Abstract
Fumonisins are agriculturally important mycotoxins produced by the maize pathogen Fusarium verticillioides. The chemical structure of fumonisins contains two tricarballylic esters, which are rare structural moieties and important for toxicity. The mechanism for the tricarballylic ester formation is not well understood. FUM7 gene of F. verticillioides was predicted to encode a dehydrogenase/reductase, and when it was deleted, the mutant produced tetradehydro fumonisins (DH4–FB). MS and NMR analysis of DH4–FB1 indicated that the esters consist of aconitate with a 3′-alkene function, rather than a 2′-alkene function. Interestingly, the purified DH4–FB1 eventually yielded three chromatographic peaks in HPLC. However, MS revealed that the metabolites of the three peaks all had the same mass as the initial single-peak DH4–FB1. The results suggest that DH4–FB1 can undergo spontaneous isomerization, probably including both cis–trans stereoisomerization and 3′- to 2′-ene regioisomerization. In addition, when FUM7 was expressed in Escherichia coli and the resulting enzyme, Fum7p, was incubated with DH4–FB, no fumonisin with typical tricarballylic esters was formed. Instead, new fumonisin analogs that probably contained isocitrate and/or oxalosuccinate esters were formed, which reveals new insight into fumonisin biosynthesis. Together, the data provided both genetic and biochemical evidence for the mechanism of tricarballylic ester formation in fumonisin biosynthesis.
1. Introduction
Fumonisins are a group of mycotoxins produced by several agriculturally important fungi, including Fusarium verticillioides (synonym F. moniliforme, teleomorph Gibberella moniliformis, synonym G. fujikuroi mating population A), which is a common fungal contaminant of maize and maize-derived products worldwide (Marasas et al. Citation2004; Wang et al. Citation2006, Citation2008). The fungus can cause ear, stalk, and seedling rots of maize, and can also occur at a high frequency in healthy corn tissues (Miller Citation2001). Fumonisin contamination in maize has been associated with diseases in some livestock and with human health problems, including esophageal cancer and neural tube defects (Marasas et al. Citation2004).
At least 28 fumonisin analogs have been characterized (Rheeder et al. Citation2002). The B-series fumonisins, FB1, FB2, FB3, and FB4, are the predominant analogs produced by wild-type isolates of F. verticillioides (, FB1 structure shown) (Nelson et al. Citation1993). The metabolites contain a linear 18-carbon chain (C-3 to C-20) that is synthesized from acetate by the activity of an iterative polyketide synthase (Fum1p) (Proctor et al. Citation1999; Zhu et al. Citation2007; Du et al. Citation2008). The amino group and C-1 and C-2 are derived from alanine (Branham and Plattner Citation1993; Blackwell et al. Citation1996; Gerber et al. Citation2009). The two methyl groups at C-12 and C-16 are derived from methionine (Plattner and Shackelford Citation1992). In FB1, the hydroxyl groups at C-5, C-10, C-14, and C-15 are derived from molecular oxygen and are formed following synthesis of the polyketide (Caldas et al. Citation1998; Ding et al. Citation2004; Proctor et al. Citation2006), while the C-3 hydroxyl originates from acetate and results from reduction of a 3-keto group (Butchko et al. Citation2003; Yi et al. Citation2005). The condensation between the 18-carbon chain and alanine involves a unique polyketide chain-releasing mechanism (Du et al. Citation2008; Gerber et al. Citation2009).
Figure 1. Chemical structure of fumonisins and analogs. DH4–FB1, tetradehydro fumonisin B1. (A) Structure of fumonism B1 and tetradehydro fumonism B1. (B) Example of possible regio- and sterioisomers of DH4–FB1.
