Abstract
Lichens can either disperse sexually through fungal spores or asexually through vegetative propagules and fragmentation. Understanding how genetic variation in lichens is distributed across a landscape can be useful to infer dispersal and establishment events in space and time as well as the conditions needed for this establishment. Most studies have sampled lichens across large spatial distances on the order of hundreds of kilometers, while here we sequence the internal transcribed spacer (ITS) for 113 samples of three Peltigera species sampling at a variety of small spatial scales. The maximum distance between sampled lichens was 3.7 km and minimum distance was approximately 20 cm. We find significant amounts of genetic diversity across all three species. For P. praetextata, two out of the three most common ITS genotypes exhibit spatial autocorrelation supporting short-range dispersal. Using rarefaction we estimate that all ITS genotypes in our sampling area have been found for P. praetextata and P. evansiana, but not P. canina. Comparing our results with other ITS data in the literature provides evidence for global dispersal for at least one sequence followed by the evolution of endemic haplotypes with wide dispersal and rare haplotypes with more local dispersal.
Introduction
Landscape genetics combines the fields of population genetics and landscape ecology to consider the effect of landscape and geographical distances on the distribution and abundance of genetic diversity (Manel et al. Citation2003; Holderegger and Wagner Citation2008). Traditional evolutionary forces such as drift, gene flow and dispersal are considered within the larger geographical context through spatially explicit surveys and modeling (Storfer et al. Citation2007; Landguth and Cushman Citation2010; Storfer et al. Citation2010). The detection of such processes is strongly influenced by the scale of sampling (Cushman and Landguth Citation2010) and must match the size of the focal organism and what is known about its dispersal ecology (Wiens and Milne Citation1989; Thompson and McGarigal Citation2002). Smaller spatial regions have been shown to increase power under certain circumstances (Cushman and Landguth Citation2010), whereas multiple scales increase the ability to identify scale-specific processes (Galpern et al. Citation2012). However, the spatial distribution of genetic diversity has most often been studied at specific, larger spatial scales, ranging from regional to continental (Hirao and Kudo Citation2004; Walser et al. Citation2005; Werth et al. Citation2007; Gauffre et al. Citation2008; Soares et al. Citation2008; Murphy et al. Citation2010), with less focus on multiple scales, smaller scale patterns, and degrees of spatial autocorrelation (the degree to which observations are non-independent across space). Here we illustrate the value of investigating genetic patterns both at smaller and larger scales using lichens as a model.
Lichens are symbiotic organisms composed of fungi (mycobionts) associated with algae, and/or cyanobacteria (photobionts), an association that is required for the establishment of the lichen phenotype (Honegger Citation1998). The need to maintain or reconstitute this interaction after dispersal makes lichens an intriguing system to use in the study of gene flow (Werth Citation2010). Lichen mycobionts can disperse as spores, but then must acquire their symbiotic partners horizontally from the new location they colonize (Hedenas et al. Citation2007). Therefore, this type of dispersal is limited to substrates that contain the necessary photobionts. Lichens can also disperse asexually as soredia, isidia, or thallus fragments containing both mycobionts and photobionts (Dal Grande et al. Citation2012).
Lichen propagule dispersal can be empirically studied in two ways. First, traps can be set to catch both sexual and asexual propagules; however, it remains challenging to identify the species of the propagules (Werth et al. Citation2006a). Some studies have successfully used molecular approaches to identify the propagules (Walser et al. Citation2001; Werth et al. Citation2006a). Walser et al. (Citation2001) found evidence of dispersal only up to 50 m in Lobaria pulmonaria. Werth et al. (Citation2006a) found evidence of both local and long distance (>200 m) dispersal, although the calculated mean dispersal distance was 34 m. The problem with these distance estimates is that only dispersal is quantified and not the successful establishment necessary for gene flow to have actually occurred. The second approach is to sequence molecular markers from individuals at various distances, testing questions related to genetic autocorrelation, isolation by distance or by landscape features (Werth et al. Citation2006b, Citation2007). This approach allows for consideration of both dispersal and establishment. In L. pulmonaria evidence of autocorrelation has been found at scales from 50 to 150 m largely attributable to asexual reproduction (Werth et al. Citation2006b).
