Abstract
A luminous mushroom from Vietnam, identified earlier as Omphalotus aff. illudens (Van, Citation2009. J Siber Fed Univ. 2:157–171), was studied. Morphological observations of collected fruiting bodies were made in the field. A culture isolated from tissues of one basidiome was studied and the internal transcribed spacer regions (ITS1-2) of the ribosomal RNA repeat were sequenced to confirm identifications. From these observations, the luminous mushrooms collected in Vietnam were identified as Neonothopanus nambi (Speg.) R.H. Petersen & Krisai. To resolve initial sequence ambiguities amplified DNA fragments were cloned and 38 different ITS sequences were obtained from a total of 98 clones, with 14 nt step differences (2% variability) across the entire spectrum of clone sequences. Commonalities in the sequences indicated that the cloned sequences represented two monokaryon mating strains from the basidiome and one possible recombinant. Cloning errors were accounted for by deletion of single occurrence nucleotide variants. Six insertions/deletions and four single nucleotide polymorphisms distinguished the two monokaryon sequences (98% similarity), which differed by 4 bp in length. As many as six multiple occurrence intragenomic polymorphisms in either monokaryon were found to be statistically significant. Possible evolutionary mechanisms for the persistence of intergenomic and intragenomic ITS polymorphisms are discussed.
Introduction
A large variety of living organisms are able to emit light, and fungi are notable among them. Approximately 80 species of luminous fungi are currently recognised (Desjardin et al. Citation2008; Vydryakova et al. Citation2009; Bondar et al. Citation2011). However, the biochemical mechanism of luminescence in fungi and its ecological function are still largely unresolved (Desjardins et al. Citation2008). In south-east Asia, luminous mushrooms, such as Omphalotus nidiformis (Berk) O.K. Mill. “Pleurotus lunaillustria” [nom. invalid.], Mycena citricolor (Berk. & M.A. Curtis) Sacc., M. manipularis (Berk.) Sacc., M. pruinosoviscida Corner, M. chlorophos (Berk. & M.A. Curtis) Sacc. and M. noctilucens Corner, are widespread in Thailand, Malaysia and Singapore (Desjardin et al. Citation2008; Isobe et al. Citation1994). Luminous Neonothopanus nambi has been identified from Thailand (Buaart et. al. Citation2008) and N. nambi is also known from South America, the Caribbean region and Australasia, where it is widespread in areas of relatively high rainfall (Corner Citation1981, Desjardin et al. Citation2008).
Tropical luminous mushrooms are widely distributed in Vietnam, especially in the southeast. Their fruiting bodies appear in the rainy season, although it is possible to find them also in the dry season in forests, often associated with rotting wood. Van (Citation2006) collected a luminous mushroom provisionally identified as Omphalotus aff. illudens in Vietnam and isolated it into pure culture, developing a method for growing fruiting bodies of the mushroom in a commercial setting. The original goal of the current study was to characterise and provide an authoritative identification of this luminous mushroom from Vietnam.
Methods
Collections
Fruiting bodies (basidiomes) were collected in Binh Phuoc and Binh Duong provinces of Vietnam. Descriptions are based on basidiomes collected on a rotten log of a rubber tree (Hevea brasiliensis) in Binh Phuoc province. A dikaryon culture with clamped mycelium (strain BIN 2379) was studied, obtained from tissue dissected from a single basidiome above. The culture is maintained at the Komarov Botanical Institute Basidiomycetes Culture Collection (BIN, St. Petersburg, Russia) on 2% beer-ale (4° balling) agar slants at 4–6°C, and in cryovials under distilled water at room temperature.
Morphological observations
Mycelium was cultivated in low pH Fungal Broth (FB) (Himedia, India), and on low pH Fungal Agar (FA) (Himedia, India), Malt Extract Agar (MEA) and Potato Dextrose Agar (PDA) (Biokar Diagnostics, France). Morphological observations in culture were made from three replicates on MEA and PDA incubated at 25°C in darkness for 8 weeks. Macromorphology was studied at 2, 4, 6 and 8 weeks, and micromorphology examined after 2 and 4 weeks using differential interface contrast (DIC) and a JSM-6390 LA scanning electron microscope (SEM) at 14 kV. Growth rates were recorded every 2 days for 3 weeks. Phenoloxidase production (laccase activity) was evaluated by spot tests at 2 and 3 weeks using syringaldazine and guaiacol as substrate-specific reagents (Marr, Citation1979) and activity recorded at 5, 15, 30 min and 1, 3, and 24 h. Reactions were evaluated as negative (− or ±), positive (+ or ++) or strong (+++).