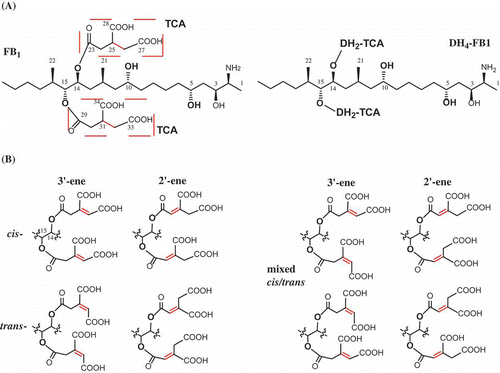
Another unusual modification during fumonisin biosynthesis is esterification of tricarballylic moieties to the C-14 and C-15 hydroxyls. Tricarballylic esters are rare structural features in natural products and are necessary for fumonisin toxicity (Yin et al. Citation1996; Humpf et al. Citation1998; Seefelder et al. Citation2003). The molecular mechanism by which fumonisin tricarballylic esters are formed has not been fully elucidated. Previous studies using 13C-substrates suggested that the precursor for the tricarballylic esters is an intermediate from the citric acid cycle (Blackwell et al. Citation1996). Data obtained from gene-deletion mutants of F. verticillioides suggest that four genes, FUM7, FUM10, FUM11, and FUM14, are required for the formation of esters (Butchko et al. Citation2006; Zaleta-Rivera et al. Citation2006). FUM7 is predicted to encode an iron-containing dehydrogenases/reductases, similar to those that catalyze the reversible reduction of an alkene to an alkane (Seibert et al. Citation1998; Kallberg et al. Citation2002). FUM7-deletion mutants produce FB analogs with an alkene function in both esters (tetradehydro-FB, DH4–FB) () (Butchko et al. Citation2006). This suggests that the enzyme (Fum7p) encoded by FUM7 is likely to catalyze reduction of the alkene. In order to determine the regio- and stereo-specificity of this biosynthetic step, we prepared the predominant component (DH4–FB1) and determined the positions of the carbon–carbon double bond in tricarballylate. We also investigated the alkene reductase activity of Fum7p through heterologous expression.
2. Materials and methods
2.1. Materials and general methods
Chemicals were purchased from Fisher Scientific or Sigma. All oligonucleotide primers for PCR were synthesized by Integrated DNA Technologies (IDT; Coralville, IA). Escherichia coli strain DH5α was used as the host for general plasmid DNA propagation, and cloning vectors were the pGEM-zf series from Promega (Madison, WI). Plasmid preparation and DNA extraction were carried out using Qiagen kits (Valencia, CA), and all other DNA manipulations were carried out according to standard methods (Sambrook et al. Citation1989).
2.2. Isolation and analysis of fumonisin analogs from F. verticillioides
Procedures for isolating the B-series fumonisins from F. verticillioides were essentially same as described previously (Bojja et al. Citation2004). For isolating and analyzing the tetradehydro tricarballylate-containing analogs from the FUM7 mutant, the procedure was modified as follows. Single colonies of the fungus growing on YPD/hygromycin (300 μg/ml) agar medium were transferred to test tubes containing 3 ml YPD/hygromycin (150 μg/ml) liquid medium and allowed to grow in a shaker (60 rpm) at room temperature for 2 days. From the 3 ml culture, 100 μl was transferred to a flask containing 25 ml YPD/hygromycin (150 μg/ml). The culture was incubated with shaking at room temperature for another 2 days. The culture was transferred to a 50-ml tube and centrifuged at 2500 rpm for 20 min. The pellet was washed three times with sterile water and finally re-suspended in 10 ml sterile water. From the suspended solution, 500 μl was transferred to a new flask containing 10 g of autoclaved Cracked Maize Kernel (CMK) medium (Proctor et al. Citation2003). After 4 weeks of growth at room temperature in the dark, metabolites were extracted from the CMK cultures with 20 ml of 50% acetonitrile (CH3CN). An aliquot of the extracts was filtered and a sample of 50 μl was injected in HPLC-ELSD to analyze the metabolites, following the method described previously (Bojja et al. Citation2004).