Landscape genetics research on lichens has been conducted at multiple scales only in L. pulmonaria (Walser et al. Citation2005; Werth et al. Citation2006a, Citation2006b, Citation2007). In the present study we focus on advancing the small spatial scale work with genetic diversity characterization of three species in the genus Peltigera. Specifically we ask: (1) What is the genetic diversity and degree of relatedness within P. praetextata, P. evansiana, and P. canina at the study sites for the ribosomal ITS Locus, and how does it compare to worldwide diversity? (2) What is the amount of sampling effort required to fully sample the genetic diversity of the three species at the ITS locus? (3) Is there evidence of spatial autocorrelation between the same genotypes in P. praetextata? (4) Are particular genotypes correlated with certain substrates in P. praetextata?
Methods
Sampling
Sampling was conducted from February 2011 to May 2012 at two field sites 113 km northeast of Toronto near Coboconk ON, Canada (44o64′47′′N, 78o98′96′′W) () and ()). These sites are minimally 3.1 km apart with the farthest lichens being 3.7 km apart. Two sites were chosen to have a range of distances between samples. All sites are located on an ecotone between limestone plain (alvar) and the start of the Canadian Shield (granite). This environment contains a series of limestone ridges, granite erratics, granite ridges, mixed-wood forest, and dead wood. Peltigera spp. were exclusively found on moss-covered limestone, granite, and dead wood within mixed-wood forests. These locations function as island-like habitats in a matrix of habitats rendered unsuitable due to yearly leaf deposition which would block the lichens’ access to sunlight. This creates a system ideal for the study of colonization and landscape genetics at small spatial scales. Sites in two regions of 1.3 km2 and 0.7 km2 (3.1 km apart) were tagged and sampled. GPS lat/long coordinates were recorded at each sample location using a Garmin Oregon Series GPS unit (model 550 software 3.70) and analyzed using Basecamp software (Version 2.1.2). One lichen thallus was sampled at each chosen location. Efforts were made to sample young tissue at lobe tips to obtain the DNA from healthy dividing cells.
Figure 1. Map of ITS genotypes successfully sampled for P. praetextata (prae), P. evansiana, and P. canina. (a) In the main sampling site, sampling was concentrated along three limestone ridges containing suitable habitat with some granite erratics. (b) The additional sampling site was located 3.1–3.7 km from the first site. Sampling was conducted along a limestone cliff face at the edge of an alvar and on several granite ridges.
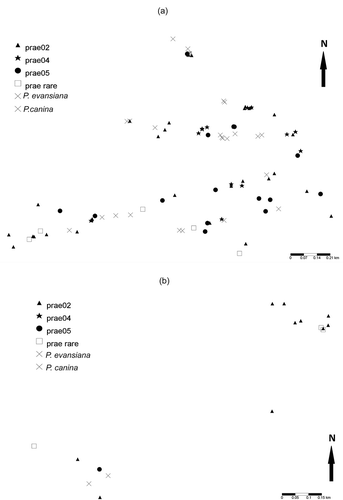
Four P. praetextata samples were initially taken from three locations in the first site and one from the second site. As these samples included three genotypes, one which had not been previously described, a more thorough study was then undertaken. The majority of sampling was carried out by conducting transects along each limestone ridge and habitat where Peltigera sp. was found. Thalli were systematically sampled every 50 m when possible or at longer distances if a thallus could not be located after 50 m. To give a greater range of distances between thalli, samples were taken at shorter spatial scales (1 m–10 m away) around randomly selected thalli. In total we collected 251 samples. However, we were able to extract viable DNA and successfully sequence only 118 samples. While this does introduce heterogeneity into our sampling design, we have no reason to suspect that successfully sequenced samples are biased toward any species or ITS genotype. Of these samples nine were species other than our three focal species (three Peltigera neckerii, three Peltigera ponogensis, one Peltigera neopolydactylon, one Peltigera didactyla, and one Peltigera rufescens). This leaves 109 samples of our three focal species (). We also collected and sequenced one sample from New Hampshire, USA and four samples from Northern Ontario, Canada.
Table 1. Number ITS genotypes found for each of the three Peltigera species present at the main field sites and at outlying locations.