DNA isolation and sequencing
Mycelium was harvested after 7 days at 28°C, and genomic DNA purified using the Genomic DNA Purification Kit (Fermentas, EU) according to the manufacturer's protocol. A region of nuclear rDNA, containing the internal transcribed spacer regions 1 and 2 and the 5.8S rRNA gene, was amplified by polymerase chain reaction (PCR) using the primer combinations ITS5-fw (5′-GGAAGTAAAAGTCGTAACAAGG-3′) and ITS4-rev (5′-TCCTCCGCTTATTGATATGC-3′) (White et al. Citation1990), in an automated temperature-cycling device (Biometra TProfessional, Biometra GmbH, Goettingen, Germany), using the following parameters: 3 min initial denaturation at 94°C, followed by 30 cycles of 1 min denaturation at 94°C, 75 s primer annealing at 58°C, 105 s extension at 72°C, and a final extension period of 10 min at 72°C. PCR reactions were prepared in 10 μl volume containing the following mix: 1 μl 10× Titanium Taq Buffer (Clontech, Mountain View, CA, USA), 0.5 μl 2 mM dNTP, 0.32 μl each of 5 μM upper and lower primers, 7.76 μl sterile distilled water, 1 μl template (10–20 ng), and 0.1 μl 50× Titanium Taq Polymerase (Clontech). Sequencing reactions were prepared using the ABI Prism® BigDye™ Terminator reaction kit v3.1 (Applied Biosystems Inc., Foster City, CA, USA) in 10 μl volume and 1/8 dilution using 5× sequencing buffer. The sequencing reaction contained the following mix: 1.75 μl 5× Sequencing Buffer, 0.5 μl BigDye V3.1 mix, 0.5 μl of 3.2 μM primer, 6.25 μl sterile distilled water, 1.0 μl (10–40 ng) PCR template, and employing the following amplification protocol: 25 cycles each of 30 s denaturation at 96°C, 15 s annealing at 50°C, and 4 min extension at 60°C. The ITS primers used for PCR were also used in sequencing. Sequences were obtained using an ABI Prism 3100 Genetic Analyzer (Applied Biosystems).
Analysis of clone sequences
The original sequences from PCR had ambiguities appearing as multiple peaks arising in one or more long stretches, apparently caused by the amplification of multiple copies of the ITS regions containing indels. Therefore, cloning was performed to obtain unambiguous reads of the ITS variants, using the TOPO TA Cloning® Kit (Invitrogen, Carlsbad, CA, USA) and following the manufacturer's protocol. Titanium Taq, a “high sensitivity” enzyme lacking “proofreading” capability (3′–5′ exonuclease activity) was used for PCR and cloning. Tandem nucleotide repeats in the predominant clone sequence were investigated using the program “Tandem Repeats Finder” (http://tandem.bu.edu/trf/trf.html; Benson Citation1999). Single occurrence sequence variants were attributed to cloning error and corrected. The statistical significance of multiple occurrence polymorphisms was investigated using an approximation of the Taylor series representation of the exponential function [p(n)≈1−e−n(n−1)/(2xsl)], where n = total number of polymorphisms and sl = sequence length. This is basically a version of the “Birthday Paradox”, which tells us the probability of two random events from a population (n) of random events co-occurring at the same location in a defined space (sl). Since we can infer that the ‘corrected’ sequences (accounting for cloning errors), which include the predominant clone sequences, are the original sequences from which sequence polymorphisms originated, we can determine the nature of the nucleotide substitutions and separately calculate the statistical significance of the two classes of substitutions: (1) transitions (purine↔purine and pyrimidine↔pyrimidine) and (2) transversions (purine↔pyrimidine). For transitions there is only one possible nt variant from the consensus sequence, so their probability of co-occurrence is p(n) where n is the total number of transitions. However, for transversions there are two possible nt variants and the actual probability for the co-occurrence of transversions would be p(n)/2, where n is the number of observed transversions.
Phylogenetic analysis
We performed phylogenetic analyses to resolve the multiplicity of different sequences obtained by cloning and confirm the identification of the Vietnam isolate by determining its phylogenetic position relative to available sequences for Omphalotus and related Omphalotaceae and Marasmiaceae selected from GenBank BLAST comparisons with the predominant clone sequence. DNA sequences were aligned using the multiple sequence alignment program MAFFT ver. 6 using the FFT-NS-i strategy (Katoh et al. Citation2002; http://align.bmr.kyushu-u.ac.jp/mafft/software/). Alignments were trimmed to approximately 10 bp immediately preceding the ITS1 and following the ITS2. Gaps were treated as missing data. Phylogenetic analysis was performed in PAUP 3.1.1 (Swofford Citation1991). A parsimony analysis was performed using a heuristic search, with a starting tree obtained via step-wise addition, with random addition of sequences with 200 replicates. Stability of clades was assessed with 1000 bootstrap replications. The final phylogenetic tree was rooted using selected taxa from the Marasmiaceae, the sister clade to the Omphalotaceae (Matheny et al., Citation2006). The alignment and resulting phylogenetic tree are deposited in TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S11779).
Observations
Basidiomes
Basidiomes () 2–5cm wide, white, concolorous, or pileus with a pale yellowish centre and reddish brown injured areas (spots) on older samples; pileus more or less circular, petaloid or shallowly infundibuliform; lamellae decurrent, rather shallow, in 2–3 ranks or branching dichotomously; texture soft, rubbery; stipe distinct, laterally attached, white, up to 6 × 4 mm.
Figures 1–6. Neonothopanus nambi (BIN 2379). Figure 1. Basidiome in natural light and in darkness (copyright Dao Thi Van). Figure 2. Colonies on MEA and PDA media after 3 weeks growth. Figure 3. Colonies on MEA after 8 weeks growth. Figure 4. Broad hyphae and multiple clamps (DIC). Figure 5. Clamp connections and ring structure in narrow hyphae (DIC). Figure 6. Clamp connection (SEM).