To prepare DH4–FB1 for spectroscopic studies, the crude extract was dried and partitioned into EtOAc-soluble and H2O-soluble extracts. The H2O extract was loaded onto a 10-g C18 cartridge and eluted with the stepwise gradient of 0, 15%, 30%, 50%, and 70% CH3CN in H2O (containing 0.025% TFA, 100 ml for each step). The 30% CH3CN fraction was loaded onto another 10-g C18 cartridge and eluted with the stepwise gradient of 15%, 25%, and 35% CH3CN in H2O (containing 0.025% TFA, 100 ml for each step). The 35% CH3CN fraction (280 mg) was subjected to a gel permeation chromatography (50 g Sephadex LH-20, in methanol) for further purification. Purified DH4–FB1 was analyzed by ESI-MS on an API Qtrap 4000 and NMR spectra (1H- and 13C-NMR and HMBC) on a Bruker DRX-500 spectrometer, at 500/125 MHz, respectively, in methanol-d6, δ in ppm relative to Me4Si, J in Hz.
2.3. FUM7 gene expression and enzyme purification
FUM7 has no introns (Proctor et al. Citation2003) and was amplified directly from the genomic DNA of F. verticillioides by PCR using a forward primer of 5′-C TCT AGA CAT ATG AGT CTT GAT CAT CAC CAG-3′ (NdeI site underlined) and a reverse primer of 5′-CCC AAG CTT CTA TGC TGC CAT GTA CAG-3′ (HindIII site underlined). After the identity of the 1275 bp fragment was confirmed by DNA sequencing, it was cloned into pET28a for expression in E. coli BL21(DE3) and pMAL-c2 for expression in E. coli TB1 as specified by the manufacturers. Fum7p was highly expressed in both systems, but only the TB1/pMAL-c2 system yielded a soluble protein. The soluble fraction of the expressed protein was loaded onto an amylose resin column (New England BioLabs) for affinity purification of the protein. The purified protein was desalted by dialysis against a buffer containing 50 mM Tris–HCl, pH 7.8, 100 mM NaCl, 10 mM MgCl2, 2 mM dithiothreitol, and 15% glycerol. The resulting preparation was stored at –80°C until it was used in assays.
2.4. In vitro activity assay for Fum7p
The reactions contained 0.4 µM Fum7p, 2.8 mM DH4–FB, 1 mM NAD(P)H, 1 mM FeCl2, and 100 mM Tris–HCl, pH 7.8, in a total volume of 100 µl. The reactions were incubated at 37°C for 30 min and stopped by adding two volumes of ethanol at 4°C. After 30 min on ice, the precipitated proteins were removed by centrifugation at 13,200 rpm at 4°C for 20 min, and the supernatant was transferred to a new tube. A 100 μl fraction of supernatant was used for HPLC–ELSD and ESI–MS analysis.
3. Results and discussion
3.1. Position and configuration of the alkene in tricarballylic esters of DH4–FB1
FUM7 mutants of F. verticillioides produce fumonisin analogs, tetradehydro fumonisins (DH4–FBs), that are four mass units less ([M + H]+ m/z 718 and m/z 702) than FB1 (m/z 722) or FB2/3 (m/z 706) (Butchko et al. Citation2006). The difference in mass between DH4–FBs and FBs is due to an alkene function present in each of the two tricarballylate esters of DH4–FBs. However, the position and configuration of the alkene function are not clear, as multiple regio- and stereoisomers of aconitate could account for the difference (). We therefore set out to determine the structure of tricarballylate esters in DH4–FB. After several purification steps, we were able to obtain DH4–FB1 that yielded as a single chromatographic peak in HPLC analysis (). MS analysis confirmed a [M + H]+ m/z 718.4 for the purified DH4–FB1, and 1H-NMR analysis was consistent with one major compound. The position of the carbon–carbon double bond in the tricarballylic esters of DH4–FB1 was identified by spectroscopic analyses, including 1D- and 2D-NMR data (), and by comparison of these data to those previously published for fumonisins (Bezuidenhout et al. Citation1988). The selected HMBC corrections from H-15 to C-22 and C-29; H-30/24 to C-29/23 and C-34/28; and H-32/26 to C-30/24, C-33/27, and C-34/28 established the tricarballylic ester on the C-15 hydroxyl with a double bond between C-31 and C-32 (3′-ene) and the tricarballylic ester on the C-14 hydroxyl with a double bond between C-25 and C-26 (3′-ene) ( and ).