Genetic diversity
DNA was extracted from each sample using Fermentas GeneJET genomic DNA purification kits with gram-negative bacteria DNA purification protocol included in the kit. The protocol was modified by removing the bacteria-specific harvest step. The 600 bp ITS region, was PCR amplified using forward (ITS1F, Gardes and Bruns Citation1993) and reverse primers (ITS4, White et al. Citation1990). PCR was carried out using BIO-RAD iProof high-fidelity PCR kit on one cycle at 98°C for 30 s; 35 cycles of 98°C for 10 s, 53°C for 35 s, and 72°C for 30 s; and one cycle at 72°C for 10 min. Enzymatic cleanup was conducted on each sample using 0.2 µl each of Exonuclease I (Fermentas) and Calf Intestinal Alkaline Phosphatase (Biolabs) per sample with one cycle at 37°C for 30 min, and one cycle at 85°C for 15 min. Sequencing was carried out using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) on one cycle at 96°C for 1 min; 55 cycles at 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min; and one cycle at 60°C for 4 min. Samples were sequenced using an Applied Biosystems 3730 DNA Analyzer (San Francisco, CA, USA) at the Centre for the Analysis of Genome Evolution and Function (CAGEF). Sequences were assembled and edited using CLC Genomics Work Bench (version 5.5) and Sequencher (4.2, Ann Arbor, MI). All other ITS sequences in GenBank, for the species found at our sampling sites, were identified using BLAST (Altschul et al. Citation1997) and aligned to our sequences to infer haplotype networks using CLC Genomics Work Bench, including gaps as single mutations.
Statistical analysis
Rarefaction analysis is commonly used in community ecology to compare species richness to sampling effort (Simberloff Citation1978). In this paper, we compare genetic richness of the ITS locus to sampling effort. We conducted individual-based rarefaction and extrapolation (Colwell et al. Citation2012) to compare between genetic richness accumulation curves. In addition to traditional richness, a Chao 1 richness estimator was used to correct for rare allele bias (Gimaret-Carpentier et al. Citation1998) using: Sobs + a2/2b, where Sobs is the number of alleles observed in the sample, a and b are the number of alleles present once or twice in the species-specific data set. Analyses for richness and Chao 1 were conducted using EstimateS (ver 9.1.0).
Peltigera praetextata was selected for two further analyses because it was by far the most common Peltigera species in our field sites, allowing for greater statistical power in spatial analyses. First, we tested if randomness alone accounts for spatial adjacency of alleles of P. praetextata using joint-count simulation analysis (Fortin and Dale Citation2005) in PASSaGE (ver 2). Rather than assuming full connectivity between all adjacent individuals, we model spatial adjacency as a Gabriel graph (Fortin and Dale Citation2005), effectively capturing the closest linkages between samples. Contrasts A/A, A/B, A/not A were all run using inverse square distance and 10,000 permutations. Second, we tested if common P. praetextata genotypes (prae02, prae04, or prae05) were correlated with type of substrate (limestone, granite, or wood). This analysis was carried out with a 3 × 3 chi-squared contingency table with substrate on one axis and genotype on the other. We compared observed values against an expectation that alleles were equally likely to be found on each substrate type keeping the number of alleles and number of substrates constant. We chose not to analyze rare alleles because of a lack of statistical power. We were only able to include a subsection of the sampled genotypes, because the substrate was not recorded for one prae02 sample, seven prae04 samples, and four prae05 samples.
Results
Genetic diversity
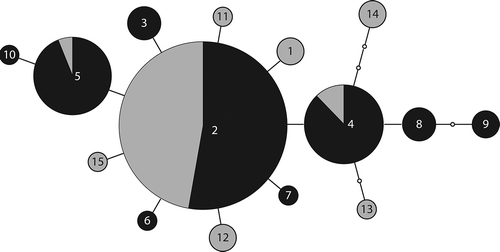
Contrary to the null hypothesis of each Peltigera species being clonal at small spatial scales, nine P. praetextata genotypes (eight of which are new to the literature), four genotypes for P. evansiana (three new) and five for P. canina (two new) () were found. For P. praetextata, ITS genotypes are mostly closely related to Prae02 (). Prae02, the most commonly sampled haplotype in our sample sites, is also the most commonly sampled haplotype in western Canada and in Europe and has also been sampled in China. Two other common haplotypes are derived from prae02 by a single mutation each. One of these haplotypes was also found in a single specimen in northern Ontario (prae05), while the other has also been sampled in New Hampshire and Japan (prae04). Twelve other haplotypes are derived from these three common haplotypes by one to three mutations and each are restricted to single localities, six of them from our sampling sites and the others from British Columbia, Europe, and Korea.
