
ABSTRACT
Understanding the molecular mechanisms behind the capacity of cancer cells to adapt to the tumor microenvironment and to anticancer therapies is a major challenge. In this context, cancer is believed to be an evolutionary process where random mutations and the selection process shape the mutational pattern and phenotype of cancer cells. This article challenges the notion of randomness of some cancer-associated mutations by describing molecular mechanisms involving stress-mediated biogenesis of mRNA-derived small RNAs able to target and increase the local mutation rate of the genomic loci they originate from. It is proposed that the probability of some mutations at specific loci could be increased in a stress-specific and RNA-depending manner. This would increase the probability of generating mutations that could alleviate stress situations, such as those triggered by anticancer drugs. Such a mechanism is made possible because tumor- and anticancer drug-associated stress situations trigger both cellular reprogramming and inflammation, which leads cancer cells to express molecular tools allowing them to “attack” and mutate their own genome in an RNA-directed manner.
Introduction
One of the currently prevailing models inspired by Darwin's central hypotheses defines cancer as an evolutionary process, fueled by random and chaotic somatic genomic rearrangements and mutations, with sequential selection of different cell populations.Citation1-4 In this model, the random mutational process generates two major types of mutations: the “driver” mutations that are causally implicated in oncogenesis by conferring a selective advantage and the “passenger” mutations that are a consequence of the chaotic mutational process but do not confer a selective advantage.Citation1-4
In contrast to organismal evolution which has a unique history, cancer can be considered in some sense as a repeated experiment.Citation4 In this context, despite intra- and inter-tumoral genetic heterogeneity, some mutations are repeatedly identified (hotspot mutations), some genes (e.g., oncogenes) are more frequently mutated than others in a cancer-type dependent manner and different genes that are mutated in specific cancer types are often involved in the same pathway or network.Citation1,5,6 In addition, it is now widely recognized that anticancer treatment resistance is the norm and some resistance mechanisms involving genetic modifications reproducibly appear in different patients.Citation7-11 It is believed that reproducible events in cancer rely on a number of random mutations that create tumor cell genetic heterogeneity, which allows for adaptation to stress situations (e.g., anticancer therapies) after selectionCitation2,7,8 (Fig. S1A). While chance and selection constitute an explanation of reproducibility, we need to look for other potential mechanisms that may contribute to the high adaptability of cancer cells.
It is not clear yet whether mutations always initiate the cancer process or whether cancer-associated and -driving mutations occur at the end of a set of, more or less, long-term adaptive responses to sustained stress situations, in which epigenetics could play an important roleCitation12-19 (Fig. S1C). In an attempt to understand the initiation of the mutational process involved in unicellular organism adaptation to stress situations, some authors have proposed that the selective pressure (e.g., a stress imposed to a cell) could induce a general increase in the cellular mutational rate.Citation20-24 This concept of “adaptive mutations” implies that high mutational rate is a direct consequence of selective pressure that induces genomic plasticity and leads to random mutations, which would be further selected depending on the resulting phenotype (Fig. S1A). A concept that goes a step forward implies “directed adaptive mutations” and postulates that selective pressure induces targeted mutations that directly contribute to cellular adaptation.Citation20-24 This concept is controversial because of the absence of proven molecular mechanisms (Fig. S1A), but worth exploring for its ability to help explain the contribution of the mutational process to the high adaptive capacity of tumor cells.
The reproducibility of specific mutations or of mutations affecting the same set of genes can in principle result from specific and reproducible causative mutational molecular mechanisms. This would narrow the number of possible mutations within a genome and consequently increase the probability of some mutations to occur. In this context, sequencing of billions of bases from tumor samples led to the recent discovery of cancer-type specific “mutational signatures” demonstrating that the appearance of mutations is context dependent and that some regions of the genome have a higher propensity for sequence changes.Citation25-27 As detailed in BOX 1, these mutational signatures rely, in part, on the molecular mechanisms involved in the mutational process (e.g., DNA repair pathway or editing enzymes).Citation25-37 There is also increasing evidence indicating that chromatin and DNA modifications and topology are associated with local mutational rate (BOX 1).Citation19,34,35,37-42 The characterization of molecular links between regional mutational rate with chromatin or DNA modifications or topology is particularly interesting as it raises the intriguing possibility that cellular mechanisms could influence the mutational rate at specific genomic loci by impacting on local chromatin and DNA modifications (Fig. S1D).
In this setting, it must be underlined that there is considerable evidence demonstrating that RNA molecules can direct chromatin and DNA modifications at specific lociCitation43-57 (BOX 2). Some recent reports have even demonstrated that RNA molecules can direct DNA-editing enzymes to specific loci and also that RNAs can serve as templates during DNA repair and for de novo DNA synthesis.Citation58-77 Based on these observations, several authors have proposed that RNA molecules could direct DNA sequence modifications.Citation68,70,73,74,78,79 Therefore, RNA molecules could be part of cellular mechanisms that influence the mutational rate at specific genomic loci (Fig. S1D). What would be the origin of these RNAs and could they really direct mutations in response to cellular environment variations?
The hypothesis defended here, inspired from the concept of “directed adaptive mutations,” is that some tumor cells produce small RNAs derived from mRNAs encoding for proteins directly engaged in cellular stress situations. It is postulated that these mRNA-derived small RNAs would target the genome regions they originate from and increase the local mutational rate of the targeted regions. Therefore, mRNA-derived small RNAs could link the cellular environment and stress situations to the mutational rate of coding genes. This article develops this hypothesis in two parts based on recent published observations. In the first part, I will describe the molecular pathways that could be involved in the biogenesis of small RNAs derived from mRNAs encoding stressed proteins and in subsequent RNA-directed mutations. Part one is divided in three sub-parts. The first subpart will show that mRNAs undergoing translation are in close physical proximity to the intended site of action of the coded proteins raising the possibility of a direct association of a specific stressor with specific proteins and the metabolism of the encoding mRNAs (). The second subpart will propose that stress-induced translationally-stalled mRNAs are used to generate mRNA-derived small RNAs (). In the third subpart, I will review the literature that highlights the role of small RNAs in targeting genomic regions and driving chromatin and/or DNA modifications as well as DNA editing ().
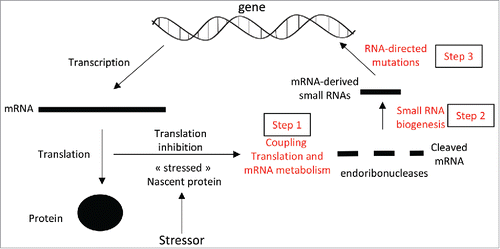
Figure 1. (A) By altering the biochemical properties of targeted proteins during translation, a molecular stressor causes the inhibition of translation of the corresponding mRNAs. Stress-induced translationally-stalled mRNAs are cleaved by stress-induced endoribonucleases (step 1). The mRNA fragments are next used as substrates for the biogenesis of small RNAs (step 2). The mRNA-derived small RNAs target the genomic region corresponding to the mRNA precursors and enhance the recruitment of proteins modulating local mutation rate in a direct- or indirect-manner (step 3).
In the second part, I will explore the possibility that the molecular pathways described above might be specific to cancer cells because of their requirement for dedicated molecular machineries that appear to be expressed in tumor cells exposed to sustained stressful environment. Cells exposed to sustained stresses express factors that are normally restricted to stem and/or germ cells and that may contribute to the biogenesis of mRNA-derived small RNAs. Sustained stress also creates an inflammatory microenvironment that mimics viral infection; many factors involved in small RNA biogenesis and small RNA-mediated effects that could contribute to cancer cell mutations are known to be involved in virus host defense and in retrotransposon inactivation. Therefore, the possibility will be discussed that mRNAs coding for stress-associated proteins are “mistaken for” virus or retrotransposon RNAs, which cause stress-induced mRNA-derived small RNAs to “attack” the corresponding genomic region in cancer cells.
In the proposed model, the probability of mutations at specific loci is increased in a stress-specific and RNA-dependent manner, which in turn leads to an increased probability of generating mutations that alleviate the stress situation (Fig. S1B). Therefore, the described molecular mechanisms may contribute to the capacity of tumor cells to adapt to the tumor microenvironment and to anticancer drug treatment. It is important to underline that RNA-directed adaptive mutations would act alongside the currently characterized cancer-associated mutational framework and would account for some of the more frequently observed mutations by increasing the probability of mutations occurring at specific loci.
From stressed-proteins to RNA-directed DNA mutations
Coupling protein activities and mRNA metabolism
mRNA localization and local translation: Proteins are transported by specialized transporter systems, some of which are associated with the endomembrane system, to specific subcellular locations where they perform their functions.Citation80,81 However, increasing evidence indicates that subcellular protein localization also relies on the subcellular targeting of their coding mRNAs and subsequent on-site mRNA translation or local translationCitation82 (). Local translation is involved, for example, in the dynamic and spatially-regulated translation of proteins playing a role in cell polarity, protrusion, and migration.Citation82 While mRNA transport relies on cytoskeleton microfilaments and microtubules, an emerging concept is that mRNA transport involves the endomembrane system that may therefore coordinate protein and mRNA flow.Citation83 Interestingly, mRNA metabolism and small RNA biogenesis pathways also involve the endomembrane system (see section entitled “mRNA-derived small RNAs”). In addition, the endomembrane system constitutes a signaling pathway platform, or hub, as transmembrane receptors can still operate when present on endosomal vesicles.Citation84 Therefore, local translation results in mRNAs being in close physical proximity to the site of their encoded protein's action and endomembrane-associated mRNAs tend to be physically close to signaling complexes, as further detailed below.
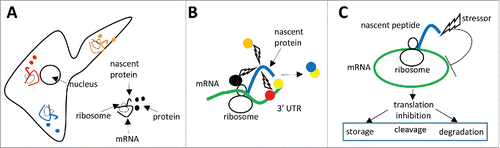
Figure 2. (A) The subcellular location of proteins (colored circles) rely on the subcellular targeting of their coding mRNAs and subsequent on-site mRNA translation or local translation. The nascent proteins are in different subcellular environment, which may impact on subsequent modifications and interactions with different partners. (B) Co-translational modifications rely on the binding of the protein-modifying enzymes to the translating ribosomes (black circle), or on local translation that positions the nascent proteins in the proximity of specific modifying enzymes (orange circle), or on the binding of protein modifying enzymes to the mRNA 3′-UTR (red circle). The binding of a protein (yellow circle) to mRNA 3′UTR may increase the probability of its interaction with the newly-synthetized protein (blue circle). (C) A stressor affecting a nascent polypeptide chain impact on the mRNA undergoing translation by inhibiting its translation, thus inducing its storage, cleavage, or degradation.