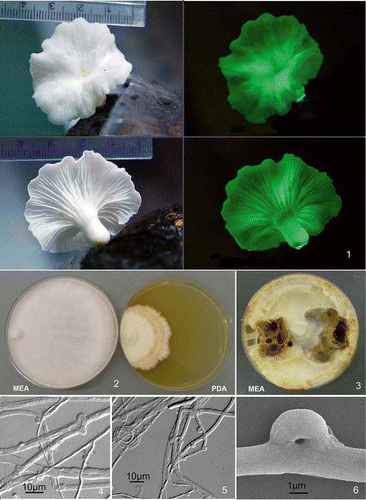
Culture
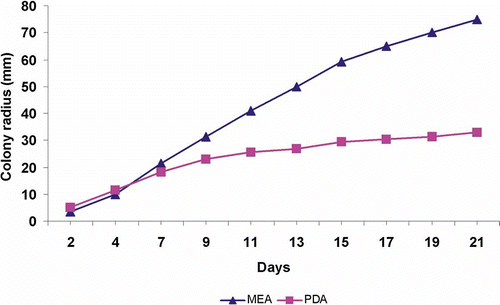
Cultures on MEA growing relatively rapidly, colonies covering 90-mm plates in 3 weeks, predominantly white, the advancing zone raised with margin even; aerial mycelium cottony with long radiating hyphae, slightly zonate (); zones of yellowish-olive-brownish to dark brown colour appearing after 4 weeks (), brown exudate becoming abundant on mycelium. Cultures on PDA growing much slower, about 33 mm in diameter in 3 weeks with plates not covered after 8 weeks (), colonies off-white to cream, forming brownish zones; the advancing zone is white and appressed, margin slightly scalloped; aerial mycelium felty with hyphae cottony or matted (). Odor lacking. Reverse unchanged on both media. Positive laccase activity within 5 min on both MEA and PDA (syringaldazine ++, guaiacol +); strong reaction (+++) with syringaldazine after 15 min, and for guaiacol +++ after 3 h. Hyphae 3–5 μm in diameter in the advancing zone, rarely branched and sparingly septate with few clamps. Aerial mycelium thin-walled, 4–6.5 μm in diameter, with septate branches narrower (1–3.5 μm in diameter); hyphal rings present; clamp connections mostly coin-shaped, occasionally multiple or sprouting, up to 4.8 × 3.0 μm ().
Figure 7. Growth rates of Neonothopanus nambi (BIN 2379) on MEA and PDA.
Luminescence
Mycelium does not produce visible luminescence in the beginning of growth and requires some nutritional limitation (Van Citation2009). Visible luminescence may be observed after 3–4 days in cultivation, and mycelium growing on different kinds of sawdust, grains, and straw is luminous for several weeks. Luminescence may be increased on agar media by cutting the mycelium.
Morphological observations of Vietnamese strain BIN 2379 from the field and in culture agreed with descriptions given by Petersen and Krisai-Greilhuber (Citation1999) for N. nambi from Malaysia and Puerto Rico, although our strain did not produce arthroconidia in cultures as described by these authors.
Molecular analysis
Our initial attempts to confirm the identification of the Vietnamese isolate by sequencing the ITS regions gave ambiguous results appearing as long stretches of multiple peaks apparently due to indels in different copies of the ITS in the rDNA repeat region. To resolve ambiguities, PCR amplicons from the Neonothopanus culture were cloned and unambiguous sequences were obtained from 98 clones, representing 38 different ITS1-2 (including 5.8S) sequence types (the unintended large number of clones resulted from the inadvertent inclusion of the Neonothopanus sample in the work stream of unrelated environmental samples). In total, there were 14 nt step differences across the entire spectrum of clone sequences. The sequences are about 750 nt long, so this represents a total sequence variation of about 2%.
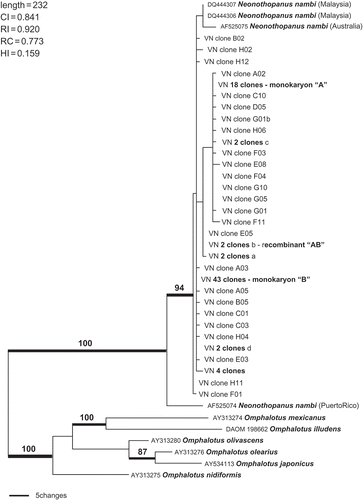
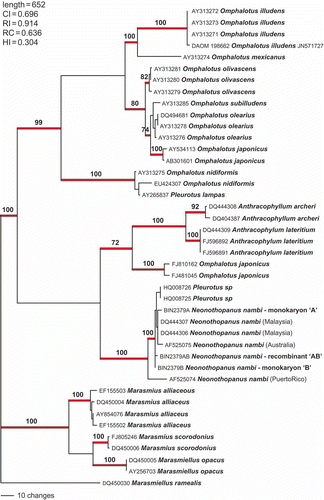
In a parsimony analysis (), the clones from the Vietnamese culture were bracketed by sequences deposited in GenBank by Kirchmair et al. (Citation2004) and Mata et al. (Citation2007) for N. nambi from Malaysia, Australia and Puerto Rico (). The genetic distance of the Puerto Rico strain from the Asian and Australian strains is evident, in that this strain had 11 novel deletions and two insertions not seen in the other strains. This could indicate that the strains from SE Asia are a different species from the type, which was described originally from Argentina (basionym: Agaricus nambi Speg., 1883). Petersen and Krisai-Greilhuber (Citation1999) cited Singer (Beih. Sydowia 7, 1973) as noting minor spore size differences among strains in this species. However, the former authors reported successful pairings of monokaryon isolates from North America and Asia. Therefore, we conclude that the investigated mushroom is N. nambi as interpreted by Petersen and Krisai-Greilhuber (Citation1999), and that it is premature to conclude that the Asian strains represent a different species.
Figure 8. Parsimony analysis of Neonothopanus and clone sequences. Outgroup comprised of six Omphalotus taxa. Numbers above branches are bootstrap values >70%. The alignment comprised 42 taxa, length = 761 bp. A total of 169 characters were included in the analysis, including 110 parsimony informative.
