
ABSTRACT
Cellular transcriptional programs driven by genetic and epigenetic mechanisms could be better understood by integrating “omics” data and subsequently modeling the gene-regulatory events. Toward this end, computational biology should keep pace with evolving experimental procedures and data availability. This article gives an exemplified account of the current computational challenges in molecular biology.
Introduction
Genetic and epigenetic mechanisms act together to organize the fine-tuned gene regulatory circuitries underlying cell-type identities and functions.Citation1,2 Misregulation of these pathways has been associated with diseases. Genetic variations are associated with a multitude of disorders, including developmental defects and cancers.Citation3 Sequence-dependent targeting of the critical transcription factors at enhancers is required to generate a gene expression program underlying cell-type specification and function.Citation4,5 In line with this need, mutations in regulatory elements such as enhancers have been shown to disrupt their ability to bind host transcription factors, leading to misregulation of gene expression.Citation6 With the advent of “epigenetics”, a number of mechanisms have been identified that regulate gene expression without influencing the genetic sequence. These mechanisms include the modification of histones, DNA methylation, and non-coding RNA, which modulate the chromatin structure and alter transcriptional outcomes.Citation1 The study of these epigenetic mechanisms at the genome-wide level in a high-throughput fashion is termed “Epigenomics”.Citation6
The arrival of next-generation sequencing technology led to many critical discoveries in multiple branches of the life sciences by providing a comprehensive overview of the genomic, transcriptomic, and epigenomic maps of cell types of interest.Citation7 These assays have facilitated the discovery of novel gene regulatory features.Citation8 These methods can be implemented toward (1) obtaining gene expression profiles (RNA-seq), (2) interrogating chromatin and transcription factor landscapes (ChIP-seq), (3) reconstructing chromatin structures and long-range chromosomal interactions (e.g., ATAC-seq, DNAse-seq,. and HiC), and (4) uncovering the methylation and other modification states of DNA (e.g., MEDIP-seq and BiS-seq). In parallel, the evolution of proteomic tools has facilitated discoveries in the gene regulation field, including the identification of the differentially expressed proteins and protein–protein interactions involved in transcriptional regulation and the discovery of novel histone modifications and their modifiers, readers, and erasers.Citation9 Computational tools for the analysis of both genomics and proteomics datasets are increasingly advancing and routinely updated.Citation9,10
These advances in the large data acquisition have also invited challenges that demand active systematic efforts to tackle them. In any cell at a given time point, a large number of transcription factors are functioning in concert with one another and the chromatin to guide the gene expression program underlying cell identity and functions.Citation11 Given the limitations of the current experimental measures, it is not possible to fully explore these dimensions. However, advanced bioinformatics and systems biology tools offer the opportunity to gain better insights into the complexity of gene regulatory events and to generate comprehensive quantitative models of these networks. Furthermore, there is a growing need to develop algorithms that integrate data sets from various “omics” studies, such as genomics and proteomics experiments, to improve the quantitative models of gene regulatory networks by integrating events, including protein–protein interactions and post-transcriptional (RNA) and translational modifications (protein). In the wake of the increasing data availability, there is a need to develop improved methods for analysis that take into account pre-existing information to allow deeper biological insights in the analysis.Citation12 Integration and making the data availability process smoother with a one-point entry access remain important challenges.Citation13 This article outlines critical and immediate computational challenges and reports possible measures to overcome them with a special focus on gene regulation.
Challenges in molecular systems biology
Modeling of gene regulatory networks
Comprehensive and integrative modeling of gene regulatory networks is one of the key challenges in current molecular systems biology. Toward this end, transcription factor binding preferences and the effects of binding on chromatin and the gene regulatory network mediated by these factors need to be understood.