Co-translational events: Contrary to common belief, protein modifications (in the general sense of the term) can occur during translation, i.e., on the nascent peptides emerging from ribosomes as they are translating mRNAs. For example, mRNAs coding for proteins that function in specific organelles are localized on these organelles and the protein's importation into the organelles occurs co-translationally.Citation85 Folding of the nascent polypeptide chains occurs co-translationally thanks to chaperones (e.g., HSP70) that associate with translating ribosomes.Citation85 Proteins undergo several modifications during translation, including removal of the first methionine, N-α-acetylation, N-myristoylation, N-glycosylation, and N-terminal O-GlcNAc glycosylation.Citation86 An increasing number of phosphorylation modifications have been demonstrated to occur on nascent polypeptide chains. For example, DYRK1A and PKA induce their own co-translational auto-phosphorylation.Citation87-89 Kinases, like PKA, p38 MAPK, and MTORC2 can also phosphorylate different nascent polypeptide targets.Citation90-93
How are these co-translational modifications executed? Certainly the implication is that protein-modifying enzymes are in close proximity to the translated mRNAs. One possibility is that the modifying enzymes are attached to the translating ribosomes, as it has been shown for enzymes involved in N-terminal modifications ().Citation85,86 Another possibility is that the precise location of the local translation process positions nascent proteins next to regulatory factors (). Finally, another possibility is that protein-modifying enzymes bind to mRNAs and modify the nascent proteins coded by the bound mRNAs ().
This latter possibility is supported by several exciting observations. First, there are an increasing number of “unconventional” non-specialized RNA-binding proteins such as metabolic enzymes and kinases that have been shown to bind mRNAs in vivo.Citation94-96 Second, it has been reported in yeast that some mRNAs are bound by proteins that interact co-translationally with the mRNA-encoded proteins.Citation97,98 A similar mechanism has recently been demonstrated in human cells, where the 3′ UTR of CD47 mRNA is bound by the HuR and SET proteins, which facilitates the interaction of SET with the newly-synthetized CD47 protein and which in turn targets CD47 to the plasma membrane.Citation99 Further evidence for mRNA-directed protein interactions include examples of co-translational protein complex assembly in several organisms.Citation100 It has been proposed that co-translational assembly might be particularly important for efficient homo-dimer formation as in the case of p53.Citation100,101 Likewise, the NFκB p50-p105 complex is assembled co-translationally.Citation100,102 Finally, recent data in Drosophila have demonstrated that the binding of the same protein on different mRNAs contributes to the coordinated actions of the mRNA-encoded proteins.Citation103 These observations physically position the mRNA molecules at the heart of protein functions.
Coupling co-translational events and mRNA metabolism: There is now considerable evidence demonstrating that co-translational events can feed-back to control mRNA metabolism. This is particularly well documented for the unfolded protein response pathway that is activated when misfolded proteins accumulate in the endoplasmic reticulum lumen.Citation104,105 Co-translational detection of misfolded proteins activates the endoplasmic reticulum transmembrane endoribonuclease, called IRE1 that cleaves the XBP1 mRNA leading to mRNA splicing and translation of the XBP1 transcription factor. The IRE1-mediated cleavage of XBP1 mRNA and of several other mRNAs localized to the endoplasmic reticulum membrane occurs co-translationally when the coded nascent chains are imported into the endoplasmic reticulum.Citation104,105 A similar coupling between co-translational folding and mRNA decay happens in the cytoplasm since the detection of misfolded nascent polypeptides induces both co-translational polypeptide ubiquitination and mRNA decay.Citation106-108 This strengthens the concept of coupling between proteostasis and mRNA metabolism.Citation109-111
Co-translational events (e.g., quality control, protein modifications, and complex assembly) are not restricted to cytoplasmic, transmembrane, or secreted proteins. They also apply to proteins with well-characterized nuclear functions. Indeed, nuclear proteins are produced in the cytosol and nuclear protein-encoding mRNAs may be translated while being anchored on the endoplasmic reticulum.Citation112 It is notable that many proteins that were originally identified as having functions at the cell periphery can act as nuclear transcriptional regulators and, conversely, many proteins with nuclear functions also have cytosolic or mitochondrial roles.Citation113-115 Finally, co-translational complex assembly has been demonstrated for nuclear proteins.Citation97,98,100,116,117
Local translation and coupling between translation and mRNA metabolism is a straightforward mechanism for ensuring local protein homeostasis. It is likely a powerful mechanism to improve functional complex assembly as it avoids potential toxic protein–protein interactions as well as protein or RNA aggregation.Citation82,100,104,106,107,109,118 However, as a direct consequence, these mechanisms imply that a stressor affecting a nascent protein will impact on the encoding mRNA by directly inhibiting its translation ( and Fig. S2).
Stress-induced translationally-stalled mRNAs can then have several destinies (). They can be degraded by exoribonucleases. Also an increasing number of endoribonucleases that have been shown to be activated under stress situations can fragment stress-associated mRNAs (see section entitled “mRNA-derived small RNAs”). While mRNA cleavage is often associated with degradation by exoribonucleases, there is increasing evidence that cleaved-RNAs are used to generate small-derived RNAs with regulatory functions (this will be described in the following section). Finally, stressed-induced translationally-stalled mRNAs can be stored in cytoplasmic granules like stress granules (Fig. S3). Interestingly, stress granules contain many enzymes involved in mRNA metabolism and small RNA biogenesis, including several endoribonucleases. These granules could also play a role in mRNA cleavage initiating the biogenesis of small RNAs deriving from stress-induced translationally-stalled mRNAs ().
mRNA-derived small RNAs
Endoribonucleases and mRNA fragments: The degradation of mRNA depends on 5′- to 3′- or 3′- to 5′-exoribonucleases. It occurs after removal of the mRNA's 5′-cap and/or 3′-polyadenylated tail, both of which are added to mRNAs during transcription in the nucleus to protect mRNAs from degradation in the cytoplasm. However, uncapped mRNAs have been detected and re-capping can occur in the cytoplasm.Citation119-121 Likewise, non-polyadenylated mRNAs have been detected and de novo polyadenylation can occur in the cytoplasm.Citation122 The ability to re-cap and re-polyadenylate cleaved mRNAs, as well as the diversity of cytoplasmic processes affecting mRNA 5′- and 3′-ends,Citation123,124 might be particularly relevant in the context of RNA cleavage by endoribonucleases since these mechanisms could contribute to generate mRNA-derived products.
Many endoribonucleases have been characterized in recent yearsCitation125-128 (Fig. S4). Remarkably, most of them play a role in cleaving mRNAs when cells are exposed to stress situations and during virus infection. For example, the IRE1 endoribonuclease, described above, cleaves mRNAs during the unfolded protein stress response. RNAse L plays a very important role in cellular innate immunity and stress response by cleaving RNAs from viruses, as well as endogenous mRNAs, and leads to the production of small RNAs that amplify the antiviral signaling pathway.Citation129,130 Regnase-1, which associates with polysomes and the reticulum endoplasmic, cleaves mRNAs coding for proteins involved in inflammation.Citation131 RNASET2 plays a role in host defenses against RNA viruses and in extracellular RNA scavenging.Citation132 RNASET2 is also detected in cytoplasmic RNA granules and is associated with the endomembrane system, autophagosomes, and lysosomes.Citation132 Endoribonuclease VCitation133 and TUDOR-SNCitation134,135 have been shown to cleave ADAR1-edited RNAs, which is believed to contribute to cellular protection from double-stranded viral RNAs. TUDOR-SN also plays a very important role in stress granule dynamics.Citation134,135 In addition, an increasing number of proteins that are structurally unrelated to RNases have been shown to behave as endoribonucleases. For example, PMR1 (polysome ribonuclease 1) cleaves polysome-associated mRNAs in a regulated manner and also localizes to stress granules.Citation125-128 The G3BP proteins are major inducers of stress granule formation and can cleave several mRNAs.Citation125-128,136 In addition to several other poorly characterized endoribonucleases,Citation125-128 there are other well-characterized endoribonucleases involved in small non-coding RNA biogenesis (Fig. S4) that have also been reported to cleave mRNAs. For example, the miRNA biogenesis factor, Drosha cleaves nuclear pre-mRNAs and/or mRNAs.Citation137-140 As Drosha has been detected in the cytoplasm during viral infection,Citation141 it is tempting to speculate that it may contribute to cytoplasmic mRNA cleavage. Likewise, some proteins of the Argonaute family (Ago and Piwi proteins) can cleave mRNA when guided by small RNAs (e.g., miRNAs or piRNAs, see below) that hybridize to target mRNAs.Citation142 Although this mechanism is involved in post-transcriptional gene silencing (PTGS), since it induces targeted mRNA degradation, the slicing activity of some Argonaute proteins can also contribute to small RNA biogenesis (see section entitled “piRNAs and piRNA-like molecules”).
RNA fragments and biogenesis of small RNAs: While endoribonuclease activity is often associated with mRNA degradation, cleaved-mRNA fragments may be used under certain circumstances to generate functional mRNA-derived small RNAs. Several pieces of evidence support this possibility. First, fragmentation of short or long mature RNAs (e.g., tRNAs, snoRNAs, vault RNAs, Y RNAs, rRNAs, and miRNAs) yields functional smaller non-coding RNAs with regulatory functions.Citation143-146 For example, tRNAs are cleaved in stress situations to generate tRNA-derived small RNAs that induce stress granule formation and inhibit translation.Citation147 Second, mRNA fragments have been identified and correspond to different parts of mRNAs, including 3′- and 5′-UTRs and internal exons.Citation120,121,143,144,148,149 For example, the YB-1 RNA-binding protein, which plays a major role in cellular stress response, associates with short RNAs, some of which are derived from mRNAs.Citation150 Third, some small RNAs, related to classical small non-coding RNAs (e.g., piRNAs) correspond to mRNA fragments (see section entitled “piRNAs and piRNA-like molecules”). Therefore, successive cleavages of RNA can produce functional RNAs all along the fragmentation cascade (). The hypothesis of this manuscript is that mRNA-derived small RNAs can function during stress. To illustrate the capability of cells to generate small RNAs derived from single-stranded RNAs like mRNAs, I will next present recent findings on piRNA biogenesis.