Figure 2. Mutational steps among ITS P. praetextata genotypes. Numbers in each node represent the numbered P. praetextata genotypes (e.g. 2 = prae02). Edges represent one transition; small nodes without numbers represent predicted, unsampled genotypes. Sizes of circles indicate the number of samples with that haplotype (ranging from 1 to 14). Dark shading indicates samples collected at our field sites while light shading indicates samples collected elsewhere (see for details).
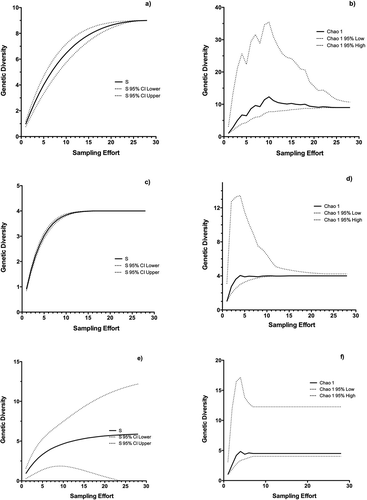
Peltigera evansiana also had one common haplotype that was shared with another location (British Columbia), while other haplotypes were endemic to our sampling location and derived from the common haplotype by a single mutation each (). While P. canina also had a putatively common ancestral haplotype that was shared with other locations (British Columbia and Europe), most samples from other localities clustered into three other lineages that branched off from hypothesized unsampled haplotypes (). One of these lineages was comprised of specimens that have been described as a distinct species, P. koponenii (Sérusiaux et al. Citation2009). Based on rarefaction analysis, we have likely found most if not all the genetic diversity for P. praetextata ((a) and (b)) and P. evansiana () and ()). There is still much uncertainty present for P. canina ((e) and (f)).
Figure 3. Mutational steps among ITS P. evansiana genotypes. See legend.
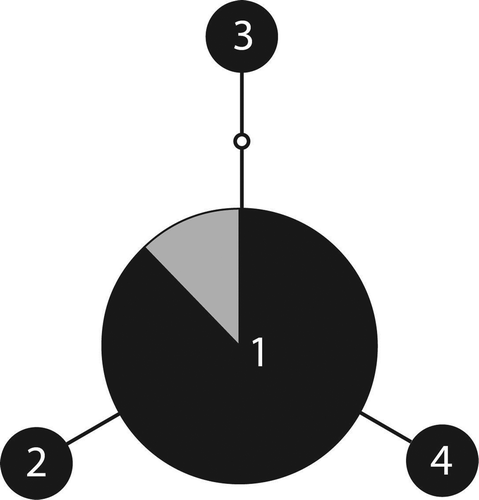
Figure 4. Mutational steps among ITS P. canina genotypes. See legend.
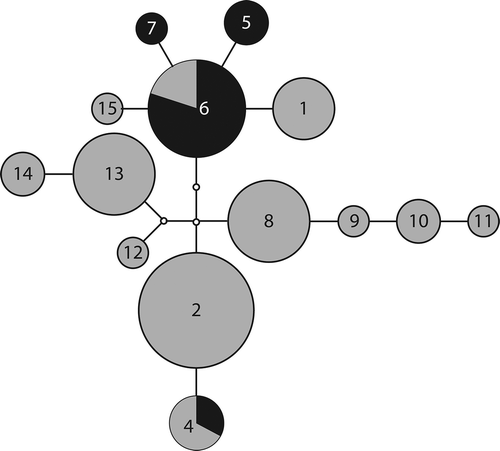
Figure 5. Rarefaction curves with extrapolation for (a), (b) P. praetextata, (c), (d) P. evansiana, (e), (f) P. canina. Species richness (S) is plotted against sampling effort on the left (a), (c), (e), while Chao1 is plotted on the right (b), (d), (f). Dotted lines represent 95% confidence intervals.