Discussion
Initially, we found it remarkable to find so many ITS sequence variants in a single basidiome of the Vietnamese mushroom. An examination of the alignment of the 98 clone sequences provides an explanation for a portion of the variance (). We discovered 10 “hallmarks” (SNP's or 1–2 base indels) that were present in one of three patterns in all of the clones. A total of 33 clones (A) had the hallmark pattern AAAAAA-AAAA (), 60 clones (B) had the hallmark pattern BBBBBB-BBBB, and we suspect these represent the two monokaryon mating strains. Five clones (AB) had the hallmark pattern BBBBBB-AAAA, possibly representing one recombinant from populations A and B, assuming that this state would be present in basidiospores in the original basidiome. This hallmark pattern would be inherited from the ITS1 from monokaryon B, and from ITS2 from monokaryon A, with the crossover event (chiasma) from homologous recombination occurring in the 5.8S region. In this scenario the “allele” having the AB hallmark pattern would have to be fixed in the rRNA array by intrachromasomal homogenization mechanisms and the recombinant favoured by biased selection pressures to germinate and grow in the environmental conditions of culture. The possible recombinant having the hallmark pattern AAAAAA-BBBB was not seen, although this is within the range of statistical probability (p = 0.125) even in the absence of biased selection pressures in culture. There remain 11 ITS sequence variants for monokaryon A with an additional SNP observed in the 18S region and two in the 5.8S, 12 ITS variants for monokaryon B with an additional SNP found in the 28S region and two in the 5.8S, and three ITS sequence variants for “recombinant AB”. Cloning and sequencing errors could explain some of this diversity, even though the electrophoretic patterns of all clone sequences were unambiguous bidirectional reads.
Figure 9. Alignment of Neonothopanus nambi clones. “Hallmarks” are nucleotide indels or single nucleotide polymorphisms inherited in the apparent recombinant, and also occurring in isolates from other geographic regions in varying recombinations. The numerical positions of the “hallmarks” in the alignment are indicted relative to the start of the ITS1. The location of multiple copy intragenomic nt variants are indicated by arrows with the copy numbers. “Singleton” clones are presumed cloning errors.
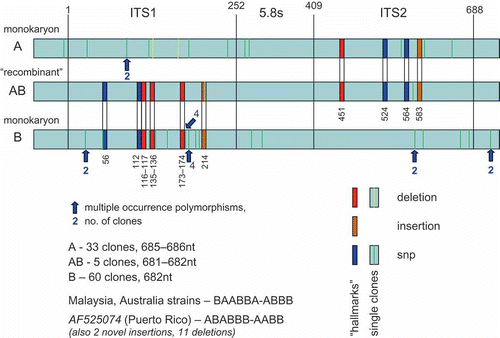
The same 10 “hallmarks” in the ITS regions observed in the Neonothopanus clones were observed in the four other sequences for Neonothopanus deposited in GenBank, although in different combinations. The strains from Malaysia and Australia had the pattern BAABBA-ABBB, and the single strain from Puerto Rico ABABBB-AABB. It is surprising that these “hallmarks” are persistent, and that they are not inherited together considering their close proximity on the ITS1 and ITS2, respectively. The redistribution of these hallmark patterns could be attributed to unequal crossing-over events in the absence of strong intergenomic gene conversion during the long evolutionary history of these geographically distant strains.
A mystery solved?
Initially, we discounted cloning errors as accounting for a significant portion of the ITS polymorphisms observed in this study. This led us to question the amount of variation in ribosomal DNA that is possible among mating populations of Neonothopanus and whether this variation may be attributed entirely to the existence of numerous “allelic” variants in the rDNA regions, or by a multinucleate condition in this species, or even by the occurrence of multiple compatible mating strains (Vydryakova et al. Citation2010). Subsequently, we came to realise that TAQ sequencing errors could substantially impact our observations, with experimentally determined nucleotide misincorporation rates ranging from 1.85 × 10−5 to 2.1 × 10−4/base/cycle (e.g. Keohavong and Thilly Citation1989; Acinas et al., Citation2005; Cummings et al., Citation2010). Previous studies have demonstrated that pyrosequencing and cloning of environmental samples can create false taxa by the propagation of nucleotide variants resulting from transcription errors during PCR (e.g. Dickie Citation2010; Tendersoo et al. Citation2010). Applying Taq error rates estimated by Kobayashi et al. (Citation1999) of 7.3 × 10−5/base/cycle) we could expect approximately 40 sequencing artefacts in the current investigation. Therefore, all 24 “singleton” (one occurrence) nt variants could be attributable to PCR errors propagated by cloning. However, the probability of random, multiple occurrences of identical sequence variants at any one site are more remote. Applying a Taylor series approximation to determine the probability of any two randomly generated polymorphisms occurring at the same location over a 684 nt (averaged) sequence, we determined the probability of a single co-occurrence among the 28 observed nt transitions as p = 0.424, and for the 10 observed transversions as p = 0.032. Consequently, we conclude that the six “multiple occurrence” variants are most likely intragenomic (i.e. intercistronic) polymorphisms and not products of PCR and cloning errors. We note there is a reasonable probability for a random co-occurrence of one transitional nt modification (p = 0.424), but a declining probability of 2, 3 or more random co-occurrences (p = 0.180, p = 0.076, and so on).
The observed PCR error rate in our study appears to be in the low range of previously published rates. Among the presumptive cloning errors in the current study eight (33%) were A∙T→G∙C transitions, eight (33%) were G∙C→A∙T transitions, six (25%) were transversions (purine↔pyrimidine substitutions) and two (8%) were deletions. In previous accounts, the majority of Taq cloning errors have been attributed to A∙T→G∙C transitions (Keohavong and Thilly Citation1989; Kobayashi et al. Citation1999; Simon and Weiss Citation2008). The six significant, multiple occurrence intragenomic polymorphisms included five transitions (three A∙T→G∙C) and one C→A transversion. The frequency of intragenomic polymorphisms in Neonothopanus was estimated as 6% (2/33) for monokaryon A and 13% (8/60) for monokaryon B (including four copies with two nt substitutions), with an additional 2-copy polymorphism in the 18S region of monokaryon B.