Figure 3. (A) The cleavage of “precursor” RNAs (primary transcripts) is required for the production of mature RNAs with biological functions. “Mature” RNAs are also cleaved and generate intermediate fragments that are themselves cleaved and modified to generate “derived-small RNAs” having cell regulatory functions. (B) Retrotransposon sequences are transcribed from both DNA strands. Precursor sense RNAs are cleaved by Zucchini. Intermediate RNA fragments are next loaded into Piwi proteins and trimmed by an exoribonuclease up to the regions where Piwi proteins protect small RNA regions from degradation. The resulting sense piRNAs then hybridize to antisense transcripts and induce their cleavage by some Piwi proteins, generating antisense piRNAs that in turn target sense transcripts, into the so-called “ping-pong” process. Several proteins (on the right) involved in piRNA biogenesis are (over)-expressed in cancer cells; protein names followed by * are classified as Cancer/Testis Antigens. (C) A stressor affecting a nascent polypeptide chain impacts on the mRNA undergoing translation by inhibiting its translation and inducing its cleavage. The generated mRNA fragments are then used to produce mRNA-derived small RNAs.
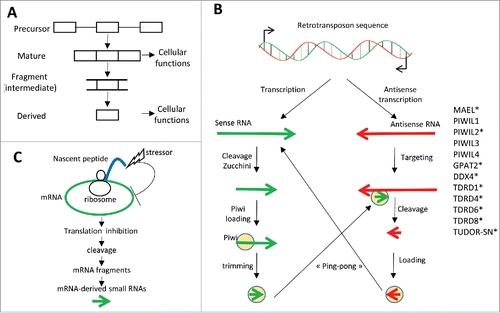
piRNAs and piRNA-like molecules: PiRNAs compose a heterogeneous group of 24–35 nucleotide-long small RNAs that are produced from single-stranded RNA precursors and that associate with Piwi proteinsCitation151-153 (Fig. S4). The biogenesis and functions of piRNAs have been particularly well characterized in drosophila and mouse germ cells, where one of their main functions is to repress retrotransposons that are repeated sequences expressed in germ cells during chromatin de-condensation (BOX 3). In summary, single-stranded RNA precursors (e.g., retrotransposon RNAs) are cleaved and generate the small piRNAs that are loaded onto Piwi proteins (). PiRNAs can next direct Piwi proteins onto complementary mature RNAs thus inducing PTGS or nascent RNAs inducing transcriptional gene silencing (TGS).Citation151-153
PiRNAs have been detected in human fetal ovary and adult testis, where they may originate from about 200 piRNA genomic clusters, some of which map to retransposon elements.Citation154,155 The potential function of human piRNAs needs further investigation. Even though the human genome contains evolutionary-conserved genes involved in the piRNA biogenesis pathway, it is important to underline that we do not know yet how much of the piRNA biogenesis pathway described in BOX 3 is conserved in human cells. Recent findings have shown first, that piRNAs are produced not only from repeat element-derived RNAs but also from other single-stranded RNAs, including mRNAsCitation53-56,154,156-172 and second that piRNAs or piRNA-like molecules are produced in different kind of somatic cells, including cancer cells,Citation54,165-172 where several proteins of the piRNA-pathway are often over-expressed (see next part). It is important to note that cancer cells do not only express major factors involved in the piRNA-pathway and produce piRNAs or piRNA-like molecules Citation55,56,156-161; in addition, manipulation of the expression of Piwi proteins and some cancer-associated piRNA-like molecules has been shown to mediate PTGS or chromatin or DNA modifications of the targeted loci. Overall, these studies support the notion that the piRNA pathway is active in the cancer cellular model tested.Citation47,55,56,158,159
In conclusion, mRNA fragments can be generated after mRNA cleavage by endoribonucleases, many of which are mobilized in stress situations. Small RNAs, including those belonging to the piRNA family, deriving from single-stranded RNAs, including mRNAs, have been reported. An interesting hypothesis is that stressed-induced translationally-stalled mRNAs can be fragmented by endoribonucleases and can initiate the biogenesis of mRNA-derived small RNAs such as piRNA-like molecules ( and Figures S5 and S6). Before describing the cancer-associated cellular context in which such molecular process may occur (see section entitled “Microenvironnement and cellular context”), I will review the literature on the functions of small RNAs in directing biochemical, topological, and sequence modifications of DNA and chromatin .
RNA-directed chromatin modifications and genome editing
RNA-mediated chromatin and DNA structure modifications: Small RNAs have been shown in many organisms to target specific genome loci and induce chromatin modifications by guiding Argonaute protein-containing complexes and triggering the local recruitment of histone- and DNA-modifying enzymes.Citation173 This mechanism is known as TGS since it often leads to the formation of compacted chromatin and repression of gene expression. For example, piRNAs generated in the cytoplasm are loaded onto Piwi proteins and imported into the nucleus, where piRNAs bind to complementary nascent transcripts and induce local chromatin compaction.Citation174 It is not clear yet how many details of these mechanisms are conserved in mammals and how widespread such regulation is in human. However, increasing evidence supports the notion that similar mechanisms do exist in human cells as detailed in BOX 2.
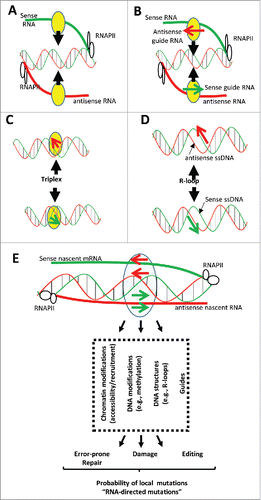
At least four mechanisms may contribute to RNA-directed modulation of chromatin. First, RNAs can be produced from promoter regions or intragenic regions and directly and locally recruit chromatin-modifying proteinsCitation173(). Second, small RNAs form Watson–Crick base-pairs with complementary nascent RNAs, leading to the formation of complexes on specific loci that enhance the recruitment of proteins involved in chromatin and/or DNA modificationsCitation173 (). A third mechanism relies on the formation of RNA:dsDNA triple-helices (triplexes) that are formed by sequence-specific binding rules where an RNA molecule binds in the major groove of the targeted double-strand DNA (dsDNA) by Hoogsteen hydrogen bonding between purine-rich strands of dsDNA and pyrimidine-rich RNA Citation175 (). As an example, it has been shown that promoters located in the intergenic space upstream of rDNA genes produce RNAs that are cleaved into 150-250 long fragments that form triplex structures with rDNA promoters and that these triplexes recruit DNA methyltransferases.Citation176 FMR1 mRNA interacts with its own promoter and induces epigenetic silencing.Citation177 It has also been proposed that miRNAs act on targeted promoters by forming RNA:dsDNA triplexes.Citation57,175 Fourth, RNAs can theoretically target specific loci and induce the formation of R-loops (). R-loops result from the Watson–Crick base-pairing of an RNA molecule to the cDNA strand displacing the second DNA strand in a single-stranded conformation.Citation41 As detailed in BOX 4, recent evidence has indicated that dedicated mechanisms favor the formation of R-loops.
Figure 4. (A) RNAs produced from intergenic, promoter, or intragenic regions directly and locally recruit proteins (in yellow) involved in chromatin or DNA modifications. (B) Small RNAs form Watson–Crick base-pairing with complementary nascent RNAs leading to the formation on specific loci of complexes enhancing the recruitment of proteins (in yellow) involved in chromatin or DNA modifications. (C) RNA:dsDNA triple-helices (or triplexes) are formed by sequence-specific binding rules where a single-stranded RNA binds in the major groove of the targeted dsDNA by Hoogsteen hydrogen bonding between a purine-rich strand of dsDNA and either a pyrimidine-rich or a purine-rich ssRNA strand. By this mechanism, RNAs direct proteins (in yellow) involved in chromatin or DNA modifications at specific loci. (D) R-loops result from the Watson–Crick base-pairing of an RNA molecule to the cDNA strand displacing the second DNA strand in a single-stranded conformation. (E) RNAs target specific genomic loci by several mechanisms and induce local chromatin and DNA modifications with potential consequences for the local accessibility of mutators and enzymes involved in DNA metabolism. RNAs also induce the formation of structures (e.g., R-loops) that induce ssDNA formation and double-stranded DNA breaks, both of which increase the local mutational rate. Finally, RNAs guide, DNA endonucleases and editing enzymes to targeted loci. Therefore, RNAs could increase the local probability of mutations (RNA-directed mutations) by inducing chromatin modifications (e.g., compaction), DNA modifications (e.g., methylation, ssDNA formation), DNA injuries (e.g., dsDNA breaks), DNA error-prone repair mechanisms, or recruitment of DNA endonucleases and editing enzymes.
The mechanisms described above demonstrate the ability of RNAs to target specific genomic loci. As a consequence, RNAs can direct chromatin or DNA, biochemical, or topological modifications (BOX 2). As these modifications can modulate the local mutational rate (BOX 1), an interesting possibility is that RNAs can target specific loci and modulate the mutational rate of the targeted regions (). Further supporting such a possibility, small RNAs have been recently shown to play a direct role in both DNA repair and editing.
RNA-directed DNA repair and editing: Increasing evidence indicates that RNAs play an important role in DNA repair. It has been shown that small non-coding RNAs are produced from sequences in the vicinity of double-stranded DNA break (DSB) sites in a Dicer-dependent manner and that RNAs contribute to DSB repair by homologous recombination.Citation58-63 It has been proposed that small RNAs form a complex with Ago2 and bind to the nascent transcripts produced around DNA break sites or directly bind to DNA at DSB sites. This complex may then induce chromatin modifications to aid DSB repair or play a more direct role by allowing the recruitment of proteins involved in DNA repair by homologous recombination. Indeed, Ago2 interacts with RAD51 and the recruitment of RAD51 on DSB sites is Dicer and Agor2 dependent.Citation58-65 Recent work has also suggested that RNA can be used as a template for homologous recombination in bacteria, yeast, and human.Citation66-70 Indeed, the assumption that homologous recombination strand exchange occurs only between two DNA molecules has recently been challenged by the discovery that RNA can be used as a template during DNA recombination and repair. It has been proposed that RAD52 anneals RNA to complementary DSB-like DNA ends and that the annealed RNA serves as a template for DNA repair by reverse transcription.Citation66-70
In this context, it is well established that RNA molecules can serve in cancer cells for de novo genomic DNA sequence synthesis as they are used as templates for reverse transcription during telomere elongation and during genomic de novo integration of retrotransposons or processed pseudogenes (Fig. S5). In this setting, it is also important to highlight that genomic neo-integration of retrotransposons is associated with high mutation rate since the neo-synthetized single-strand DNA (ssDNA) is targeted by editing enzymes during the reverse-transcription process, which is believed to limit retroelement expansion.Citation71,72 Therefore, if RNAs can be used as templates during homologous recombination, this may go with DNA editing.