Table 1. Selected HMBC correlations (H→C) for DH4–FB1 and 1H–NMR (500 MHz) and 13C–NMR (125 MHz) spectroscopic data (methanol-d4) for DH4–FB1. δ in ppm. J in Hz.
Figure 2. Isolation and analysis of DH4–FB1 from the FUM7 mutant. (A) HPLC analysis of crude extract; (B) HPLC analysis of the initially purified DH4–FB1; (C) HPLC analysis of the isomerized DH4–FB1; (D) MS analysis of the initially purified DH4–FB1; and (E) MS analysis of the isomerized DH4–FB1. A proposed cis–trans stereoisomerization of DH4–FB1 is included.
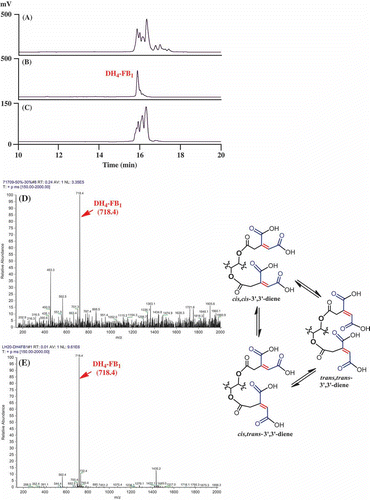
Interestingly, when the purified DH4–FB1 was incubated at room temperature for 2 days, additional chromatographic peaks were observed in the sample in HPLC analysis. To exclude potential degradation products, we attempted to further separate the preparation using a gel permeation chromatography (Sephadex LH-20). Surprisingly, the DH4–FB1 fraction continued to yield three major chromatographic peaks, instead of one peak, in HPLC analysis, but yielded only one signal [M + H]+ m/z 718.4, in MS analysis (). These results indicate that the three peaks in the preparation represent three isomers of DH4–FB1. One probable explanation for formation of the isomers is that the original preparation of purified single 3′,3′-diene stereoisomer of DH4–FB1 was converted to other cis–trans stereoisomers () and/or 2′-ene regioisomers during the 2-day incubation period. For the didehydrotricarballylate (aconitate) moieties of DH4–FB1, both regio- and stereoisomerization could occur either through a C-3′ to C-4′ bond rotation of the aconitate carbonium ion following a protonation of the double bond from an acid and then deprotonation, or through an allylic rearrangement by elimination of a proton at C-2′ of the aconitate carbonium ion (Klinman and Rose Citation1971). These mechanisms are reasonable considering that all DH4–FB1 solvents contained an acid (0.025% TFA). These results are also consistent with the previous study that observed broad NMR peaks for the tricarballylic carbons in DH4–FB (Butchko et al. Citation2006).