Spatial autocorrelation
In the genotype self-comparisons (A/A), we find evidence from joint count analysis that prae02 and prae04 are each autocorrelated (random probability, p = 0.0001). No other combinations give results with less than a 0.05 random probability (). This may suggest that prae02 and prae04 have shorter dispersal distances than is expected by chance alone. However, for prae05, which is as abundant as prae04, autocorrelation is not different from chance.
Table 2. Joint-count test and random probability values for P. praetextata for all comparisons. “Rare” refers to all other P. praetextata genotypes that are not prae02,04, or 05.
Substrate
Substrate analysis reveals that although the three most common alleles were found on each of the substrates, prae02 and prae05 genotypes seem to be associated with limestone substrates, while prae04 genotypes are associated with wood substrate (p = 0.046; ).
Table 3. Observed and expected number of P. praetextata genotypes present on each substrate.
Discussion
Spatial dispersal
Joint count analysis reveals evidence of autocorrelation in two common genotypes of ITS. Such results are mirrored in maps of ITS genotypes, where prae02 and prae04 seem to be in closer proximity than what would be expected due to chance alone () and ()). These results provide evidence that at least a significant portion of dispersal seems to be local (<100 m). This estimate is similar to previous estimates of dispersal distances based on landscape level sampling of L. pulmonaria (Werth et al. Citation2006b). These results could also be due to the fact that certain genotypes prefer certain types of habitat.
We found evidence that certain genotypes were associated with particular substrates. This matches previous findings that substrate can be very important in the establishment process (Werth et al. Citation2006a). However, there are significant levels of spatial autocorrelation among genotypes of the same species. This may violate the chi-squared test’s assumptions of independence of samples. Therefore, more sampling at less geographically correlated locations would be ideal to further investigate the question of substrate associations.
Biogeography
We found much higher diversity for ITS than we expected given previous work on Peltigera (Brodo et al. Citation2001). Specifically, we detected many ITS genotypes in all three Peltigera species that were not found in previous work on this system (O’Brien et al. Citation2009; Fedrowitz et al. Citation2011; Kaasalainen et al. Citation2013). While working with one 600 bp genetic region greatly underestimates the total diversity that may be present, it has revealed lineage relationships which may be exploited for studies at larger spatial scales. For two of the species sampled here, we found that ancestral common haplotypes were shared with samples in British Columbia, as well as Europe in two cases and China in one, with many rare haplotypes that are one or two mutational steps from the common haplotype. This pattern of high haplotype diversity and low nucleotide diversity is a signature of recent range expansion from small founder populations (Avise Citation2000). If continental or intercontinental dispersal is ongoing, it must be rare because the other common P. praetextata haplotypes in our sample sites have only been found at other sites within the same region (with the exception of a single sample from Japan). Similarly, work conducted on L. pulmonaria found significant genetic differentiation between continents also suggesting that long distance dispersal is rare (Walser et al. Citation2005). The situation with P. canina is somewhat more complicated, with four distinct geographically limited lineages. However, 11 of 12 samples from our sites belonged to a single lineage which exhibited the same signature of population expansion as the other two species. In addition to biogeographical inferences, it would be desirable to make inferences concerning time periods of dispersion. Considering the northern ranges of these species we suspect most of the variation we observed occurred in postglacial expansion (Hewitt Citation2000) from a single refugial population, but testing such a hypothesis is beyond the scope of our present data. This system could be useful for testing long-standing biogeographic questions regarding why lichen distributions are large but often have low density within their range (Werth Citation2011), as well as possible dispersal from glacial refugia after the last ice age and global landscape genetic patterns of Peltigera.
Sampling
Rarefaction and extrapolation analysis is more common in community ecology. Here we use it in determining the sampling effort needed to find all genetic diversity present in lichen species at small spatial scales. Our rarefaction curves saturate between 10 and 20 samples for P. evansiana and P. praetextata; however, while the curve for P. canina does saturate, uncertainty still remains high because we were able to sample only 12 individuals. Our results do not give a definite estimate of sample numbers needed to capture most of the diversity in one lichen species for one genetic region, as this can be largely species dependent. However, once some sampling has been conducted, rarefaction can inform the researcher if more sampling is required. This could be quite useful in landscape genetics studies, where sampling across large spatial regions can be time consuming and costly.