Systematic prediction of transcription factor binding sites
DNA binding factors can function both as activators and repressors of transcription, and their dynamics at various loci determine the ultimate transcriptional response. A set of transcription factors have also been described as so-called “pioneer factors” that can bind closed chromatin regulatory regions and induce subsequent activities to create accessible and active chromatin landscapes, thereby increasing gene expression.Citation14 By contrast, settler factors function on genomic loci made accessible by pioneer factors to drive subsequent transcriptional changes.Citation14
Most transcription factors bind in a highly sequence-specific manner, and the consensus sequence (i.e., the motif) of these factors is unique to them or their families.Citation15 Interestingly, although motif sequences appear numerous times in the genome, only a small fraction of these motifs are actually utilized. Motif analysis can be complemented with the analysis of secondary DNA structures and sequential features in the binding sites and flanking regions to considerably increase accuracy of transcription factor binding prediction.Citation16,17 Thus, efforts should be made in these directions to improve the computational methods and allow predictions of transcription factor binding with high accuracy (). Furthermore, the chromatin state of the target sites should be added as an additional feature in this analysis. This analysis would allow the cell-type specific binding patterns of transcription factors to be deciphered.Citation18
Figure 1. Computational modeling of cell type-specific transcription factor binding sites. Step 1: Construction of a database of classification models for every transcription factors by modelling features of bound and unbound motifs. For the flanking regions of these motifs, information is extracted from DNA structure (DNAshape), sequence features, and chromatin state dynamics to build the classifying model (i.e., SVM, RBF, or Regression). If the CV accuracy of the classification model is greater than 75% (arbitrary cutoff), the model is retained in the database. Step 2: Utilize the constructed model database to predict cell type-specific binding: for a set of genomic regions of interest, information is collected from DNAshape, sequence features, and chromatin states. Every classification model in the database is scanned. The factor for which the AUC is higher potentially binds these genomic regions. Thus, factors for which the AUC scores are higher are reported.
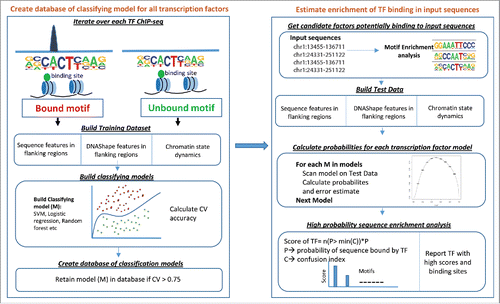
Classifying transcription factors as pioneers and settlers is very important because although both of these factors may bind similar target sites, they function via different mechanisms and for different purposes. Recently developed algorithms such as PIQCitation14 represent great advances in recognizing transcription factor binding sites and identifying pioneer-settler pairs by modeling the shape and the magnitude of chromatin accessibility at factor binding sites. These binding events can be validated using a ChIP-seq assay and used to calculate the accuracy of the model in an unbiased manner.Citation19
Deciphering transcriptional responsiveness to transcription factor binding
An expected consequence of transcription factor binding is the transcriptional regulation of target genes. However, it has been extremely difficult to determine the actual transcriptional response of a transcription factor binding to its target site in the native environment. The reasons for such difficulty are multifold; first, analysts face the challenge of assigning a target gene with a transcription factor binding event. In today's genomics standards, in cases when the binding of a factor occurs at a promoter, it is assumed that the associated gene is regulated. However, if the binding of the factor is distal, then in most cases the nearest gene is considered to be regulated, which generates noise in the computational analysis.Citation20 In such cases, researchers would benefit from the availability of genome-wide maps of long-range chromosomal maps (e.g., Hi-C) and enhancer marks (e.g., H3K27ac) for the same cell type. A recently developed machine learning-implemented computational method (TargetFinder) predicts potential target genes of distal factor binding based on a range of features learned from the Hi-C data.Citation21 A second issue is that the levels of binding do not correlate well with the actual transcriptional response even when the real target genes are deciphered.Citation22 Emerging evidence suggests that both the DNA binding and transcriptional responses are a function of the DNA sequence and structure as well as the epigenetic state.Citation23 Therefore, the current big issue in quantitative genomics is to decipher the determinants of transcriptional factor targeting and transcriptional output.
Mathematical modeling of gene regulatory networks
With the identification of genome-wide targets of a given transcription factor and its influence on gene expression, one ultimately aims to generate a quantitative model of the gene regulatory networks underlying cell identity or functions. The reconstruction of gene regulatory networks from these minimal information datasets remains a challenging task. Methods for this systems biological characterization have evolved in recent years from traditional co-expression networks to dynamic modeling approaches.Citation24
Reconstruction of networks based on gene expression profiles
Co-expression networks are reconstructed on the assumption that genes that are temporally co-expressed are functionally related and are regulating the transcriptional state of genes that respond sequentially thereafter. Popular tools such as WGCNA use this principle to reconstruct networks.Citation25 Conversely, ARACNE also reconstructs gene regulatory network based on gene expression profiles but removes indirect nodes in the network using data processing inequality (DPI) to obtain results that are close to real networks.Citation26
Although this model is helpful for the prediction of gene functions, it does not provide resolution of singular gene regulatory events.Citation27
Combination of binding and transcriptional readout factor-based methods
Popular tools such as GeneProf and EpiRegNetCitation28,29 are based on studies in which the binding of a transcription factor/epigenetic regulator and/or the transcriptional response and/or the co-expression in a time course data set can be integrated to elucidate sequential networks. Because these methods employ co-expression or information from a ChIP-sequencing or motif analysis, a combinatorial response of multiple active transcription factors cannot be determined.