3.2. Activity of heterologously expressed Fum7p
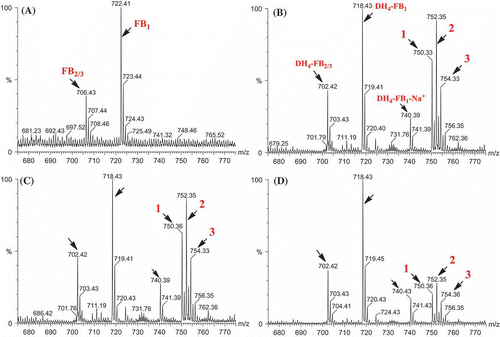
Previous in vivo feeding experiments suggested that the tetradehydro fumonisins (DH4–FB) produced by FUM7-deletion mutants of F. verticillioides are probably shunt metabolites of the biosynthetic pathway, because strains of the fungus with a wild-type FUM7 were unable to convert the DH4–FB into FB (Butchko et al. Citation2006). To obtain direct evidence, we heterologously expressed FUM7 and examined whether purified Fum7p could catalyze conversion of DH4–FB to FB in vitro. Fum7p produced in the TB2/pMALc2 expression system was partly soluble as a fusion protein with MBP. The protein purified on an amylose affinity column yielded approximately 88.2 kDa band on SDS–PAGE (), which is consistent with the expected size for the fusion protein. Fum7p was incubated with purified DH4–FB in the presence of NADPH and Fe2+, which are cofactors usually required for activity of the class of dehydrogenases/reductases to which Fum7p is most similar (Seibert et al. Citation1998; Kallberg et al. Citation2002). The MS analysis of the reaction mixtures showed that fumonisins with typical tricarballylic esters were not produced and that the [M + H]+ ions of 718.43 (DH4–FB1) and 702.42 (DH4–FB2/3) remained in the reaction mixtures, under the conditions tested (). The results are consistent with the in vivo feeding experiments (Butchko et al. Citation2006) and suggest that free DH4–FB are not direct substrates of Fum7p. Interestingly, although fumonisins with typical tricarballylic esters were not detected, several novel metabolites were produced in the reactions. One (compound 1) of these had a molecular ion of 750.33, which is the same as that expected for an FB1 analog with two oxalosuccinate esters ( and ). Another novel metabolite (compound 3) had a molecular ion of 754.33, which is consistent with an FB1 analog with two isocitrate (or citrate) esters. Finally, a third metabolite (compound 2) with a molecular ion of 752.35, which is the same as that expected for an FB1 analog with one isocitrate (or citrate) ester and one oxalosuccinate ester. These ions were not present in the initial DH4–FB isolated from the FUM7-deleted mutant. Interestingly, these ions were present, but at a lower level, in the reaction mixture containing boiled Fum7p. When NADH was used in the place of NADPH, the intensity of the ions was significantly reduced (). The results suggest that Fum7p promotes the conversion of DH4–FB into analogs with isocitrate (or citrate) and oxalosuccinate side chains. This conversion could be realized through a hydration of aconitate to generate isocitrate (or citrate), followed by a dehydrogenation of isocitrate to produce oxalosuccinate (). The hydration could be a non-enzymatic step, whereas the dehydrogenation should be facilitated by the dehydrogenase activity of Fum7p.
Figure 3. SDS-PAGE of Fum7p produced in E. coli. Lane 1, purified Fum7p; Lane 2, size markers.

Figure 4. MS analysis of the reaction mixtures containing Fum7p and tetradehydro fumonisins. (A) Standard fumonisins; (B) Fum7p + DH4–FB + NADPH; (C) Fum7p + DH4–FB + NADPH + FeCl2; and (D) Fum7p + DH4–FB + NADH + FeCl2. The peaks corresponding to FB1, FB2/3, as well as the DH4–FB analogs are indicted with arrows. The proposed structure for compounds 1, 2, and 3 are shown in . 1, fumonisin analog with two oxalosuccinate esters; 2, fumonisin analog with one oxalosuccinate ester and one isocitrate ester; and 3, fumonisin analog with two isocitrate esters.