Corrected sequences for the two monokaryons and one presumed recombinant were determined after elimination of presumed cloning errors, and are identical to the predominant clones (). Sequences for the two monokaryons differed by six indels and four SNPs (98% overall similarity) differing in length by four bps. The predominant clone sequences were deposited in GenBank with the location of the intragenomic polymorphisms indicated in the GenBank entries (monokaryon “A” GenBank JN571729, monokaryon “B” GenBank JN571728, recombinant “AB” GenBank JN571726).
The predominant clone sequences were used to perform a final phylogenetic analysis incorporating all available ITS sequences for the Omphalotaceae () with the outgroup comprising sequences for closely related species from the sister clade, Marasmiaceae (Matheny et al. Citation2006). There is high bootstrap support for the reliability of most of the terminal branches, including the clade that features all of the Neonothopanus isolates. Two Chinese isolates identified as Pleurotus (HQ008725 and HQ008726) are included in this clade and are concluded to be N. nambi. Otherwise, bootstrap support is lacking to determine the position of Neonothopanus within the Omphalotaceae clade which includes the genera Omphalotus, Neonothopanus, and Anthracophylum, and likely also some species currently accommodated in Marasmiellus, Gymnopus, Marasmius, etc. We conclude that additional genes and taxa will be required to resolve relationships at the family and subfamily levels.
Figure 10. Phylogenetic analysis of available sequences for the Omphalotaceae. The outgroup comprises nine species of presumed Marasmiaceae. Numbers above branches are bootstrap values >70%. The alignment comprised 42 taxa; length 652 bp. A total of 359 characters were included in the analysis; 296 were parsimony informative.
Intergenomic and intragenomic ITS heterogeneity
Ribosomal genes are tandemly arrayed and believed to undergo strong concerted evolution resulting in the homogenization of gene copies across the entire genome (Liao Citation1999, with numerous references cited). A number of DNA recombination, repair and replication mechanisms are responsible for both intrachromasomal and interchromasomal homogenization of repetitive sequence arrays, such as gene conversion by mismatch repair (Hillis et al., Citation1991) and unequal crossing-over between repeating units (Coen and Dover Citation1983). The resultant rDNA sequence uniformity together with their high copy number is a major advantage for phylogenetic reconstruction and has led to the widespread use of ITS in phylogenetic analyses and for barcoding (e.g. Druzhinina et al., Citation2005, Cräutlein et al., Citation2011).
There are numerous studies that demonstrate considerable intergenomic variation within species in the Basidiomycota. Hughes et al. (Citation2009), for example, suggested a value of 2–3% sequence divergence in the ITS as an upper limit to determine whether sequences represent the same biological species. There also are a number of references to intrastrain ITS heterogeneity indicating that the latter may not be an exceptionally unusual phenomenon, particularly in the Basidiomycota. Wang and Yao (Citation2005) reported 2–5 different ITS types in 10 clones of each of four Ganoderma species, with SNP's or indels occurring in the ITS1, 5.8s and ITS2 regions. Apparent intragenomic ITS heterogeneities also have been reported for Sclerotium rolfsii (Okabe et al., Citation2001), Trichaptum abietinum (Ko and Jung, Citation2002), Rhizoctonia solani (Pannecoucque and Hofte Citation2009), Pleurotus nebrodensis (Huang et al., Citation2010) and in Laetiporus and Wolfiporia (Lindner and Banik Citation2011). The same phenomenon has been observed in a wide range of other fungi, e.g. Fusarium (O'Donnell and Cigelnik Citation1997), Ascochyta (Fatehi and Bridge Citation1998), Oomycota, e.g. Peronosclerospora (Yao et al. Citation1992), and Pythium (Kageyama et al. Citation2007; Belbahri et al., Citation2008), and indels giving rise to as many as 144 nt differences within a single strain of Rhizopus microsporus (Mucoromycotina) were observed by Woo et al. (Citation2010).
What could explain intragenomic ITS polymorphisms observed in the Neonothopanus isolate and some previous studies? Intrachromosomal homogenization proceeds much more rapidly than interchromasomal recombination (Liao Citation1999), and ITS repeats usually reside in close proximity which should allow for efficient homogenization. Internal sequence repeats such as simple repetitive sequence motives are believed to shape ITS evolution (Booton et al., Citation1999), subject to molecular processes such as replication slippage (Levinson and Gutman Citation1987), unequal crossing over (Smith Citation1976) and biased gene conversion (Hillis et al Citation1991). Xylaria, for example, is more prone to transcriptional ITS modifications than other Ascomycetes due to the presence of a short palindrome sequence of 11 bp repeated in a tandem or quasi-tandem fashion (Platas et al., Citation2001, Citation2004). We examined the most common Neonothopanus clone sequence (43 identical clones) for the presence of tandem repeats that could be the basis for transcriptional modifications. We found only two tandem quasi-repeats (75% matches) of 8 and 13 nts comprising 3.4 and 1.9 copies, respectively. Simple sequence repeats can be the site for initiation of gene conversion due to slipped strand misspairing events (e.g. Hibner at al., Citation1991). Conversely to causing ITS sequence heterogeneities, the lack of short tandem repeats may impede intrachromasomal gene conversion attributable to the process of replication slippage.