Mathematical modeling integrating biological knowledge
The knowledge-based modeling protocol is the most robust approach to reconstruct realistic gene regulatory events. Biological knowledge can be integrated into a mathematical model that will provide a measure of accuracy to the reconstruction of the network.Citation30,31 For example, if the binding sites of a transcription factor (X) target (Y) are deciphered by either ChIP-seq or prediction and the target exhibits significantly changed expression during the progression of a particular biological process, then the network node X–Y can be reconstructed. The summation of all nodes for all active transcription factors can be modeled using a probabilistic approach, such as Bayesian modeling. A similar approach is implemented in MARA (Motif Activity Response Analysis); however, this approach is limited to investigating promoter regions and offers no possibility of integrating ChIP-seq and RNA-seq-derived information.Citation32
Therefore, given the current lack of standard methods to reconstruct gene regulatory networks, it is essential to develop methods that integrate information from ChIP sequencing and RNA sequencing experiments in combination with motif energies to reconstruct gene regulatory events.
Integration of multiple level datasets
Most biological processes involve a combinatorial contribution of transcriptional regulation, alternative splicing, post-translational protein modification, and complex protein–protein interactions. Thus, to deeply understand a biological process it is necessary to integrate data sets spanning these readouts, including transcriptomics, epigenomics, and proteomics.Citation33
Integration of datasets arising from similar biological contexts
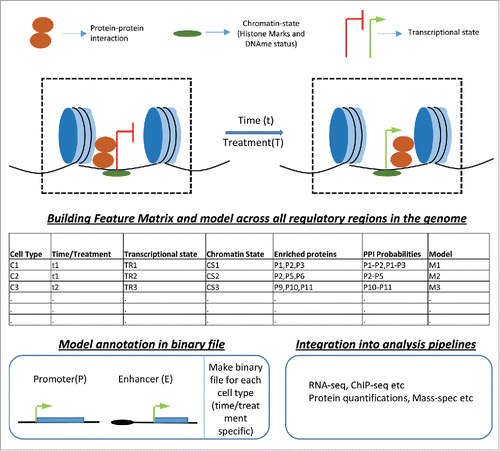
A range of data sets exist for cell types that have been extensively studied, such as ES cells. For these cell types, ChIP-seq for all available transcription factors and histone modifications as well as DNA methylation maps could be integrated to generate a comprehensive overview of the genome-wide and locus-wide transcription factor, epigenetic and transcriptional landscapes. These datasets have proven very useful for obtaining a better understanding of how a transcription factor communicates with different flavors of regulatory factors and chromatin feature combinations at distinct loci.Citation34 Furthermore, genetic data sets, such as DNA structures, the sequence conservation index and sequence patterns, can be integrated. All of this information could be made available in a cell type-specific binary file, which could be used for subsequent genomics studies ().
Figure 2. Integration of multilevel datasets. For every regulatory region (i.e., promoter and enhancer) in any cell type, data from different experiments, such as protein–protein interaction, chromatin state and transcription state in a temporal (t)/treatment (T)-specific manner, should be recorded in files. These files should be integrated in every genomic analysis to provide regulatory events enriched at the sites critical for a given analysis.