There is also increasing evidence that RNAs can be used as guides for targeting editing enzymes and DNA endonucleases to specific loci. For example, in several examples of lower organisms, including ciliates, RNAs can target genomic regions and lead to DNA elimination by enhancing endonuclease recruitment.Citation73,74 Bacterial DNA contains DNA elements, named CRISPRs that generate small RNAs (guide RNAs) that recognize virus DNAs through R-loop formation and lead to virus DNA cleavage by the Cas9 nuclease.Citation178 Human genome editing is now routinely performed by co-expressing Cas9 endonuclease and a guide RNA targeting a specific genomic region. Remarkably, RNA-guided DSBs made by this system is associated with increasing local mutational rate, with some of the mutations being generated by APOBEC-editing enzymes.Citation32,179
It has recently been shown that the human Activation-induced cytidine deaminase (AID) editing enzyme is also guided by RNAs to specific genomic regions. AID mediates somatic class-switch recombination and hyper-mutation in B cells. Class-switch recombination is a deletion-recombination event that allows antibodies to diversify. Several studies have shown that the targeting of AID to specific loci is an RNA-mediated mechanism.Citation75-77 A recent report has demonstrated that the switch regions, which are intronic repeated sequences within immunoglobin loci, produce guide RNAs that form G-quadruplex structures to which AID binds; then, these RNAs guide AID to the complementary genomic switch regions in a sequence-specific manner, probably by forming R-loop structures. These structures lead to the displacement of ssDNA that is the preferential substrate for AID de-amination activity. Deaminated DNA next engages the base excision and mismatch repair machineries to generate DSBs that lead to deletion-recombination.Citation76 This mechanism may also occur outside the immunoglobin loci (off-targets) in regions potentially forming G-quadruplex structures; the AID and APOBEC enzymes have been shown to associate with different classes of small RNAs.Citation76,180,181 Therefore, a variety of RNA guides might be able to target editing enzymes at different loci ().
In conclusion, there is large body of evidence indicating that RNAs target specific genomic loci by several mechanisms (). RNAs thus induce local chromatin modifications with potential consequences for the local accessibility of mutators and enzymes involved in DNA metabolism (BOX 1 and 2, ). RNAs also induce DNA conformational changes (e.g., R-loops) that induce DNA DSBs and single-stranded DNA (ssDNA) formation, both of which increase the local mutational rate (BOX 4). Finally, RNAs have the ability to guide DNA endonucleases and editing enzymes to targeted loci. Therefore, RNA may contribute to the genesis of directed mutations and these observations already led several authors to propose that RNAs direct DNA sequence modifications.Citation68,70,73,74,78,79
Obviously, DNA sequence modifications are highly toxic for cells. The molecular pathway described above is unlikely to occur in normal somatic cells as it requires several molecular tools (e.g., enzymes) that are generally not co-expressed in these cells. In the next part, I will describe the tumor-specific cellular context (e.g., exposition to sustained stress) that could allow tumor cells to express the factors required for the biogenesis of mRNA-derived small RNAs and for RNA-directed mutations.
Microenvironment and cellular context
Sustained stress and transcriptomic plasticity
Cancer/testis antigens: It is now widely accepted that normal somatic cells retain the capacity to change their fate through gene expression reprogramming, through dedifferentiation (from a given differentiated state into a more primordial state) or trans-differentiation (from a given differentiated state to another one). In this context, it is believed that cell “plasticity” plays a major role in the cellular adaptation to stressful conditions: (i) by generating a gene expression pattern that drives toward an adapted phenotype; and (ii) by allowing some cells to turn back into less mature cells to provide new stem cells for repopulation in damaged tissues.Citation12-19 Whether the cancer cell of origin is a stem cell or a differentiated cell, it is well established that cancer cells that are exposed to stressful conditions share many features with stem cells, including the expression of stem cell-restricted factors.Citation12-19,182
A particular class of genes that may contribute to cancer stem-ness is the so-called cancer/testis antigens (CTAs). CTAs are proteins (∼200) that are expressed preferentially in adult male germ cells and that are detected in tumor cells.Citation182 Interestingly, several genes involved in piRNA biogenesis are members of the CTA family and a substantial number of the piRNA biogenesis factors are over-expressed in cancer cells ().Citation161,182-193 The expression in cancer cells of factors involved in piRNA biogenesis factors is in agreement with the many reports indicating expression of piRNA-like molecules in cancer cells.Citation55,56,156-161 Although caution must be taken in classifying the identified small RNAs as bona fide piRNAs, the manipulation of the expression of Piwi proteins and some cancer-associated piRNA-like molecules support the notion that the piRNA pathway is active in cancer cells.Citation47,55,56,158,159 While the expression of key piRNA biogenesis factors is obviously required for piRNA production in cancer cells, other factors are likely to be involved.
Retrotransposons and antisense transcription: As shown in , amplification of piRNAs requires sense and antisense RNA precursors. In this setting, there are numerous evidences indicating that antisense transcription within coding genes is more common than originally thought. A recent report has demonstrated the dysregulation of antisense transcripts overlapping cancer-related genes in cancer cells.Citation194-196,197 Furthermore, retrotransposon expression may play an important role in promoting antisense transcription in cancer cells. It is now widely accepted that retrotransposons that are normally expressed in germ cells only, are re-expressed (and active) in cancer somatic cells, where their expression level is further enhanced by many stressesCitation198-201 (Fig. S5). The expression of retrotransposons in stressed cancer cells is likely to play a major role in cellular adaptation, notably by increasing the transcriptome diversity.Citation198-201 Several reports have shown that retrotransposons drive antisense transcription in several gene loci.Citation194-196 Several other reports have also demonstrated that antisense transcripts can be produced from pseudogenes.Citation202-204 Therefore, as described in more details in Figure S5, natural antisense transcripts, retrotransposon-driven antisense transcripts, and pseudogene-derived antisense RNAs could contribute to the biogenesis of piRNAs or piRNA-like molecules derived from mRNAs in cancer cells. This would explain why piRNAs corresponding to coding genes and pseudogenes have been detected.Citation54,165-172
In conclusion, sustained stresses that impact on chromatin remodeling trigger cell reprogramming (e.g., dedifferentiation) and retrotransposon expression.Citation12-19,198-200,201 These stresses may be responsible for the expression of stem-cell restricted factors and CTAs, including piRNA biogenesis factors, in cancer cells. The same molecular mechanisms may also contribute to the production of antisense transcripts that could play an important role in the biogenesis of mRNA-derived small RNAs ( and Figs. S5, S6, and S7).
Figure 5. (A) Sustained stress situations trigger chromatin remodeling and the expression of retrotransposons, increasing the transcriptome diversity by impacting for example on antisense transcription. Gene expression reprogramming induced by sustained stresses and retrotransposons can lead to the expression of stem cell-restricted factors and Cancer/Testis Antigens (CTAs), some of which participate in small RNA biogenesis pathways. Antisense transcription of coding genes and of pseudogenes combined with the expression of small RNA biogenesis factors would contribute to the biogenesis of mRNA-derived small RNAs. (B) Tumor cells are in an inflammatory microenvironment due to the release or cellular secretion of DAMP molecules from dead, dying, stressed, or senescent cells. This inflammatory microenvironment mimics a virus infection, which triggers the cell-autonomous innate immune response. (C) In a stressed cell (donor cell), endoribonucleases (e.g., RNase L, IRE1) cleave stress-associated mRNAs in stress granules or on the endoplasmic reticulum. In the context of the tumor inflammatory microenvironment, mRNA fragments may trigger the cellular innate immune response through the activation of receptors like RIG-I and may be released or secreted into the extracellular space by multi-vesicular bodies (MVBs) and extracellular vesicles or by autophagy. A stressed “recipient cell” can capture extracellular RNAs, which (in the context of the tumor inflammatory microenvironment) may be “mistaken” for virus RNAs. The confusion of captured exogenous RNAs for virus RNAs (non-self RNAs), as well as endogenous antisense transcripts produced by the recipient cancer cell, could license mRNA-derived small RNAs to target the corresponding genomic loci for editing.
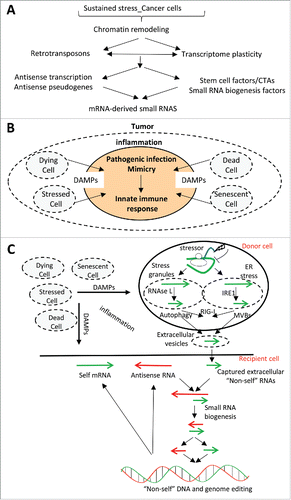
Innate cell-autonomous autoimmunity
Virus mimicry: A major challenge in the understanding of the potential RNA-directed mutational process described above will be to characterize the mechanisms that license some RNAs to target genomic loci. A possible explanation is that the pathogenic infection-like microenvironment of cancer cells triggers an “autoimmunity process” leading cancer cells to edit some parts of their own genome that they mistake for parasite genomic sequences. Several exciting observations support this possibility.