Conclusion
Overall we have provided the first landscape genetics work on a system that could be useful to study the distribution and abundance of genetic variation at small and larger spatial scales. We show spatial autocorrelation for certain P. praetextata genotypes and have found variation that appears to be phylogenetically related with distributions at large spatial scales. Those sequences that appear to be several mutational steps from the presumed ancestor are also the rarest. There is potential for a larger scale landscape genetic study that could help test the hypothesis of a range expansion after the most recent glaciation and also help distinguish between present day dispersal limitation and recent range contraction. As well, we recommend considering more genes for more fine scale genetic resolution and even moving to genome scan studies using next generation sequencing techniques such as RAD tag (Baird et al. Citation2008) and genotyping by sequencing (Elshire et al. Citation2011). When studied across an environmental gradient such work could lead to identification of adaptive loci.
Acknowledgments
We thank Sayed Khan for help with field sampling. We especially thank Andy Hon and Zhen Li for invaluable technical work and suggestions throughout DNA extraction, PCR, and sequencing processes. Sequencing work was conducted by the staff at the CAGEF, University of Toronto, who are also thanked for prompt sequencing analysis and helpful suggestions.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 25(17):3389–3402.
- Avise JC. 2000. Phylogeography: the history and formation of species. Cambridge (MA): Harvard University Press.
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, CreskoW A, Johnson EA. 2008. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 3(10):e3376.
- Brodo IM, Sharnoff SD, Sharnoff S. 2001. Lichens of North America. Yale (CN): Yale University Press.
- Colwell RK, Chao A, Gotelli NJ, Lin SY, Mao CX, Chazdon RL, Longino JT. 2012. Models and estimators linking individual-based and sample-based rarefaction, extrapolation and comparison of assemblages. J Plant Ecol. 5(1):3–21.
- Cushman SA, Landguth EL. 2010. Scale dependent inference in landscape genetics. Landsc Ecol. 25(6):967–979.
- Dal Grande F, Widmer I, Wagner HH, Scheidegger C. 2012. Vertical and horizontal photobiont transmission within populations of a lichen symbiosis. Mol Ecol. 21(13):3159–3172.
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. 2011. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 6(5):e19379.
- Fedrowitz K, Kaasalainen U, Rikkinen J. 2011. Genotype variability of Nostoc symbionts associated with three epiphytic Nephroma species in a boreal forest landscape. Bryologist. 114(1):220–230.
- Fortin M-J, Dale M. 2005. Spatial analysis: a guide for ecologists. Cambridge (MA): Cambridge University Press.
- Galpern P, Manseau M, Wilson P. 2012. Grains of connectivity: analysis at multiple spatial scales in landscape genetics. Mol Ecol. 21(16):3996–4009.
- Gardes M, Bruns TD. 1993. ITS primers with enhanced specificity for basidiomycetes‚ application to the identification of mycorrhizae and rusts. Mol Ecol. 2(2):113–118.
- Gauffre B, Estoup A, Bretagnolle V, Cosson JF. 2008. Spatial genetic structure of a small rodent in a heterogeneous landscape. Mol Ecol. 17(21):4619–4629.
- Gimaret-Carpentier C, Pélissier R, Pascal J-P, Houllier F. 1998. Sampling strategies for the assessment of tree species diversity. J Veg Sci. 9(2):161–172.
- Hedenas H, Blomberg P, Ericson L. 2007. Significance of old aspen (Populus tremula) trees for the occurrence of lichen photobionts. Biol Conserv. 135(3):380–387.
- Hewitt G. 2000. The genetic legacy of the Quaternary ice ages. Nature. 405(6789):907–913.
- Hirao AS, Kudo G. 2004. Landscape genetics of alpine-snowbed plants: comparisons along geographic and snowmelt gradients. Heredity. 93(3):290–298.
- Holderegger R, Wagner HH. 2008. Landscape genetics. Bio Sci. 58(3):199–207.