Challenges for the integration of multilevel datasets
Many attempts have been made to comprehensively collect and integrate multiple data types for a particular cell type and process. For example, the TCGA (The Cancer Genome Atlas project) is aimed toward the integration of the quantification of mRNA, miRNA, protein, DNA methylation, copy number alterations, and somatic chromosomal aberrations in cancer cells.Citation35 For this integrative analysis, it is necessary to implement careful statistical analysis due to the challenges in normalizing quantitative data from different experiments. Recently, penalized regression models were used to integrate “omics” data from three sources (genetic variations, DNA methylation, and gene expression) to reveal genes that contributed to bladder cancer.Citation36 Attempts have also been made to implement network-based approaches that integrate protein–DNA and protein–protein interactions.Citation37 The recently published tool “Network Analyst” also uses a network model to integrate differential gene expression data with protein–protein interaction maps.Citation38 However, because the techniques and their readouts are not directly comparable due to their different sensitivities and varied dynamic ranges, it has been difficult to generate a comprehensive computational model for the regulation of gene expression.Citation39 Therefore, it is necessary to develop robust statistical procedures capable of integrating cell type-specific omics data and to construct a database of these models to facilitate integrative research.
Optimizing current tools and coping with data availability
An additional computational challenge in large-scale genomics studies is the need to routinely update the procedures used to analyze the genomic data. For instance, peak calling is a critical step in the analysis of ChIP-seq data sets in which there is a need for standardized methods that ideally should take into account several parameters, such as differential peak profiles, abundance of the factor/mark on chromatin, and antibody affinity. Methods to sensitively incorporate these inputs will markedly improve the information gain from genomics datasets. Additionally, better alignment and analysis tools need to be developed to analyze the repetitive DNA sequences that constitute a large part of the genome.Citation40 Furthermore, different laboratories use different tools to analyze similar data sets, which makes it harder to perform comparative genomics. In summary, the genomics community faces critical challenges in light of the absence of strict standards for experimental and computational procedures, which can cause severe problems in the proper interpretation of large-scale data and the reproducibility of the findings.Citation41
Recently, single-cell genomics has become an extremely powerful tool to identify distinct cell populations based on distinct expression and/or chromatin profiles. In addition to single-cell RNA-seq, it is possible to perform ATAC-seq to interrogate chromatin accessibility and Hi-C to analyze higher order chromatin structures at the single-cell level.Citation42 These data are generally very noisy and require a large number of replicates to reach conclusions. Thus, integrating “omics” data from single cells remains a challenge for varied reasons, including heterogeneity and complicated experimental and computational methods. A number of laboratories are investing in the development of tools to improve data analysis and interpretation from these datasets.Citation43
An important discussion in recent years has been how to handle the accumulating large-scale data sets more efficiently, and efforts have been made to create one point entry for all of these databases. Many databases, such as CISTROME, ENCODE, and NCBI epigenomics, have been made available. Furthermore, consortia such as STATegra are actively engaged in developing methods for the integration of “omics” data.Citation44 However, many of the databases are not updated on a real-time basis, which pushes researchers to manually search for the required datasets. These databases should also encourage the integrated analysis of existing data sets with an easy workflow.
Conclusion
