2)↔H–C(3), and H–C(4) ↔H2–C(5)↔H2–C(6), led to the establishment of the core structure of 8 () (Yoshikawa et al., Citation2009). Additionally, the chemical shifts of C(2) (δ(C) 60.1) and C(3) (δ(C) 57.7) were relatively upfield compared to regular oxygenated CH groups, indicating the presence of an epoxy group between C(2) and C(3). This was further supported by HR-Q-TOF-MS data, which indicated the presence of three rather than four O-atoms in this molecule. The relative stereochemistry of 8 was determined by the analysis of ROESY spectrum. The presence of NOE correlations Me(7)↔H-C(2)↔H-C(3), and Me(8)↔H-C(3)↔Me(9) indicated that H–C(2), H–C(3) and Me(7) were in β-orientation, while H–C(6) was α-oriented (). Thus, the structure of compound 8 was established as 5-(2-hydroxypropan-2-yl)-2-methyl-7-oxabicyclo [4.1.0]heptan-2-ol, named as phomopene.
Table 5. 1H- and 13C-NMR data of 8 (600 and 150 MHz, resp. in (CD3)2CO; δ in ppm, J in Hz)
3.2. Biological study
The antibacterial activities of compounds 1–8 were tested against bacteria (Escherichia coli (CMCC (B) 44103), Bacillus subtilis (CMCC (B) 63501), Bacillus pumilus (CMCC (B) 63202), and Staphylococcus aureus (CMCC (B) 26003)) and yeast (Candida albicaus (AS 2.538)) using the Oxford plate assay system. Two replicates were performed for each compound at a concentration of 1 mg/ml with a loading volume of 30 μl. Compounds 1–8 had no effects on the growth of tested bacteria or yeast at 30 μg/plate.
Phomolide D (=5,8-dihydroxy-10-propyloxecane-2,7-dione; 1). Colorless oil. = +1.7 (c = 0.3, CHCl3); UV (CHCl3) λ max (log ε): 205 (2.27), 209 (2.23), 216 (2.29), 233 (2.31), 244 (2.29); IR (KBr)max: 3444, 2924, 2853, 1738, 1172 cm−1. 1H- and 13C-NMR: see and ; HR-Q-TOF-MS: 267.1359 ([M+Na]+, C12H20NaO5
+; calc. 267.1208).
Phomolide E (=(3Z*,5E*,7S*,8S*,10S*)-7,8-dihydroxy-10-((S*)-1-hydroxypropyl)-7,8,9,10-tetrahydro-2H-oxecin- 2-one; 2). Colorless oil. = −60.3 (c = 0.37, CHCl3); UV (CHCl3) λ max (log ε): 206 (2.37), 219 (2.40), 236 (2.57), 247 (2.67); IR (KBr)max: 3417, 2924, 2854, 1712, 1250, 1161, 1057 cm−1. 1H- and 13C-NMR: see and ; HR-Q-TOF-MS: 265.1272 ([M+Na]+, C12H18 NaO5
+; calc. 265.1052).
Phomolide F (=(3Z*,5E*,7S*,8S*,10R*)-7,8-dihydroxy- 10-(2-hydroxypropyl)-7,8,9,10-tetrahydro-2H-oxecin-2-one; 3). Colorless oil = −48.7 (c = 0.23, CHCl3); UV (CHCl3) λ max (log ε): 208 (2.54), 214 (2.72), 239 (2.86), 248 (2.88), 251 (2.88); IR (KBr)max: 3437, 2924, 2854, 1745, 1712, 1259, 1161, 1081 cm−1. 1H- and 13C-NMR: see and ; HR-Q-TOF-MS: 265.1378 ([M+Na]+, C12H18NaO5
+; calc. 265.1052).
Phomolide G (=(4S*,7S*,8S*,10R*,E*)-4,7,8-trihydroxy-10-propyl-3,4,7,8,9,10-hexahydro-2H-oxecin-2-one; 4). White powder. = −10.4 (c = 0.24, MeOH); UV (MeOH) λ max (log ε): 207 (3.13), 217 (3.38), 222 (3.23), 228 (3.41); IR (KBr)max: 3353, 2924, 2854, 1743, 1712 cm−1. 1H- and 13C-NMR: see and ; HR-Q-TOF-MS: 267.1253 ([M+Na]+, C12H20NaO5
+; calc. 267.1208).
Phomolide H (=(7S*,8S*,10R*,E*)-4,7-dihydroxy-8-methoxy-10-propyl-3,4,7,8, 9,10-hexahydro-2H-oxecin-2-one; 5). White powder. = −17.5 (c = 0.76, MeOH); UV (MeOH) λ max (log ε): 210 (3.06), 218 (3.19), 227 (3.23); IR (KBr)max: 3354, 2924, 2853, 1738, 1713, 1384, 1260, 1164, 1093, 1033 cm−1. 1H and 13C NMR: see and ; HR-Q-TOF-MS: 281.1532 ([M+Na]+, C13H22NaO5
+; calc. 281.1365).