Most ITS mutations are likely to be selectively neutral, and fixation of a neutral allele is inefficient; thus, the elimination of a singular neutral allele variant within the multicopy ITS regions may take an exceedingly long time (Liao Citation1999). Only if mutation is slow relative to gene conversion will homogenous arrays result (Elder and Turner Citation1995). High mutation rates relative to rates of fixation by gene conversion will result in the coexistence of multiple alleles within a genome. Many proteins are involved in DNA recombination, replication and repair and have roles in the various steps of gene homogenization. Abdulkarim and Hughes (Citation1996) found that specific recombination enzymes are required for gene conversion, whereas mismatch repair enzymes paradoxically interfere with the homogenization process. Therefore, one or more inherited modifications among the numerous proteins involved in DNA transcription also could impact intrachromosomal gene homogenization in fungi.
Relatively slow rates of concerted evolution may be an innate characteristic of the internally transcribed regions of rRNA. Slow rates of concerted evolution in ITS regions relative to the external transcribed spacer region (ETS) facilitate the coexistence of multiple ITS “alleles” within the genome (Polanco et al. Citation1998, Okuyama et al. Citation2005). Finally, not all of the ITS copies may reside in proximity on the same chromosome (Pasero and Marilley Citation1993), and then would not be subjected to the mechanisms of concerted evolution. If this is the case, ITS heterogeneities may be more common than indicated in the published literature, and more extensive sampling of the ITS may be required for phylogenetic studies and to ensure reliability as a PCR-based diagnostic marker for barcoding.
Recommendations for sequencing heterozygous fungi
Direct sequencing is cost-effective and usually performed, at least initially, to identify fungi. In direct sequencing, single nt variants caused by random PCR error are usually obscured by the dominant sequence and do not impact on the resulting “consensus” sequence. However, if the organism is heterozygous, this will be evident in the occurrence of multiple peaks, or unreadable electrophoretic patterns in the case of frame shifts, and neither allele can be correctly interpreted. In the current study, we found six indels differentiating two compatible mating strains of N. nambi, which resulted in unreadable electrophoretic patterns caused by frame shifts after direct sequencing of the basidiome. Nilsson et al. (Citation2008) reported a wide range of intraspecific variation in the ITS regions of fungi based on sequences in the International Nucleotide Species Databases (INSD), ranging 0–24% and averaging 3.33% for the Basidiomycota. We can expect this level of ITS within-species heterogeneity to cause problems in direct sequencing of nuclear genes which are frequently heterozygous. Ideally, single spore cultures would be used for sequencing. However, as in the current study, these are often not available.
Cloning can resolve mixtures of alleles arising from environmental samples or heterozygous materials (e.g. Lindner and Banik Citation2009). When cloning is necessary the use of high-fidelity polymerases with 3′–5′ exonuclease “proofreading” capabilities, such as Pfu polymerase, are strongly recommended to resolve ambiguities as they can reduce cloning errors by a factor of close to 10 (Kobayashi et al. Citation1999). However, since cloning errors cannot be completely eliminated, at least three clones should be sequenced for homozygous organisms and single nt variants discounted as cloning errors. For heterozygous species, six to 10 clones may be required to resolve the differing alleles, creating separate alignments for the “alleles” and discounting “singletons” as probable PCR errors propagated by cloning.
Acknowledgements
The authors are grateful to Dr Scott Redhead and Dr Keith Seifert for critical reading of the manuscript. We also thank the anonymous external reviewers for their thorough and very constructive critique of the submitted manuscript. Research was partly supported by Russian Federal Target Program (grant 02.740.11.0766). Academician I.I. Gitelson (Institute of Biophysics SB RAS) initiated and facilitated the international collaboration.
References
- Abdulkarim , F and Hughes , D. 1996 . Homologous recombination between the tuf genes of Salmonella typhimurium . J Mol Biol. , 260 : 506 – 522 .
- Acinas , SG , Sarma-Rupavtarm , R , Klepac-Ceraj , V and Polz , MF. 2005 . PCR-induced sequence artefacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample . Appl Environ Microbiol. , 71 : 8966 – 8969 .
- Belbahri , L , McLeod , A , Paul , B , Calmin , G , Moralejo , E , Spies , CFJ , Botha , WJ , Clemente , A , Descals , E , Sanchez-Hernandez , E and Lefort , F. 2008 . Intraspecific and within-isolate sequence variation in the ITS rRNA gene region of Pythium mercuriale sp. nov. (Pythiaceae) . FEMS Microbiol Lett. , 284 : 17 – 27 .
- Benson , G. 1999 . Tandem repeats finder: a program to analyse DNA sequences . Nucleic Acid Res. , 27 : 573 – 580 .
- Bondar , VS , Puzyr , AP , Purtov , KV , Medvedeva , SE , Rodicheva , EK and Gitelson , JI. 2011 . The luminescent system of the luminous fungus Neonothopanus nambi . Doklady Biochem Biophys. , 438 : 138 – 140 . doi: 10.1134/S1607672911030082
- Booton , GC , Kaufman , L , Chandler , M , Oguto-Ohwayo , R , Duan , WR and Fuerst , PA. 1999 . Evolution of the ribosomal RNA internal transcribed spacer one (ITS-1) in cichlid fishes of the lake Victoria region . Mol Phylogenet Evol. , 11 : 273 – 282 .