- Honegger R. 1998. The lichen symbiosis – what is so spectacular about it? Lichenologist. 30(3):193–212.
- Kaasalainen U, Fewer DP, Jokela J, Wahlsten M, Sivonen K, Rikkinen J. 2013. Lichen species identity and diversity of cyanobacterial toxins in symbiosis. New Phytol. 198(3):647–651.
- Landguth EL, Cushman SA. 2010. cdpop: a spatially explicit cost distance population genetics program. Mol Ecol Resour. 10(1):156–161.
- Manel S, Schwartz MK, Luikart G, Taberlet P. 2003. Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol. 18(4):189–197.
- Murphy MA, Evans JS, Storfer A. 2010. Quantifying Bufo boreas connectivity in Yellowstone National Park with landscape genetics. Ecology. 91(1):252–261.
- O’Brien HE, Miadlikowska J, Lutzoni F. 2009. Assessing reproductive isolation in highly diverse communities of the lichen-forming fungal genus Peltigera. Evolution. 63(8):2076–2086.
- Sérusiaux E, Goffinet B, Miadlikowska J, Vitikainen VO. 2009. Taxonomy, phylogeny and biogeography of the the lichen genus Peltigera in Papua New Guinea. Fungal Divers. 38(1):185--224.
- Simberloff D. 1978. Use of rarefaction and related methods in ecology. In: Wheeler JB, Hoersch HM, McGlinchey EJ, Pulver SG, editors. Biological data in water pollution assessment: quantitative and statistical analyses. Philadelphia (PA): American Society for Testing and Materials. p. 150–165.
- Soares TN, Chaves LJ, de Campos Telles MP, Diniz-Filho JA, Resende LV. 2008. Landscape conservation genetics of Dipteryx alata (“baru” tree: Fabaceae) from Cerrado region of central Brazil. Genetica. 132(1):9–19.
- Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits LP. 2007. Putting the “landscape” in landscape genetics. Heredity. 98(3):128–142.
- Storfer A, Murphy MA, Spear SF, Holderegger R, Waits LP. 2010. Landscape genetics: where are we now? Mol Ecol. 19(17):3496–3514.
- Thompson CM, McGarigal K. 2002. The influence of research scale on bald eagle habitat selection along the lower Hudson River, New York (USA). Landsc Ecol. 17(6):569–586.
- Walser J, Zoller S, Büchler U, Scheidegger C. 2001. Species-specific detection of Lobaria pulmonaria (lichenized ascomycete) diaspores in litter samples trapped in snow cover. Mol Ecol. 10(9):2129–2138.
- Walser JC, Holderegger R, Gugerli F, Hoebee SE, Scheidegger C. 2005. Microsatellites reveal regional population differentiation and isolation in Lobaria pulmonaria, an epiphytic lichen. Mol Ecol. 14(2):457–467.
- Werth S. 2010. Population genetics of lichen-forming fungi – a review. Lichenologist. 42(5):499–519.
- Werth S. 2011. Biogeography and phylogeography of lichen fungi and their photobionts. In: Fontaneto D, editor. Biogeography of microscopic organisms: is everything small everywhere? Cambridge (UK): Cambridge University Press. p. 191–208.
- Werth S, Gugerli F, Holderegger R, Wagner HH, Csencsics D, Scheidegger C. 2007. Landscape-level gene flow in Lobaria pulmonaria, an epiphytic lichen. Mol Ecol. 16(8):2807–2815.
- Werth S, Wagner HH, Gugerli F, Holderegger R, Csencsics D, Kalwij JM, Scheidegger C. 2006a. Quantifying dispersal and establishment limitation in a population of an epiphytic lichen. Ecology. 87(8):2037–2046.
- Werth S, Wagner HH, Holderegger R, Kalwij JM, Scheidegger C. 2006b. Effect of disturbances on the genetic diversity of an old-forest associated lichen. Mol Ecol. 15(4):911–921.
- White TJ, Bruns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. San Diego (CA): Academic Press. p. 315–322.
- Wiens JA, Milne BT. 1989. Scaling of landscapes in landscape ecology, or, landscape ecology from a beetle’s perspective. Landsc Ecol. 3(2):87–96.