With the growth of experimental procedures for the high-throughput mapping of molecular events as well as increasing data availability and computational resources, it is necessary for computational biologists to put systematic efforts into quality checks, the development of robust but standard analytical tools, the integration of all existing datasets and the development of predictive models that make the best use of the existing genomics data resources. The current challenge in computational biology is to decipher the cell type-specific transcription factor, chromatin and transcriptional landscapes and to integrate these data to evolve quantitative models of gene regulatory networks. At the same time, it is extremely important to routinely update computational tools to deal with the rapidly evolving experimental methods and data availability. An important aspect is to develop standard experimental and computational methods for similar data sets worldwide and to provide an easy platform for integrative analysis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33(Suppl):245-254; PMID:12610534; http://dx.doi.org/10.1038/ng1089
- Rockman MV, Kruglyak L. Genetics of global gene expression. Nat Rev Genet 2006; 7:862-872; PMID:17047685; http://dx.doi.org/10.1038/nrg1964
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet 2009; 10:241-251; PMID:19293820; http://dx.doi.org/10.1038/nrg2554
- Sakabe NJ, Savic D, Nobrega MA. Transcriptional enhancers in development and disease. Genome Biol 2012; 13:238; PMID:22269347; http://dx.doi.org/10.1186/gb-2012-13-1-238
- Thakurela S, Sahu SK, Garding A, Tiwari VK. Dynamics and function of distal regulatory elements during neurogenesis and neuroplasticity. Genome Res 2015; 25:1309-1324; PMID:26170447; http://dx.doi.org/10.1101/gr.190926.115
- Romanoski CE, Glass CK, Stunnenberg HG, Wilson L, Almouzni G. Epigenomics: Roadmap for regulation. Nature 2015; 518:314-316; PMID:25693562; http://dx.doi.org/10.1038/518314a
- Galperin MY, Koonin EV. Who's your neighbor? New computational approaches for functional genomics. Nat Biotechnol 2000; 18:609-613; PMID:10835597; http://dx.doi.org/10.1038/76443
- Satterlee JS, Beckel-Mitchener A, Little R, Procaccini D, Rutter JL, Lossie AC. Neuroepigenomics: resources, obstacles, and opportunities. Neuroepigenetics 2015; 1:2-13; PMID:25722961; http://dx.doi.org/10.1016/j.nepig.2014.10.001
- Altelaar AF, Munoz J, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet 2013; 14:35-48; PMID:23207911; http://dx.doi.org/10.1038/nrg3356
- Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell 2013; 155:27-38; PMID:24074859; http://dx.doi.org/10.1016/j.cell.2013.09.006
- Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 2014; 15:69-81; PMID:24342920; http://dx.doi.org/10.1038/nrg3623
- Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet 2010; 11:476-486; PMID:20531367; http://dx.doi.org/10.1038/nrg2795
- Gomez-Cabrero D, Abugessaisa I, Maier D, Teschendorff A, Merkenschlager M, Gisel A, Ballestar E, Bongcam-Rudloff E, Conesa A, Tegnér J. Data integration in the era of omics: current and future challenges. BMC Syst Biol 2014; 8(Suppl 2):I1; PMID:25032990; http://dx.doi.org/10.1186/1752-0509-8-S2-I1
- Sherwood RI, Hashimoto T, O'Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, Gifford DK. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol 2014; 32:171-178; PMID:24441470; http://dx.doi.org/10.1038/nbt.2798
- Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 2014; 15:272-286; PMID:24614317; http://dx.doi.org/10.1038/nrg3682
- Dror I, Golan T, Levy C, Rohs R, Mandel-Gutfreund Y. A widespread role of the motif environment in transcription factor binding across diverse protein families. Genome Res 2015; 25:1268-1280; PMID:26160164; http://dx.doi.org/10.1101/gr.184671.114
- Zhou T, Yang L, Lu Y, Dror I, Dantas Machado AC, Ghane T, Di Felice R, Rohs R. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res 2013; 41:W56-62; PMID:23703209; http://dx.doi.org/10.1093/nar/gkt437
- Ernst J, Kellis M. Interplay between chromatin state, regulator binding, and regulatory motifs in six human cell types. Genome Res 2013; 23:1142-1154; PMID:23595227; http://dx.doi.org/10.1101/gr.144840.112
- Wagner M, Jung J, Koslowski M, Türeci Ö, Tiwari VK, Sahin U. Chromatin immunoprecipitation assay to identify genomic binding sites of regulatory factors. Methods Mol Biol 2016; 1366:53-65; PMID:26585127; http://dx.doi.org/10.1007/978-1-4939-3127-9_6
- Yao L, Berman BP, Farnham PJ. Demystifying the secret mission of enhancers: linking distal regulatory elements to target genes. Crit Rev Biochem Mol Biol 2015; 50:550-573; PMID:26446758; http://dx.doi.org/10.3109/10409238.2015.1087961