3.3. Mechanism for the tricarballylic ester formation
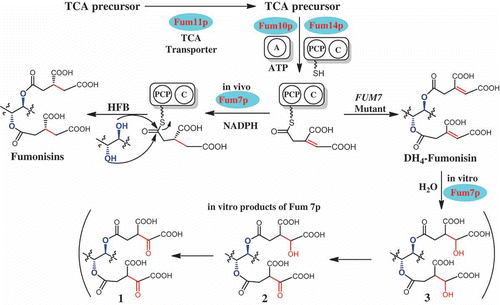
Previous studies have shown that FUM7, FUM10, FUM11, and FUM14 are involved in formation of the tricarballylic esters of fumonisins (Butchko et al. Citation2006; Zaleta-Rivera et al. Citation2006). Among the four genes, the function of FUM14 has been demonstrated both in vivo and in vitro (Butchko et al. Citation2006; Zaleta-Rivera et al. Citation2006). FUM14 is predicted to encode an unusual nonribosomal peptide synthetase (NRPS) containing two domains, peptidyl carrier protein (PCP) and condensation domain. Fum14p catalyzes the transfer of the activated tricarballylic acid to the hydroxyl groups on C-14 and C-15 to form the two ester bonds. FUM10-deletion mutants produce hydrolyzed FB3 and FB4 (HFB3 and HFB4, respectively), analogs of FB3 and FB4 that lack the esters (Butchko et al. Citation2006). FUM10 is predicted to encode an acyl-CoA synthetase or the adenylation domain of NRPS (Proctor et al. Citation2003). Therefore, FUM10 is likely responsible for in vivo activation of the tricarballylic acid. FUM11-deletion mutants produce the wild-type complement of fumonisins as well as a significant amount of partially hydrolyzed fumonisins, fumonisin analogs that lack one of the ester functions (Butchko et al. Citation2006). FUM11 is predicted to encode a tricarboxylate transporter and is likely to function in vivo to supply the tricarboxylic acid precursors for esterification (Proctor et al. Citation2003). Two lines of evidence support its involvement of FUM7 in the tricarballylic ester formation. First, FUM7 mutants produce DH4–FBs with an alkene function in both esters rather than the wild-type complement of FBs (Butchko et al. Citation2006). Second, the deduced amino acid sequence of FUM7 is most similar to a class of dehydrogenases/reductases that catalyze alkene-to-alkane reductions (Seibert et al. Citation1998; Kallberg et al. Citation2002). Given the evidence, it makes sense to propose that Fum7p catalyzes the hydrogenation of DH4–FB to form FB (Du et al. Citation2008; Huffman et al. Citation2010). However, the regio- and stereo-specificity of the reduction was not clear. In addition, the timing of the reduction remained unknown, because the reduction could take place after the DH4–FB product has been released from the carrier domain (PCP) of Fum14p, or the reduction could take place while aconitate is still covalently linked to the PCP after the substrate has been activated by Fum10p and loaded to the PCP of Fum14p (). In this study, we have determined the position of the carbon–carbon double bond in DH4–FB1, the predominant DH4–FB produced by FUM7 mutants of F. verticillioides. In addition, our results indicate that alkene function in the tricarballylic esters of DH4–FB1 undergo isomerization that leads to the formation of a mixture of regio- and stereoisomers. If this isomerization occurs in vivo, it seems probable that Fum7p would have a broad specificity to reduce the mixture into regular fumonisins with saturated tricarballylate esters, because wild-type F. verticillioides does not produce DH4–FBs. Furthermore, the results of the heterologous expression of FUM7 and assessment of in vitro activity of the resulting enzyme suggest that the Fum7p-catalyzed reduction reaction most likely takes place while the aconitate substrate is covalently linked to the PCP of Fum14p enzyme.
Figure 5. A proposed mechanism for in vivo formation of tricarballylic esters in fumonisin biosynthesis and the observed in vitro activity of Fum7p. TCA, tricarboxylic acid; A, adenylation domain encoded by FUM10; PCP, peptidyl carrier protein domain in the protein encoded by FUM14; C, condensation domain in the protein encoded by FUM14; and HFB, hydrolyzed fumonisins.
In conclusion, this work provides molecular insights into the biosynthetic mechanism of the rare structural moieties of fumonisins. Because the toxicity of DH4–FBs relative to FBs is not known and tricarballylic esterification is an essential step for the maturation of the mycotoxins, an understanding of the complexity of this process is useful for efforts in agriculture to reduce mycotoxin contamination.