Cancer cells exist within an inflammatory environment that resembles a site of virus infection resulting in part from cell death. In this setting, several recent reports have indicated that anticancer therapies induce an immune response mimicking those induce by pathogens.Citation205-209 For example, DNA demethylating agents, which kill cancer cells, induce the production of double-stranded RNAs (derived in particular from retrotransposon sequences), which triggers an immune cell response and immunogenic cancer cell death.Citation205-209
In addition, the cancer cell inflammatory microenvironment relies on the release or cellular secretion of damage-associated molecular pattern molecules (DAMPs) recognized by pattern recognition receptors that initiate an immune and inflammation response, as they do in the presence of pathogen-associated molecular pattern molecules (PAMPs) deriving from microorganismsCitation210,211 (). While DAMP-mediated mechanisms contribute to induce immunogenic cancer cell death,Citation207-211 they may also activate some of the molecular tools that are normally dedicated to fight against RNA or DNA from parasites. For example, pathogenic infection-like microenvironment of cancer cells may explain why these cells over-express several DNA-editing enzymes. Indeed, the expression of these enzymes is normally more or less restricted to immune and sometimes to stem cells. However, many reports have shown that the expression of DNA-editing enzymes can be enhanced in various somatic cell types by virus infection, by stresses and more importantly by inflammation; this may explain why many cancer-associated mutations are generated by (over)-expressed DNA-editing enzymes.Citation28-31,35-37,71,72,75,212-215
Extracellular RNAs: Recent discoveries have indicated that cancer cells release RNAs within extracellular vesicles. It was believed that extracellular vesicles allow cells to discard unwanted or damaged material through their release to the extracellular environment. However, extracellular vesicles also play a role in cellular communication.Citation216-222 In this setting, much evidence now shows that RNAs can be exchanged from one cell to another through extracellular vesicles generated from multi-vesicular bodies and sharing features with the assembly and release of virus particles.Citation216-222 The presence of RNA within these vesicles is likely to rely on mRNA metabolism being tightly connected to the endomembrane system.Citation223,224 Meanwhile, mitochondria and stress granules can be degraded and cleared by autophagy, which is a selective mechanism enhanced during stress situations (e.g., induced by DAMPs) allowing to degrade specific cytoplasmic contents.Citation225,226 Autophagy occurs by de novo formation of a double membrane that engulfs cytoplasmic contents within autophagosomes. The autophagosomes fuse with lysosomes and sometimes with multivesicular bodies, which can lead to the secretion of the autophagosome's content.Citation226,227
Therefore, several mechanisms contribute to the secretion of RNAs into the extracellular space (). Remarkably, stresses induce cleavage of mRNAs by endoribonucleases, some of those (e.g., RNase L and IRE1) are involved in production of virus- and cellular-cleaved RNAs that are detected as “non-self” and activate antiviral cellular response.Citation104,129,130,228,229 It would therefore be interesting to test whether stress-induced mRNA fragments are secreted by cancer cells (as a result of autophagy or within extracellular vesicles) and whether they induce an immune response from neighboring cancer cells that have the ability to incorporate extracellular RNAs.Citation230
In conclusion, cancer cells are within an inflammatory environment that resembles a pathogenic infection-like microenvironment, which likely contributes to activate antiviral host defense responseCitation205-209 and molecular tools, including editing enzymes that are normally dedicated to mutate the genome of parasites but that have been shown to be major mutators of the genome of cancer cells. The unexpected release of mRNA fragments into the extracellular space by cancer cells could, within the cancer inflammatory environment, further mimic a virus infection as endoribonucleases cleaving mRNAs in stress situations can also cleave parasite RNAs and induce the biogenesis of RNAs that activate antiviral cellular response. Virus-like captured exogenous RNAs and antisense transcripts produced by cancer cells may contribute to produce small RNAs licensed to target genomic loci (). Conceptually, this RNA-dependent mechanism is supported by the recently proposed model where small RNAs, including piRNAs and endo-siRNAs, play a general role in “genome immunity” by recognizing, eliminating, or mutating viruses and transposons that may otherwise colonize the genome.Citation161,231-236
Conclusion
Cancer cells live in a stressed microenvironment, particularly when they are exposed to anticancer treatments aimed at killing cells. Several mechanisms exist that allow cells to survive and adapt to stress situations. In this context, the concept of a “continuous adaptation spectrum” has recently been proposed, where organisms adapt by progressing through the adaptation spectrum as necessary starting with physiological changes (gene expression) and epigenetic changes and ending with structural re-arrangements of the genome (e.g., gene copy number) and changes in DNA sequences.Citation237 RNA's many facets make it likely that small RNAs play a role in this “continuous adaptation spectrum.”
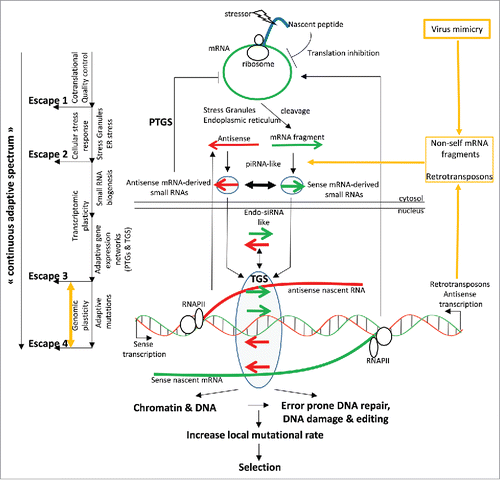
During the translation of an mRNA, a perturbation (or “local stressor”) affecting the nascent protein can inhibit translation (). A “local stressor” is defined as anything that can disturb events occurring during the translation of an mRNA (Fig. S2). This could be (i) a molecule that interferes directly with a nascent protein target; (ii) a protein modification; (iii) a mutation affecting the folding or activity of the protein coded by the translated mRNA; (iv) a mutation affecting a protein interacting with the nascent protein; and (v) or the alteration of the concentration of a protein, interacting with the nascent protein that is important for its folding or its assembly within a complex. Mechanisms involving translation-inhibition of local stress-associated mRNAs are part of the well-characterized co-translational quality control process, which can momentarily limit the synthesis of proteins in an unfavorable local environment (). However, if the mRNAs encoding the stress targets are continuously locally delivered and if the stress situation persists, translationally-stalled mRNAs may aggregate and form stress granules activating survival pathways (the well characterized so-called “cellular stress response”). The cellular stress response, which includes mRNA cleavage by endoribonucleases, either alleviates the stress situation or induces cellular senescence or apoptosis ().
Figure 6. (A) Stressor can affect a nascent polypeptide chain and trigger translation inhibition, which can momentarily limit the synthesis of proteins in an unfavorable local environment (“co-translational quality control,” “escape 1”). However, if the stress persists in inducing, for example, an unfolded protein response (ER stress) and if translationally-stalled mRNAs accumulate, they may form stress granules activating a cellular stress response that can collectively either alleviate the stress situation or induce cellular senescence or apoptosis (“Cellular stress response,” “escape 2”). Sustained-stress situations within a cell population induce cell reprogramming (dedifferentiation), leading in particular to retrotransposon expression and antisense transcription. In these conditions, stress-associated mRNAs could be used as substrates to generate mRNA-derived small RNAs through either the “piRNA” or the “endo-siRNA” pathways. Antisense small RNAs could trigger the degradation of the corresponding mRNAs, thereby providing a post-transcriptional adaptive regulatory loop for limiting the accumulation of translationally-stalled mRNAs (PTGS, “escape 3”). mRNA-derived small RNAs may also target the corresponding genomic loci and induce chromatin and DNA modifications leading to transcriptional gene silencing, thereby providing a transcriptional adaptive regulatory loop for limiting the synthesis of stress-associated mRNAs (TGS, “escape 3”). In the context of the virus infection-like microenvironment, which induces the expression of DNA metabolic enzymes that contributing to “genomic plasticity,” including DNA-editing enzymes, small RNAs may increase the local mutational rate of the corresponding and targeted genomic loci through different mechanisms, which would increase the probability of mutation appearance contributing to alleviate the stress situation (escape 4).
Within a population of cells exposed to the same stress, the cellular microenvironment can become overloaded with dying and senescent cells, which can activate local inflammation. In addition, cellular reprogramming plays an important role within a damaged tissue (i.e., the tumor exposed to killing agents) as it allows some cells to express gene networks driving a potential adaptive phenotype (e.g., trans-differentiation); it also allows some cells to de-differentiate and to provide new stem cells for repopulation. Therefore, long-term stress exposure of a cell population results in cell reprogramming and expression of stem and germ cell-restricted factors, including retrotransposons and factors involved in small RNA biogenesis. In this situation, stressed-induced mRNA fragments may initiate the production of mRNA-derived small RNAs, either within the same cell or after being released and captured by a neighboring cell.
Sense mRNA fragments combined with antisense transcripts can trigger the production of sense and anti-sense small RNAs (e.g., piRNAs and endo-siRNAs, , Figs. S5 and S6). Antisense small RNAs could trigger the degradation of the corresponding mRNAs, therefore providing a post-transcriptional adaptive regulatory loop (i.e., PTGS) for limiting the accumulation of translationally-stalled mRNAs (). Both sense and antisense mRNA-derived small RNAs may also target the corresponding genomic loci and induce chromatin and DNA modifications leading to TGS, therefore providing a transcriptional adaptive regulatory loop for limiting the synthesis of stress-associated mRNAs (). Small RNAs present on DNA may increase the local mutational rate of the corresponding genomic loci through direct or indirect mechanisms. Therefore, they may increase the probability of generating mutations that alleviate the stress ().
In this scenario, RNAs allow a “continuous adaptation spectrum” whose progression is driven by the sustained stress situation. The proposed model relies on the recently recognized “plasticity” of cancer cells that, when exposed to sustained stress situations, adapt by “reactivating” various pathways and retrotransposon expression patterns that are normally restricted to stem and/or germ cells. However, these reprogramming events occur in a microenvironment that mimics viral infection, which activates molecular tools involved in cell immunity, including DNA-editing enzymes. The combination of these processes may provide tumor cells with the conditions for RNA-directed adaptive mutations and cell-autonomous autoimmunity (Fig. S7).