- Whalen S, Truty RM, Pollard KS. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet 2016; 48:488-496; PMID:27064255; http://dx.doi.org/10.1038/ng.3539
- MacQuarrie KL, Fong AP, Morse RH, Tapscott SJ. Genome-wide transcription factor binding: beyond direct target regulation. Trends Genet 2011; 27:141-148; PMID:21295369; http://dx.doi.org/10.1016/j.tig.2011.01.001
- Segal E, Widom J. From DNA sequence to transcriptional behaviour: a quantitative approach. Nat Rev Genet 2009; 10:443-456; PMID:19506578; http://dx.doi.org/10.1038/nrg2591
- Chai LE, Loh SK, Low ST, Mohamad MS, Deris S, Zakaria Z. A review on the computational approaches for gene regulatory network construction. Comput Biol Med 2014; 48:55-65; PMID:24637147; http://dx.doi.org/10.1016/j.compbiomed.2014.02.011
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9:559; PMID:19114008; http://dx.doi.org/10.1186/1471-2105-9-559
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 2006; 7(Suppl 1):S7; PMID:16723010; http://dx.doi.org/10.1186/1471-2105-7-S1-S7
- Hsu CL, Juan HF, Huang HC. Functional analysis and characterization of differential coexpression networks. Sci Rep 2015; 5:13295; PMID:26282208; http://dx.doi.org/10.1038/srep13295
- Halbritter F, Vaidya HJ, Tomlinson SR. GeneProf: analysis of high-throughput sequencing experiments. Nat Methods 2012; 9:7-8; PMID:22205509; http://dx.doi.org/10.1038/nmeth.1809
- Wang LY, Wang P, Li MJ, Qin J, Wang X, Zhang MQ, Wang J. EpiRegNet: constructing epigenetic regulatory network from high throughput gene expression data for humans. Epigenetics 2011; 6:1505-1512; PMID:22139581; http://dx.doi.org/10.4161/epi.6.12.18176
- Frohlich H. biRte: Bayesian inference of context-specific regulator activities and transcriptional networks. Bioinformatics 2015; 31:3290-3298; PMID:26112290; http://dx.doi.org/10.1093/bioinformatics/btv379
- Hecker M, Lambeck S, Toepfer S, van Someren E, Guthke R. Gene regulatory network inference: data integration in dynamic models-a review. Bio Systems 2009; 96:86-103; PMID:19150482; http://dx.doi.org/10.1016/j.biosystems.2008.12.004
- Balwierz PJ, Pachkov M, Arnold P, Gruber AJ, Zavolan M, van Nimwegen E. ISMARA: automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res 2014; 24:869-884; PMID:24515121; http://dx.doi.org/10.1101/gr.169508.113
- Joyce AR, Palsson BO. The model organism as a system: integrating ‘omics’ data sets. Nat Rev Mol Cell Biol 2006; 7:198-210; PMID:16496022; http://dx.doi.org/10.1038/nrm1857
- Pataskar A, Jung J, Smialowski P, Noack F, Calegari F, Straub T, Tiwari VK. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J 2016; 35:24-45; PMID:26516211; http://dx.doi.org/10.15252/embj.201591206
- Wang Z, Jensen MA, Zenklusen JC. A practical guide to the cancer genome atlas (TCGA). Methods Mol Biol 2016; 1418:111-141; PMID:27008012; http://dx.doi.org/10.1007/978-1-4939-3578-9_6
- Pineda S, Real FX, Kogevinas M, Carrato A, Chanock SJ, Malats N, Van Steen K. Integration analysis of three omics data using penalized regression methods: an application to bladder cancer. PLoS Genet 2015; 11:e1005689; PMID:26646822; http://dx.doi.org/10.1371/journal.pgen.1005689
- Bebek G, Koyuturk M, Price ND, Chance MR. Network biology methods integrating biological data for translational science. Brief Bioinform 2012; 13:446-459; PMID:22390873; http://dx.doi.org/10.1093/bib/bbr075
- Xia J, Gill EE, Hancock RE. Network analyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc 2015; 10:823-844; PMID:25950236; http://dx.doi.org/10.1038/nprot.2015.052
- Koestler DC, Jones MJ, Kobor MS. The era of integrative genomics: more data or better methods? Epigenomics 2014; 6:463-467; PMID:25431938; http://dx.doi.org/10.2217/epi.14.44
- Nakato R, Shirahige K. Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation. Brief Bioinform 2016; PMID:26979602; http://dx.doi.org/10.1093/bib/bbw023
- Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 2012; 13:36-46; PMID:22124482; http://dx.doi.org/10.1038/nrg3117
- Pott S, Lieb JD. Single-cell ATAC-seq: strength in numbers. Genome Biol 2015; 16:172; PMID:26294014; http://dx.doi.org/10.1186/s13059-015-0737-7
- Schwartzman O, Tanay A. Single-cell epigenomics: techniques and emerging applications. Nat Rev Genet 2015; 16:716-726; PMID:26460349; http://dx.doi.org/10.1038/nrg3980
- Hernandez-de-Diego R, Boix-Chova N, Gómez-Cabrero D, Tegner J, Abugessaisa I, Conesa A. STATegra EMS: an experiment management system for complex next-generation omics experiments. BMC Syst Biol 2014; 8(Suppl 2):S9; PMID:25033091; http://dx.doi.org/10.1186/1752-0509-8-S2-S9