Phomonol (=1-(3,4-dihydroxy-6-propyltetrahydro-2H-pyran-2-yl)propan-2-one; 6). Colorless oil. = +6.0 (c = 0.3, CHCl3); UV (CHCl3) λ max (log ε): 215 (2.23), 220 (2.04), 229 (1.93), 234 (1.91), 248 (1.88); IR (KBr)max: 3417, 2956, 2924, 2854, 1743, 1712, 1359, 1085 cm−1. 1H- and 13C-NMR: see ; HR-Q-TOF-MS: 239.1503 ([M+Na]+, C11H20NaO4
+; calc. 239.1259).
Phomotone (=3-hydroxy-4-(hydroxymethyl)-7-methoxy-6-methylisobenzofuran-1(3H)-one; 7). White powder. = +1.7 (c = 0.53, MeOH); UV (MeOH) λmax (log ε): 212 (3.69), 223 (4.00), 290 (2.91); IR (KBr) max: 3286, 2924, 2854, 1744, 1074 cm−1. 1H- and 13C-NMR: see ; HR-Q-TOF-MS: 247.0881 ([M+Na]+, C11H12NaO5
+; calc. 247.0582).
Phomopene (=5-(2-hydroxypropan-2-yl)-2-methyl-7-oxabicyclo[4.1.0]heptan-2-ol; 8). Colorless oil. = −1.3 (c = 0.46, CHCl3); UV (CHCl3) λmax (log ε): 205 (1.78), 229 (1.73), 241 (2.06), 243 (2.07); IR (KBr) max: 3441, 2925, 2854, 1382, 1158, 1123 cm−1. 1H- and 13C-NMR: see ; HR-Q-TOF-MS: 209.1313 ([M+Na]+, C10H18NaO3
+; calc. 209.1313).
Acknowledgements
This work was financially supported by the Key Grant of Chinese Ministry of Education (No. 306010), the National Science Fund (No. 31070053), the National 863 Program (No. 2007AA091503).
Related Research Data
References
- Beno , MA , Cox , RH , Wells , JM , Cole , RJ , Kirksey , JW and Christoph , GG . 1977 . Structure of a new [11] cytochalasin, cytochalasin H or kodo-cytochalasin-1 . J Am Chem Soc. , 99 : 4123 – 4130 .
- Cole , RJ , Wells , JM , Cox , RH and Cutler , HG . 1981 . Isolation and biological properties of deacetylcytochalasin H from Phomopsis sp . J Agric Food Chem. , 29 : 205 – 206 .
- Brady , SF , Bondi , SM and Clardy , J . 2001 . The guanacastepenes: a highly diverse family of secondary metabolites produced by an endophytic fungus . J Am Chem Soc. , 123 : 9900 – 9901 .
- Grove , JF . 1953 . Gladiolic acid, a metabolic product of Penicillium gladioli. II. Structure and fungistatic activity . Biochem J. , 54 ( 4 ) : 664 – 673 .
- Ichihara , A , Sawamura , S , Kawakami , Y and Sakamura , S . 1985 . Dihydrogladiolic acid, another phytotoxin from Phoma asparagi Sacc . Agric Biol Chem. , 49 ( 6 ) : 1891 – 1892 .
- Lu , C and Shen , Y . 2003 . A new macrolide antibiotic with antitumor activity produced by Streptomyces sp. CS, a commensal microbe of Maytenus hookeri . J Antibiot (Tokyo) , 56 ( 4 ) : 415 – 418 .
- Rivero-Cruz , JF , Macias , M , Cerda-Garcia-Rojas , CM and Mata , R . 2003 . A new phytotoxic nonenolide from Phoma herbarum . J Nat Prod. , 66 ( 4 ) : 511 – 514 .
- Strobel , G and Daisy , B . 2003 . Bioprospecting for microbial endophytes and their natural products . Microbiol Mol Biol Rev. , 67 ( 4 ) : 491 – 502 .
- Tan , RX and Zou , WX . 2001 . Endophytes: a rich source of functional metabolites. Nat Prod Rep . 18 : 448 – 459 .
- Yoshikawa , M , Morikawa , T , Oominami , H and Matsuda , H . 2009 . Absolute stereostructures of olibanumols A, B, C, H, I, and J from olibanum, gum-resin of Boswellia carterii, and inhibitors of nitric oxide production in lipopolysaccharide-activated mouse peritoneal macrophages . Chem Pharm Bull (Tokyo) , 57 : 957 – 964 .
- Zhang , HW , Song , YC and Tan , RX . 2006 . Biology and chemistry of endophytes . Nat Prod Rep. , 23 : 753 – 771 .