- Buaart , SW , Saksirirat , S , Sirimungkararat , A and Hiransalee . 11 Oct 2008 2008 . “ Ribosomal DNA sequences of luminescent mushroom (Neonothopanus nambi Speg.) and effect of culture filtrate on entomopathogenic nematode (Steinernema carpocapsae) ” . In Proceedings of the 3rd Annual Meeting of Thai Mycological Association and Mycology Conference 11 Oct 2008 , 77 Thailand
- Coen , ES and Dover , GA. 1983 . Unequal exchanges and the coevolution of X and Y rDNA arrays in Drosophila melanogaster . Cell , 33 : 849 – 855 .
- Corner , EJH. 1981 . The agaric genera Lentinus, Panus and Pleurotus . Beih Nova Hedwigia , 69 : 1 – 169 .
- Cräutlein , M von , Korpelainen , H , Pietiläinen , M and Rikkinen , J. 2011 . DNA barcoding: a tool for improved taxon identification and detection of species diversity . Biodivers Conserv , 20 : 373 – 389 .
- Cummings , SM , McMullan , M , Joyce , DA and van Oosterhout , C. 2010 . Solutions for PCR, cloning and sequencing errors in population genetic analysis . Conserv Genet , 11 : 1095 – 1097 .
- Desjardin , DE , Oliveira , AG and Stevani , CV. 2008 . Fungi bioluminescence revisited . Photochem Photobiol Sci. , 7 : 170 – 182 .
- Dickie , IA. 2010 . Insidious effects of sequencing errors on perceived diversity in molecular surveys . New Phytol. , 188 : 916 – 918 .
- Druzhinina , IS , Kopchinskiy , AG , Komon , M , Bissett , J , Szakacs , G and Kubicek , CP. 2005 . An oligonucleotide barcode for species identification in Trichoderma and Hypocrea . Fungal Gen Biol. , 42 : 813 – 828 .
- Elder , JF Jr and Turner , BJ. 1995 . Concerted evolution of repetitive DNA sequences in eukaryotes . Q Rev Biol. , 70 : 297 – 320 .
- Fatehi , J and Bridge , P. 1998 . Detection of multiple rRNA-ITS regions in isolates of Ascochyta . Mycol Res. , 102 : 762 – 766 .
- Hibner , BL , Burke , WD and Eickbush , TH. 1991 . Sequence identity in an early chorion multigene family is the result of localised gene conversion . Genetics , 128 : 595 – 606 .
- Hillis , DM , Moritz , C , Porter , CA and Baker , RJ. 1991 . Evidence for biased gene conversion in concerted evolution of ribosomal DNA . Science , 251 : 308 – 310 .
- Huang , C , Xu , J , Gao , W , Chen , Q , Wen , H and Zhang , J. 2010 . A reason for overlap peaks in direct sequencing of rRNA gene ITS in Pleurotus nebrodensis . FEMS Microbiol Lett. , 305 : 14 – 17 .
- Hughes , KW , Petersen , RH and Lickey , EB. 2009 . Using heterozygosity to estimate a percentage DNA sequence similarity for environmental species’ delimitation across basidiomycete fungi . New Phytol. , 182 : 795 – 798 .
- Isobe , M , Takahashi , H , Usami , K , Hattori , M and Nishigohri , Y. 1994 . Bioluminescence mechanism on new systems . Pure Appl Chem , 66 : 765 – 772 .
- Kageyama , K , Senda , M , Asano , T , Suga , H and Ishiguro , K. 2007 . Intra-isolate heterogeneity of the ITS region of rDNA in Pythium helicoides . Mycol Res. , 111 : 416 – 423 .
- Katoh , M , Misawa , K , Kuma , K and Miyata , T . 2002 . MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform . Nucleic Acids Res. , 30 : 3059 – 3066 .
- Keohavong , P and Thilly , WG. 1989 . Fidelity of DNA polymerases in DNA amplification . Proc Nat Acad Sci USA , 86 : 9253 – 9257 .
- Kirchmair , M , Morandell , S , Stolz , D , Poder , R and Sturmbauer , C. 2004 . Phylogeny of the genus Omphalotus based on nuclear ribosomal DNA-sequences . Mycologia , 96 : 1253 – 1260 .
- Ko , KS and Jung , HS. 2002 . Three nonorthologous ITS1 types are present in a polypore fungus Trichaptum abietinum . Mol Phylogenet Evol. , 23 : 112 – 122 .
- Kobayashi , N , Tamura , K and Aotsuka , T. 1999 . PCR error and molecular population genetics . Biochem Genet. , 37 : 317 – 321 .
- Levinson , G and Gutman , GA. 1987 . Slipped strand misspairing: a major mechanism for DNA sequence evolution . Mol Biol Evol. , 4 : 203 – 221 .
- Liao , DQ. 1999 . Concerted evolution: molecular mechanism and biological implications . Am J Hum Genet. , 64 : 24 – 30 .
- Lindner , DL and Banik , MT. 2009 . Effects of cloning and root-tip size on observations of fungal ITS sequences from Picea glauca roots . Mycologia , 101 : 157 – 165 .
- Lindner , DL and Banik , MT. 2011 . Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in genus Laetiporus . Mycologia , 103 : 731 – 740 .
- Marr , CD. 1979 . Laccase and tyrosinase oxidation of spot test reagents . Mycotaxon , 9 : 244 – 276 .
- Mata , J L , Hughes , KW and Petersen , RH. 2007 . An investigation of Omphalotaceae (Fungi: Euagarics) with emphasis on the genus Gymnopus . Sydowia , 59 : 181 – 289 .