Acknowledgment
The authors thank Kurt Wulser and Sara Basiaga for technical support.
Funding
This work was supported in part by National Natural Science Foundation of China [grant number 31329005] and National Science Foundation [grant number MCB-0614916].
References
- Bezuidenhout SC, Gelderblom WCA, Gorst-Allman CP, Horak RM, Marasas WFO, Spiteller G, Vleggaar R. 1988. Structure elucidation of the fumonisins, mycotoxins from Fusarium moniliforme. J Chem Soc Chem Commun. 11:743–745.
- Blackwell BA, Edwards OE, Fruchier A, ApSimon JW, Miller JD. 1996. NMR structural studies of fumonisin B1 and related compounds from Fusarium moniliforme. Adv Exp Med Biol. 392:75–91.
- Bojja RS, Cerny RL, Proctor RH, Du L. 2004. Determining the biosynthetic sequence in the early steps of the fumonisin pathway by use of three gene-disruption mutants of Fusarium verticillioides. J Agric Food Chem. 52:2855–2860.
- Branham BE, Plattner RD. 1993. Alanine is a precursor in the biosynthesis of fumonisin B1 by Fusarium moniliforme. Mycopathologia. 124:99–104.
- Butchko RA, Plattner RD, Proctor RH. 2003. FUM13 encodes a short chain dehydrogenase/reductase required for C-3 carbonyl reduction during fumonisin biosynthesis in Gibberella moniliformis. J Agric Food Chem. 51:3000–3006.
- Butchko RA, Plattner RD, Proctor RH. 2006. Deletion analysis of FUM genes involved in tricarballylic ester formation during fumonisin biosynthesis. J Agric Food Chem. 54:9398–9404.
- Caldas ED, Sadilkova K, Ward BL, Jones AD, Winter CK, Gilchrist DG. 1998. Biosynthetic studies of fumonisin B1 and AAL Toxins. J Agric Food Chem. 46:4734–4743.
- Ding Y, Bojja RS, Du L. 2004. Fum3p, a 2-ketoglutarate-dependent dioxygenase required for C-5 hydroxylation of fumonisins in Fusarium verticillioides. Appl Environ Microbiol. 70:1931–1934.
- Du L, Zhu X, Gerber R, Huffman J, Lou L, Jorgenson J, Yu F, Zaleta-Rivera K, Wang Q. 2008. Biosynthesis of sphinganine-analog mycotoxins. J Ind Microbiol Biotechnol. 35:455–464.
- Gerber R, Lou L, Du L. 2009. A PLP-dependent polyketide chain releasing mechanism in the biosynthesis of mycotoxin fumonisins in Fusarium verticillioides. J Am Chem Soc. 131:3148–3149.
- Huffman J, Gerber R, Du L. 2010. Recent advancements in the biosynthetic mechanisms for polyketide-derived mycotoxins. Biopolymers. 93:764–776.
- Humpf HU, Schmelz EM, Meredith FI, Vesper H, Vales TR, Wang E, Menaldino DS, Liotta DC, Merrill Jr AH. 1998. Acylation of naturally occurring and synthetic 1-deoxysphinganines by ceramide synthase: formation of N-palmitoyl-aminopentol produces a toxic metabolite of hydrolyzed fumonisin, AP1, and a new category of ceramide synthase inhibitor. J Biol Chem. 273:19060–19064.
- Kallberg Y, Oppermann U, Jornvall H, Persson B. 2002. Short-chain dehydrogenases/reductases (SDRs). Eur J Biochem. 269:4409–4417.
- Klinman JP, Rose IA. 1971. Mechanism of aconitate isomerase reaction. Biochem. 10:2259–2266.