Anticancer drug resistance has become a major clinical and public health problem. Understanding the molecular mechanisms behind the capability of cancer cells to adapt to the tumor microenvironment and to anticancer therapies, which are basically nothing other than new stressful situations for the tumor cells, is a major challenge toward the design of new therapeutic strategies. There is an urgent need to develop “evolution-based methods” that take into account cancer cell plasticity as recently proposed Citation7-11 and to develop strategies restricting the genomic plasticity of cancer cells.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
BOX 1: Context-dependent mutations
The appearance of mutations (from single-base mutations to complex genomic rearrangements) relies on the complex interplay between DNA repair and DNA injuries, or damage, caused by endogenous or exogenous mutators. Mutation probability increases by increasing DNA damage and/or by the modulation of DNA repair efficiency and fidelity, as some DNA repair mechanisms are error-prone processes, notably because of the transient formation of ssDNA during the DNA repair process.Citation25-27,32-34 Supporting a model where mutation pattern depends on specific DNA damage and/or repair mechanisms, genome-wide analysis of cancer mutations led to the recent discovery of more or less specific mutational signatures in each cancer type analyzed.Citation25-27 In addition, it is now widely accepted that contextual DNA sequence, local DNA structure, DNA replication timing, chromatin status, and genome topology influence both DNA damage and DNA repair. DNA methylation is a good example of a direct link between DNA context and mutations. Indeed, DNA methylation by cytosine-5-methyltransferases, that plays an important role in genome imprinting and transcription regulation, increases the mutational probability owing to spontaneous deamination, further modifications of methylated cytosines, and active demethylation processes.Citation38 Mutation spectra in cancers and mechanistic studies have demonstrated that chromatin organization is a major determinant of variation in regional mutation rate since chromatin modifications can impact on (i) DNA accessibility by DNA damage agents and/or by the DNA repair machineries; and (ii) some specific histones modifications that recruit DNA repair machineries; iii) and some DNA repair complexes (e.g., transcription-coupled repair) that cooperate with the transcriptional machinery.Citation19,35,39 Local DNA structures, like G-quadruplex structures and R-loops (see also BOX 4), render chromosomes fragile by increasing DNA DSBs and forming ssDNA. Indeed, these structures are resolved by endonucleases of the DNA repair process pathway leading to DSBs and may block DNA polymerase progression during replication causing fork collapse and subsequently DSBs.Citation36,37 DSBs can then result in translocation events and can increase local mutational rate because of error-prone DNA synthesis that occur during repair of DSBs or because of unrepaired lesions in regions of ssDNA created around break sites. The formation of ssDNA in local DNA structures, like R-loops, also contributes to an increase in the mutational rate, as ssDNA is more accessible to DNA-damaging agents and is the preferential substrate of DNA-editing enzymes, like APOBECs (apolipoprotein B mRNA editing enzymes) and AID (activation-induced deaminase).Citation35-37 Indeed, editing enzymes induce deamination of cytosines in ssDNA leading to uracils, which can either be repaired or lead to cytosine to thymine transition. By mutating DNA sequences, editing enzymes of the AID/APOBEC family play a major role in physiological functions such as driving immunoglobulin diversity and inhibiting retrotransposon and virus propagation.Citation71,72,75,212-214 Both APOBECs and AID enzymes play a major role in generating mutations in cancer cells.Citation28-31
BOX 2: RNA-directed chromatin modifications
Increasing evidence supports the notion that RNAs direct chromatin modifications in human cells. Indeed, many proteins involved in RNA-mediated chromatin modifications, including the Dicer and Argonaute proteins, have been detected in nuclei and have been shown to play a role in chromatin modifications in mammalian somatic cells.Citation43-47 There are also many examples of long non-coding RNAs (lncRNAs, >200 nts) that play a role in histone and DNA modification, acting either in trans (i.e., on different loci from their production site) or in cis (i.e., they tether proteins involved in chromatin modification on the loci or in proximity to the loci where they are produced).Citation48 For example, production of the p15 antisense lncRNA controls the silencing of the sense p15 gene in cis by triggering heterochromatin formation in a Dicer-dependent manner.Citation49 In addition, the transfection of designed small RNAs, similar to naturally occurring ones, induces targeted-gene expression modification (either repression or activation), chromatin modifications, or DNA methylation in human cell lines.Citation50 For example, several repeated sequences in the human genome have been shown to produce small RNAs that, when transfected into cells, induce locus-specific histone and DNA modifications.Citation51,52 Likewise transfection of various human cells with piRNA-like molecules results in targeted histone and DNA modifications.Citation47,53-56 Also, some miRNAs seem to target specific gene promoters and to modulate gene transcription activity and local chromatin modifications, even though more experiments are needed to ascertain whether these effects are direct.Citation57
BOX 3: piRNA biogenesis
The biogenesis and functions of piRNAs have been particularly well characterized in drosophila and mouse germ cells, where one of their main functions is to repress retrotransposons, which are repeated sequences expressed in germ cells likely due to chromatin decondensation at specific stages of gametogenesis. In these organisms, single-strand piRNA precursors transcriptionally derived from retrotransposon sequences are exported to the cytoplasm where they are cleaved by the Zucchini endoribonuclease, which contains an N-terminal transmembrane domain attached to mitochondria, in the vicinity of which several steps of piRNA biogenesis occurCitation151-153 (). Maelstrom is another mouse endoribonuclease that cleaves piRNA precursors.Citation238 The cleaved piRNA intermediates are next loaded into Piwi proteins and trimmed by a 3′ to 5′ exoribonuclease, which degrades the RNAs until the region of the RNAs protected by the Piwi protein is reached. In drosophila, this primary process can next initiate a second mechanism of piRNA production known as “ping-pong” that relies on the presence of sense and antisense piRNA precursors. First, an antisense piRNA, produced as described above, is used to cleave a targeted sense transcript, whose product produces a mature sense piRNA, which can in turn, induces cleavage of an antisense piRNA precursor (). This “ping-pong” amplification mechanism allows the cell to adapt the production of piRNAs depending on the abundance of the precursors.Citation151-153 Overall, it is estimated that the piRNA biogenesis pathway is composed of 50 proteins most of which have been conserved during evolution.Citation151-153 These include several RNA helicases like DDX4 (MVH) and Mov10L1, as well as a large family of proteins that contain Tudor domains (TDRDs), including TDRD1 to TDRD12 that function as scaffolds and platforms in the piRNA biogenesis pathway, which mostly occurs in cytoplasmic granules in the vicinity of mitochondria. TDRD proteins are key regulators of the piRNA biogenesis pathway as recently shown for TUDOR-SN or TRDR11Citation151-153 and contribute to the selectivity of the piRNA biogenesis pathway. Indeed, it has been shown in mice that TDRD4 (or RNF17) and TDRD1 play a major role in selecting appropriate piRNA precursors, as depletion of TDRD1 or TDRD4 leads to the amplification of mRNA-derived piRNAs.Citation239,240 Notably, recent reports in mice have suggested that some piRNAs can trigger mRNA cleavage, which in turn initiates a ping-pong like process.Citation153,162,163 These observations are supported by the identification of piRNA-like molecules mapping to protein-coding genes in several species including human, as piRNA-like molecules derived from mRNA 3′ UTRs or from mRNA internal exons have been reported.Citation53,54,154,162-169 Several recent reports have indicated that piRNA-like molecules also derive from pseudogenes and are capable of inducing the cleavage of their parental gene's transcripts.Citation166,170-172 Collectively, these observations support the idea that mRNAs are used to generate piRNAs or piRNA-like molecules, which seems to occur in cancer cells.
BOX 4: R-loops and G-quadruplexes
RNAs can theoretically target specific loci and induce the formation of R-loops (). R-loops result from the Watson–Crick base-pairing of an RNA molecule to the cDNA strand which displaces the second DNA strand into a single-stranded conformation.Citation41 Until recently, it was postulated that R-loops only occur during transcription, when the nascent RNA hybridizes to the cDNA strand, and during replication, where DNA copying is initiated by the transcription of RNA primers.Citation41 It was believed that RNA invasion into dsDNA is limited by the stability of nucleosome-bound dsDNA structures. However, recent evidence has suggested that the presence of R-loops does not depend solely on transcription and some intriguing recent results have suggested that dedicated mechanisms favor the formation of R-loops.Citation41 In particular, factors involved in homologous recombination may support the formation of RNA:DNA hybrids. For example, the bacterial RecA and the human RECQL5 proteins have both been shown to promote RNA:dsDNA hybrids in vitro.Citation241,242 It has recently been shown in yeast that Rad51 (the homolog of RecA), which is known to promote ssDNA invasion of a homologous DNA region at DNA DSBs, also promotes RNA:DNA hybrid formation in vivo.Citation243 As described in the part “RNA-directed DNA repair and editing,” this mechanism may play a role in the recently discovered mechanism of DSB homologous recombination repair in which RNAs are used as templates. This opens the possibility that some proteins may promote RNA invasion into dsDNA and induce the formation of R-loops.Citation243 Another mechanism that may favor the invasion of dsDNAs by RNA relies on specific DNA structures that expose short ssDNA regions. In this context, a strong association between R-loop formation and G-quadruplex structures has been reported. G-quadruplex structures are hairpins and stacks of Hoogsteen base pair-stabilized guanine tetrads that can form within G-rich DNA strands and displace the C-rich complementary single strand.Citation37,244 The strong association between R-loops and G-quadruplexes may result either from RNA:DNA hybrid favoring G-quadruplex formation within the G-rich displaced ssDNA, or conversely, from G-quadruplex occurring first and the displaced C-rich ssDNA being more prone to be hybridized by complementary RNAs. If the interacting RNAs contain G-tracts, it may contribute to the stabilization of the G-quadruplex structures since it has been shown that intermolecular G-quadruplexes between RNA and DNA further stabilize the G-rich hybrids.Citation245
1221491_Supplemental_Material.docx
Download MS Word (358 KB)Acknowledgment
I thank Cyril Bourgeois, Franck Mortreux, Martin Dutertre, Marina Touillaud, and Julian Venables for a critical reading of the manuscript.
References
- Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science 2015; 349:1483-1489.
- Gerlinger M, McGranahan N, Dewhurst SM, Burrell RA, Tomlinson I, Swanton C. Cancer: evolution within a lifetime. Annu Rev Genet 2014; 48:215-236.
- Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science 2011; 331:1553-1558.
- Sidow A, Spies N. Concepts in solid tumor evolution. Trends Genet 2015; 31:208-214.
- Krogan NJ, Lippman S, Agard DA, Ashworth A, Ideker T. The cancer cell map initiative: defining the hallmark networks of cancer. Mol Cell 2015; 58:690-698.
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014; 505:495-501.
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13:714-726.
- Wood KC. Mapping the Pathways of Resistance to Targeted Therapies. Cancer Res 2015; 75:4247-4251.
- Enriquez-Navas PM, Wojtkowiak JW, Gatenby RA. Application of Evolutionary Principles to Cancer Therapy. Cancer Res 2015; 75:4675-4680.
- Hughes D, Andersson DI. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet 2015; 16:459-471.
- Willyard C. Cancer therapy: an evolved approach. Nature 2016; 532:166-168.
- Ben-David U. Genomic instability, driver genes and cell selection: Projections from cancer to stem cells. Biochim Biophys Acta 2015; 1849:427-435.
- Campos-Sanchez E, Cobaleda C. Tumoral reprogramming: Plasticity takes a walk on the wild side. Biochim Biophys Acta 2015; 1849:436-447.
- Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: [What does not kill me strengthens me]. Br J Cancer 2015; 112:1725-1732.
- Goding CR, Pei D, Lu X. Cancer: pathological nuclear reprogramming? Nat Rev Cancer 2014; 14:568-573.
- Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 2014; 54:716-727.
- Bhat R, Bissell MJ. Of plasticity and specificity: dialectics of the microenvironment and macroenvironment and the organ phenotype. Wiley Interdiscip Rev Dev Biol 2014; 3:147-163.
- Johnstone SE, Baylin SB. Stress and the epigenetic landscape: a link to the pathobiology of human diseases? Nat Rev Genet 2010; 11:806-812.
- Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell 2013; 153:38-55.
- Rosenberg SM, Queitsch C. Medicine. Combating evolution to fight disease. Science 2014; 343:1088-1089.
- Foster PL. Stress responses and genetic variation in bacteria. Mutat Res 2005; 569:3-11.
- Wright BE. Stress-directed adaptive mutations and evolution. Mol Microbiol 2004; 52:643-650.
- Rosenberg SM. Evolving responsively: adaptive mutation. Nat Rev Genet 2001; 2:504-515.
- Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature 1988; 335:142-145.
- Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 2014; 15:585-598.
- Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev 2014; 24:52-60.
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature 2013; 500:415-421.
- Henderson S, Fenton T. APOBEC3 genes: retroviral restriction factors to cancer drivers. Trends Mol Med 2015; 21:274-284.
- Rebhandl S, Huemer M, Greil R, Geisberger R. AID/APOBEC deaminases and cancer. Oncoscience 2015; 2:320-333.