- Matheny , PB , Curtis , JM , Hofstetter , V , Aime , MC , Moncalvo , J-M , Ge , ZW , Yang , ZL , Slot , JC , Ammirati , JF , Baroni , TJ , Bougher , NL , Hughes , KW , Lodge , DJ , Kerrigan , RW , Seidl , MT , Aanen , DK , DeNitis , M , Daniele , GM , Desjardin , DE , Kropp , BR , Norvell , LL , Parker , A , Vellinga , EC , Vilgalys , R and Hibbett , DS. 2006 . Major clades of Agaricales: a multilocus phylogenetic overview . Mycologia , 98 : 982 – 995 .
- Nilsson , RH , Kristiansson , E , Ryberg , M , Hallenberg , N and Larsson , K-H. 2008 . Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification . Evol Bioinform. , 4 : 193 – 201 .
- O'Donnell , K and Cigelnik , E. 1997 . Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous . Mol Phylogenet Evol. , 7 : 103 – 116 .
- Okabe , I , Arawaka , M and Matsumoto , N. 2001 . ITS polymorphism within a single strain of Sclerotium rolfsii . Mycoscience , 42 : 107 – 113 .
- Okuyama , Y , Fujii , N , Wakabayashi , M , Kawakita , A , Ito , M , Watanabe , M , Murakami , N and Kato , M. 2005 . Nonuniform concerted evolution and chloroplast capture: heterogeneity of observed introgression patterns in three molecular data partition phylogenies of Asian Mitella (Saxifragaceae) . Mol Biol Evol. , 22 : 285 – 296 .
- Pannecoucque , J and Höfte , M. 2009 . Detection of rDNA ITS polymorphism in Rhizoctonia solani AG 2–1 isolates . Mycologia , 101 : 26 – 33 .
- Pasero , P and Marilley , M. 1993 . Size variation of rDNA clusters in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe . Mol Gen Genet. , 236 : 448 – 452 .
- Petersen , R.H and Krisai-Greilhuber , I. 1999 . Type specimen studies in Pleurotus . Persoonia , 17 ( 2 ) : 201 – 219 .
- Platas , G , Acero , J , Borkowski , JA , González , V , Portal , MA , Rubio , V , Sánchez , J , Salazar , O and Peláez , F. 2001 . Presence of a simple tandem repeat DNA sequence in the ITS region of the Xylariales . Curr Microbiol. , 43 : 43 – 50 .
- Platas , G , Ruibal , C and Collado , J. 2004 . Size and sequence heterogeneity in the ITS1 of Xylaria hypoxylon isolates . Mycol Res. , 108 : 71 – 75 .
- Polanco , C , González , AF and Dover , GA. 1998 . Multigene family of ribosomal DNA in Drosophila melanogaster reveals contrasting patterns of homogenization for IGS and ITS spacer regions: a possible mechanism to resolve this paradox . Genetics , 149 : 243 – 256 .
- Simon , UK and Weiss , M. 2008 . Intragenomic variation of fungal ribosomal genes is higher than previously thought . Mol Biol Evol. , 25 : 2251 – 2254 .
- Smith , GP. 1976 . Evolution of repeated DNA sequences by unequal crossover . Science , 191 : 528 – 535 .
- Swofford , DL. 1991 . PAUP: Phylogenetic Analysis Using Parsimony , Champaign , Illinois : Version 3.1 Computer program distributed by the Illinois Natural History Survey .
- Tendersoo , L , Nilsson , RH , Abarenkov , K , Jairus , T , Sadam , A , Saar , I , Bahram , M , Bechem , E , Chuyong , G and Kõljalg , U. 2010 . 454 pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases . New Phytol. , 188 : 291 – 301 .
- Van DT. 2006. The successful cultivation of a new luminous mushroom Omphalotus aff. Illudent http://springbrook.info/research/external/omphalotus.af.illudent/ (http://springbrook.info/research/external/omphalotus.af.illudent/) (Accessed: 25 November 2010 ).
- Van , DT. 2009 . Physiological and cultural properties of the luminous fungus Omphalotus aff. Illudent . J Siber Fed Univ. , 2 : 157 – 171 .
- Vydryakova , GA , Psurtseva , NV , Belova , NV , Pachenova , NV and Gitelson , II. 2009 . [Luminous mushrooms and prospects of their use.] . Mikol Fitopatol. , 43 ( 5 ) : 369 – 376 . [Russian]
- Vydryakova , GA , Shoukouhi , P , Psurtseva , NV , Dao , TV and Bissett , J. 26 September – 1 October, 2010 2010 . “ Unusual ITS heterogeneity in the luminous mushroom Neonothopanus nambi from Vietnam ” . In Proceedings of the 12th international conference on culture collections 26 September – 1 October, 2010 , 27 Florianopolis , , Brazil
- Wang , D-M and Yao , Y-J. 2005 . Intrastrain internal transcribed spacer heterogeneity in Ganoderma species . Can J Microbiol. , 51 : 113 – 121 .
- White , T , Bruns , T , Lee , S and Taylor , J. 1990 . “ Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics ” . In PCR protocols. A guide to methods and applications , Edited by: Innis , MA , Gelfand , DH , Sninsky , JJ and White , TJ . 315 – 322 . San Diego : Academic Press .
- Woo , PCY , Leung , S-Y , To , KKW , Chan , JFW , Ngan , AHY , Cheng , VCC and Lau , SKP. 2010 . Internal transcribed spacer region sequence heterogeneity in Rhizopus microsporus: implications for molecular diagnosis in clinical microbiology laboratories . J. Clin Microbiol. , 48 : 208 – 214 .
- Yao , CL , Fredriksen , RA and Magill , CW. 1992 . Length heterogeneity in ITS2 and the methylation status of CCGG and GCGC sites in the rRNA genes of the genus Peronosclerospora . Curr Genet. , 22 : 219 – 229 .