- Marasas WF, Riley RT, Hendricks KA, Stevens VL, Sadler TW, Gelineau-van Waes J, Missmer SA, Cabrera J, Torres O, Gelderblom WC, et al. 2004. Fumonisins disrupt sphingolipid metabolism, folate transport, and neural tube development in embryo culture and in vivo: a potential risk factor for human neural tube defects among populations consuming fumonisin-contaminated maize. J Nutr. 134:711–716.
- Miller JD. 2001. Factors that affect the occurrence of fumonisin. Environ Health Perspect. 109(Suppl. 2):321–324.
- Nelson PE, Desjardins AE, Plattner RD. 1993. Fumonisins, mycotoxins produced by Fusarium species: biology, chemistry, and significance. Annu Rev Phytopathol. 31:233–252.
- Plattner RD, Shackelford DD. 1992. Biosynthesis of labeled fumonisins in liquid cultures of Fusarium moniliforme. Mycopathol. 117:17–22.
- Proctor RH, Brown DW, Plattner RD, Desjardins AE. 2003. Co-expression of 15 contiguous genes delineates a fumonisin biosynthetic gene cluster in Gibberella moniliformis. Fungal Genet Biol. 38:237–249.
- Proctor RH, Desjardins AE, Plattner RD, Hohn TM. 1999. A polyketide synthase gene required for biosynthesis of fumonisin mycotoxins in Gibberella fujikuroi mating population A. Fungal Genet Biol. 27:100–112.
- Proctor RH, Plattner RD, Desjardins AE, Busman M, Butchko RA. 2006. Fumonisin production in the maize pathogen Fusarium verticillioides: genetic basis of naturally occurring chemical variation. J Agric Food Chem. 54:2424–2430.
- Rheeder JP, Marasas WF, Vismer HF. 2002. Production of fumonisin analogs by Fusarium species. Appl Environ Microbiol. 68:2101–2105.
- Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor (NY): Laboratory Press.
- Seefelder W, Humpf HU, Schwerdt G, Freudinger R, Gekle M. 2003. Induction of apoptosis in cultured human proximal tubule cells by fumonisins and fumonisin metabolites. Toxicol Appl Pharmacol. 192:146–153.
- Seibert V, Kourbatova EM, Golovleva LA, Schlomann M. 1998. Characterization of the maleylacetate reductase MacA of Rhodococcus opacus 1CP and evidence for the presence of an isofunctional enzyme. J Bacteriol. 180:3503–3508.
- Wang J, Zhou Y, Liu W, Zhu X, Du L, Wang Q. 2008. Fumonisin level in corn-based food and feed from Linxian County: a high-risk area for esophageal cancer in China. Food Chem. 106:241–246.
- Wang Q, Wang J, Yu F, Zhu X, Zaleta-Rivera K, Du L. 2006. Mycotoxin fumonisins: health impacts and biosynthetic mechanism. Prog Nat Sci. 16:7–15.
- Yi H, Bojja RS, Fu J, Du L. 2005. Direct evidence for the function of FUM13 in 3-ketoreduction of mycotoxin fumonisins in Fusarium verticillioides. J Agric Food Chem. 53:5456–5460.
- Yin JJ, Smith MJ, Eppley RM, Troy AL, Page SW, Sphon JA. 1996. Effects of fumonisin B1 and (hydrolyzed) fumonisin backbone AP1 on membranes: a spin-label study. Arch Biochem Biophys. 335:13–22.
- Zaleta-Rivera K, Xu C, Yu F, Butchko RA, Proctor RH, Hidalgo-Lara ME, Raza A, Dussault PH, Du L. 2006. A bidomain nonribosomal peptide synthetase encoded by FUM14 catalyzes the formation of tricarballylic esters in the biosynthesis of fumonisins. Biochem. 45:2561–2569.
- Zhu X, Yu F, Li XC, Du L. 2007. Production of dihydroisocoumarins in Fusarium verticillioides by swapping ketosynthase domain of the fungal iterative polyketide synthase Fum1p with that of lovastatin diketide synthase. J Am Chem Soc. 129:36–37.