- Swanton C, McGranahan N, Starrett GJ, Harris RS. APOBEC enzymes: Mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov 2015; 5:704-712.
- Knisbacher BA, Gerber D, Levanon EY. DNA Editing by APOBECs: A Genomic Preserver and Transformer. Trends Genet 2016; 32:16-28.
- Chen J, Furano AV. Breaking bad: The mutagenic effect of DNA repair. DNA Repair (Amst) 2015; 32:43-1.
- Krokan HE, Sætrom P, Aas PA, Pettersen HS, Kavli B, Slupphaug G. Error-free versus mutagenic processing of genomic uracil-relevance to cancer. DNA Repair (Amst) 2014; 19:38-47.
- Chan K, Gordenin DA. Clusters of multiple mutations: incidence and molecular mechanisms. Annu Rev Genet 2015; 49:243-267.
- Javadekar SM, Raghavan SC. Snaps and mends: DNA breaks and chromosomal translocations. FEBS J 2015; 282:2627-2645.
- Sollier J, Stork CT, García-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 2014; 56:777-785.
- van Kregten M, Tijsterman M. The repair of G-quadruplex-induced DNA damage. Exp Cell Res 2014; 329:178-183.
- Meng H, Cao Y, Qin J, Song X, Zhang Q, Shi Y, Cao L. DNA methylation, its mediators and genome integrity. Int J Biol Sci 2015; 11:604-617.
- Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012; 488:504-507.
- Roukos V, Misteli T. The biogenesis of chromosome translocations. Nat Cell Biol 2014; 16:293-300.
- Sollier J, Cimprich KA. Breaking bad: R-loops and genome integrity. Trends Cell Biol 2015; 25:514-522.
- Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, Balasubramanian S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat Biotechnol 2015; 33:877-881.
- Gagnon KT, Li L, Chu Y, Janowski BA, Corey DR. RNAi factors are present and active in human cell nuclei. Cell Rep 2014; 6:211-221.
- Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat Struct Mol Biol 2006; 13:793-797.
- Janowski BA, Huffman KE, Schwartz JC, Ram R, Nordsell R, Shames DS, Minna JD, Corey DR. Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat Struct Mol Biol 2006; 13:787-792.
- Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004; 305:1289-1292.
- Siddiqi S, Terry M, Matushansky I. Hiwi mediated tumorigenesis is associated with DNA hypermethylation. PLoS One 2012; 7:e33711.
- Goff LA, Rinn JL. Linking RNA biology to lncRNAs. Genome Res 2015; 25:1456-1465.
- Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 2008; 451:202-206.
- Wang J, Place RF, Portnoy V, Huang V, Kang MR, Kosaka M, Ho MK, Li LC. Inducing gene expression by targeting promoter sequences using small activating RNAs. J Biol Methods 2015; 2:e14.
- Lim JW, Snider L, Yao Z, Tawil R, Van Der Maarel SM, Rigo F, Bennett CF, Filippova GN, Tapscott SJ. DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum Mol Genet 2015; 24:4817-4828.
- Pohlers M, Calabrese JM, Magnuson T. Small RNA expression from the human macrosatellite DXZ4. G3 (Bethesda) 2014; 4:1981-1989.
- Fu A, Jacobs DI, Zhu Y. Epigenome-wide analysis of piRNAs in gene-specific DNA methylation. RNA Biol 2014; 11:1301-1312.
- Cichocki F, Lenvik T, Sharma N, Yun G, Anderson SK, Miller JS. Cutting edge: KIR antisense transcripts are processed into a 28-base PIWI-like RNA in human NK cells. J Immunol 2010; 185:2009-2012.
- Fu A, Jacobs DI, Hoffman AE, Zheng T, Zhu Y. PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis 2015; 36:1094-1102.
- Wu D, Fu H, Zhou H, Su J, Zhang F, Shen J. Effects of Novel ncRNA Molecules, p15-piRNAs, on the Methylation of DNA and Histone H3 of the CDKN2B Promoter Region in U937 Cells. J Cell Biochem 2015; 116:2744-2754.
- Toscano-Garibay JD, Aquino-Jarquin G. Transcriptional regulation mechanism mediated by miRNA-DNA#DNA triplex structure stabilized by Argonaute. Biochim Biophys Acta 2014; 1839:1079-1083.
- Francia S. Non-Coding RNA: Sequence-specific guide for chromatin modification and DNA damage signaling. Front Genet 2015; 6:320.
- Yang YG, Qi Y. RNA-directed repair of DNA double-strand breaks. DNA Repair (Amst) 2015; 32:82-85.
- d'Adda di Fagagna F. A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol 2014; 24:171-178.
- Chowdhury D, Choi YE, Brault ME. Charity begins at home: non-coding RNA functions in DNA repair. Nat Rev Mol Cell Biol 2013; 14:181-189.
- Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d'Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012; 488:231-235.
- Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang YG, Qi Y. A role for small RNAs in DNA double-strand break repair. Cell 2012; 149:101-112.
- Oliver C, Santos JL, Pradillo M. On the role of some ARGONAUTE proteins in meiosis and DNA repair in Arabidopsis thaliana. Front Plant Sci 2014; 5:177.
- Gao M, Wei W, Li MM, Wu YS, Ba Z, Jin KX, Li MM, Liao YQ, Adhikari S, Chong Z, et al. Ago2 facilitates Rad51 recruitment and DNA double-strand break repair by homologous recombination. Cell Res 2014; 24:532-541.
- Wei L, Nakajima S, Böhm S, Bernstein KA, Shen Z, Tsang M, Levine AS, Lan L. DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc Natl Acad Sci U S A 2015; 112:E3495-3504.
- Keskin H, Meers C, Storici F. Transcript RNA supports precise repair of its own DNA gene. RNA Biol 2016; 13:157-165.
- Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, Storici F. Transcript-RNA-templated DNA recombination and repair. Nature 2014; 515:436-439.
- Shen Y, Nandi P, Taylor MB, Stuckey S, Bhadsavle HP, Weiss B, Storici F. RNA-driven genetic changes in bacteria and in human cells. Mutat Res 2011; 717:91-98.
- Storici F, Bebenek K, Kunkel TA, Gordenin DA, Resnick MA. RNA-templated DNA repair. Nature 2007; 447:338-341.
- Richardson SR, Narvaiza I, Planegger RA, Weitzman MD, Moran JV. APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. Elife 2014; 3:e02008.
- Koito A, Ikeda T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front Microbiol 2013; 4:28.
- Sabin LR, Delas MJ, Hannon GJ. Dogma derailed: the many influences of RNA on the genome. Mol Cell 2013; 49:783-794.
- Nowacki M, Haye JE, Fang W, Vijayan V, Landweber LF. RNA-mediated epigenetic regulation of DNA copy number. Proc Natl Acad Sci U S A 2010; 107:22140-22144.
- Kenter AL, Kumar S, Wuerffel R, Grigera F. AID hits the jackpot when missing the target. Curr Opin Immunol 2016; 39:96-102.
- Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, Chaudhuri J. Non-coding RNA generated following lariat debranching mediates targeting of AID to DNA. Cell 2015; 161:762-773.
- Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, Economides AN, Basu U. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature 2014; 514:389-393.
- Shapiro JA. The basic concept of the read-write genome: Mini-review on cell-mediated DNA modification. Biosystems 2016; 140:35-37.
- Mattick JS, Mehler MF. RNA editing, DNA recoding and the evolution of human cognition. Trends Neurosci 2008; 31:227-233.
- Hung MC, Link W. Protein localization in disease and therapy. J Cell Sci 2011; 124:3381-3392.
- Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol 2014; 31:2-10.
- Buxbaum AR, Haimovich G, Singer RH. In the right place at the right time: visualizing and understanding mRNA localization. Nat Rev Mol Cell Biol 2015; 16:95-109.
- Jansen RP, Niessing D, Baumann S, Feldbrugge M. mRNA transport meets membrane traffic. Trends Genet 2014; 30:408-417.
- Irannejad R, Tsvetanova NG, Lobingier BT, von Zastrow M. Effects of endocytosis on receptor-mediated signaling. Curr Opin Cell Biol 2015; 35:137-143.
- Breiman A, Fieulaine S, Meinnel T, Giglione C. The intriguing realm of protein biogenesis: Facing the green co-translational protein maturation networks. Biochim Biophys Acta 2015; 1864:531-550.
- Giglione C, Fieulaine S, Meinnel T. N-terminal protein modifications: Bringing back into play the ribosome. Biochimie 2015; 114:134-146.
- Walte A, Rüben K, Birner-Gruenberger R, Preisinger C, Bamberg-Lemper S, Hilz N, Bracher F, Becker W. Mechanism of dual specificity kinase activity of DYRK1A. FEBS J 2013; 280:4495-4511.
- Keshwani MM, Klammt C, von Daake S, Ma Y, Kornev AP, Choe S, Insel PA, Taylor SS. Cotranslational cis-phosphorylation of the COOH-terminal tail is a key priming step in the maturation of cAMP-dependent protein kinase. Proc Natl Acad Sci U S A 2012; 109:E1221-1229.
- Hovland R, Doskeland AP, Eikhom TS, Robaye B, Doskeland SO. cAMP induces co-translational modification of proteins in IPC-81 cells. Biochem J 1999; 342(Pt 2):369-377.
- Rao PS, Satelli A, Zhang S, Srivastava SK, Srivenugopal KS, Rao US. RNF2 is the target for phosphorylation by the p38 MAPK and ERK signaling pathways. Proteomics 2009; 9:2776-2787.
- Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, Roux PP, Su B, Jacinto E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J 2010; 29:3939-3951.
- Tobias IS, Kaulich M, Kim PK, Simon N, Jacinto E, Dowdy SF, King CC, Newton AC. Protein kinase Czeta exhibits constitutive phosphorylation and phosphatidylinositol-3,4,5-triphosphate-independent regulation. Biochem J 2016; 473:509-523.
- Dai N, Christiansen J, Nielsen FC, Avruch J. mTOR complex 2 phosphorylates IMP1 cotranslationally to promote IGF2 production and the proliferation of mouse embryonic fibroblasts. Genes Dev 2013; 27:301-312.
- Castello A, Hentze MW, Preiss T. Metabolic Enzymes enjoying new partnerships as RNA-binding proteins. Trends Endocrinol Metab 2015; 26:746-757.
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012; 149:1393-1406.
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 2012; 46:674-690.
- Duncan CD, Mata J. Widespread cotranslational formation of protein complexes. PLoS Genet 2011; 7:e1002398.
- Duncan CD, Mata J. Cotranslational protein-RNA associations predict protein-protein interactions. BMC Genomics 2014; 15:298.
- Berkovits BD, Mayr C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015; 522:363-367.
- Wells JN, Bergendahl LT, Marsh JA. Co-translational assembly of protein complexes. Biochem Soc Trans 2015; 43:1221-1226.
- Nicholls CD, McLure KG, Shields MA, Lee PW. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J Biol Chem 2002; 277:12937-12945.
- Mordmuller B, Krappmann D, Esen M, Wegener E, Scheidereit C. Lymphotoxin and lipopolysaccharide induce NF-kappaB-p52 generation by a co-translational mechanism. EMBO Rep 2003; 4:82-87.
- Hansen HT, Rasmussen SH, Adolph SK, Plass M, Krogh A, Sanford J, Nielsen FC. Christiansen J8. Drosophila Imp iCLIP identifies an RNA assemblage coordinating F-actin formation. Genome Biol 2015; 16:123.
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 2015; 10:173-194.
- Plumb R, Zhang ZR, Appathurai S, Mariappan M. A functional link between the co-translational protein translocation pathway and the UPR. eLife 2015; 4:e07426.
- Lykke-Andersen J, Bennett EJ. Protecting the proteome: Eukaryotic cotranslational quality control pathways. J Cell Biol 2014; 204:467-476.
- Pechmann S, Willmund F, Frydman J. The ribosome as a hub for protein quality control. Mol Cell 2013; 49:411-421.
- Inada T, Makino S. Novel roles of the multi-functional CCR4-NOT complex in post-transcriptional regulation. Front Genet 2014; 5:135.
- Walters RW, Parker R. Coupling of Ribostasis and Proteostasis: Hsp70 Proteins in mRNA Metabolism. Trends Biochem Sci 2015; 40:552-559.
- Liu B, Han Y, Qian SB. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol Cell 2013; 49:453-463.
- Pelechano V, Wei W, Steinmetz LM. Widespread Co-translational RNA Decay Reveals Ribosome Dynamics. Cell 2015; 161:1400-1412.
- Reid DW, Nicchitta CV. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat Rev Mol Cell Biol 2015; 16:221-231.
- Twiss JL, Merianda TT. Old dogs with new tricks: intra-axonal translation of nuclear proteins. Neural Regen Res 2015; 10:1560-1562.
- Comel A, Sorrentino G, Capaci V, Del Sal G. The cytoplasmic side of p53s oncosuppressive activities. FEBS Lett 2014; 588:2600-2609.
- Monaghan RM, Whitmarsh AJ. Mitochondrial Proteins Moonlighting in the Nucleus. Trends Biochem Sci 2015; 40:728-735.
- Trowitzsch S, Viola C, Scheer E, Conic S, Chavant V, Fournier M, Papai G, Ebong IO, Schaffitzel C, Zou J, et al. Cytoplasmic TAF2-TAF8-TAF10 complex provides evidence for nuclear holo-TFIID assembly from preformed submodules. Nat Commun 2015; 6:6011.
- Carre C, Shiekhattar R. Human GTPases associate with RNA polymerase II to mediate its nuclear import. Mol Cell Biol 2011; 31:3953-3962.
- Weatheritt RJ, Gibson TJ, Babu MM. Asymmetric mRNA localization contributes to fidelity and sensitivity of spatially localized systems. Nat Struct Mol Biol 2014; 21:833-839.
- Mercer TR, Dinger ME, Bracken CP, Kolle G, Szubert JM, Korbie DJ, Askarian-Amiri ME, Gardiner BB, Goodall GJ, Grimmond SM, et al. Regulated post-transcriptional RNA cleavage diversifies the eukaryotic transcriptome. Genome Res 2010; 20:1639-1650.
- Kiss DL, Oman K, Bundschuh R, Schoenberg DR. Uncapped 5′ ends of mRNAs targeted by cytoplasmic capping map to the vicinity of downstream CAGE tags. FEBS Lett 2015; 589:279-284.
- Affymetrix, E.T.P. & Cold Spring Harbor Laboratory, E.T.P. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature 2009; 457:1028-1032.
- Norbury CJ. Cytoplasmic RNA: a case of the tail wagging the dog. Nat Rev Mol Cell Biol 2013; 14:643-653.
- Abdelhamid RF, Plessy C, Yamauchi Y, Taoka M, de Hoon M, Gingeras TR, Isobe T, Carninci P. Multiplicity of 5′ cap structures present on short RNAs. PLoS One 2014; 9:e102895.
- Chang H, Lim J, Ha M, Kim VN. TAIL-seq: genome-wide determination of poly(A) tail length and 3′ end modifications. Mol Cell 2014; 53:1044-1052.
- Li WM, Barnes T, Lee CH. Endoribonucleases–enzymes gaining spotlight in mRNA metabolism. FEBS J 2010; 277:627-641.
- Tomecki R, Dziembowski A. Novel endoribonucleases as central players in various pathways of eukaryotic RNA metabolism. RNA 2010; 16:1692-1724.
- Schoenberg DR. Mechanisms of endonuclease-mediated mRNA decay. Wiley Interdiscip Rev RNA 2011; 2:582-600.
- Arraiano CM, Mauxion F, Viegas SC, Matos RG, Seraphin B. Intracellular ribonucleases involved in transcript processing and decay: precision tools for RNA. Biochim Biophys Acta 2013; 1829:491-513.
- Malathi K, Dong B, Gale M, Jr, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007; 448:816-819.
- Onomoto K, Yoneyama M, Fung G, Kato H, Fujita T. Antiviral innate immunity and stress granule responses. Trends Immunol 2014; 35:420-428.
- Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, Uehata T, Tartey S, Akira S, Suzuki Y, et al. Regnase-1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell 2015; 161:1058-1073; PMID:26000482; http://dx.doi.org/10.1016/j.cell.2015.04.029
- Luhtala N, Parker R. T2 Family ribonucleases: ancient enzymes with diverse roles. Trends Biochem Sci 2010; 35:253-259.
- Morita Y, Shibutani T, Nakanishi N, Nishikura K, Iwai S, Kuraoka I. Human endonuclease V is a ribonuclease specific for inosine-containing RNA. Nat Commun 2013; 4:2273.
- Weissbach R, Scadden AD. Tudor-SN and ADAR1 are components of cytoplasmic stress granules. RNA 2012; 18:462-471.
- Scadden AD. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol 2005; 12:489-496.
- Irvine K, Stirling R, Hume D, Kennedy D. Rasputin, more promiscuous than ever: a review of G3BP. Int J Dev Biol 2004; 48:1065-1077.
- Burger K, Gullerova M. Swiss army knives: non-canonical functions of nuclear Drosha and Dicer. Nat Rev Mol Cell Biol 2015; 16:417-430.
- Karginov FV, Cheloufi S, Chong MM, Stark A, Smith AD, Hannon GJ. Diverse endonucleolytic cleavage sites in the mammalian transcriptome depend upon microRNAs, Drosha, and additional nucleases. Mol Cell 2010; 38:781-788.
- Johanson TM, Keown AA, Cmero M, Yeo JH, Kumar A, Lew AM, Zhan Y, Chong MM. Drosha controls dendritic cell development by cleaving messenger RNAs encoding inhibitors of myelopoiesis. Nat Immunol 2015; 16:1134-1141.
- Chong MM, Zhang G, Cheloufi S, Neubert TA, Hannon GJ, Littman DR. Canonical and alternate functions of the microRNA biogenesis machinery. Genes Dev 2010; 24:1951-1960.
- Shapiro JS, Schmid S, Aguado LC, Sabin LR, Yasunaga A, Shim JV, Sachs D, Cherry S, tenOever BR. Drosha as an interferon-independent antiviral factor. Proc Natl Acad Sci U S A 2014; 111:7108-7113.
- Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 2013; 14:447-459.
- Tuck AC, Tollervey D. RNA in pieces. Trends Genet 2011; 27:422-432.
- Jackowiak P, Nowacka M, Strozycki PM, Figlerowicz M. RNA degradome–its biogenesis and functions. Nucleic Acids Res 2011; 39:7361-7370.
- Desvignes T, Batzel P, Berezikov E, Eilbeck K, Eppig JT, McAndrews MS, Singer A, Postlethwait JH. miRNA Nomenclature: A View Incorporating Genetic Origins, Biosynthetic Pathways, and Sequence Variants. Trends Genet 2015; 31:613-626.
- Okamura K, Ladewig E, Zhou L, Lai EC. Functional small RNAs are generated from select miRNA hairpin loops in flies and mammals. Genes Dev 2013; 27:778-792.
- Emara MM, Ivanov P, Hickman T, Dawra N, Tisdale S, Kedersha N, Hu GF, Anderson P. Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J Biol Chem 2010; 285:10959-10968.
- Peach SE, York K, Hesselberth JR. Global analysis of RNA cleavage by 5′-hydroxyl RNA sequencing. Nucleic Acids Res 2015; 43:e108.
- Bracken CP, Szubert JM, Mercer TR, Dinger ME, Thomson DW, Mattick JS, Michael MZ, Goodall GJ. Global analysis of the mammalian RNA degradome reveals widespread miRNA-dependent and miRNA-independent endonucleolytic cleavage. Nucleic Acids Res 2011; 39:5658-5668.
- Liu TT, Arango-Argoty G, Li Z, Lin Y, Kim SW, Dueck A, Ozsolak F, Monaghan AP, Meister G, DeFranco DB, et al. Noncoding RNAs that associate with YB-1 alter proliferation in prostate cancer cells. RNA 2015; 21:1159-1172.
- Iwasaki YW, Siomi MC, Siomi H. PIWI-Interacting RNA: Its Biogenesis and Functions. Annu Rev Biochem 2015; 84:405-433.
- Fu Q, Wang PJ. Mammalian piRNAs: Biogenesis, function, and mysteries. Spermatogenesis 2014; 4:e27889.
- Czech B, Hannon GJ. One loop to rule them all: The ping-pong cycle and piRNA-guided silencing. Trends Biochem Sci 2016; 41(4):324-337.
- Williams Z, Morozov P, Mihailovic A, Lin C, Puvvula PK, Juranek S, Rosenwaks Z, Tuschl T. Discovery and characterization of piRNAs in the human fetal ovary. Cell Rep 2015; 13:854-863.
- Roovers EF, Rosenkranz D, Mahdipour M, Han CT, He N, Chuva de Sousa Lopes SM, van der Westerlaken LA, Zischler H, Butter F, Roelen BA, et al. Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell Rep 2015; 10:2069-2082.
- Zhang H, Ren Y, Xu H, Pang D, Duan C, Liu C. The expression of stem cell protein Piwil2 and piR-932 in breast cancer. Surg Oncol 2013; 22:217-223.
- Huang G, Hu H, Xue X, Shen S, Gao E, Guo G, Shen X, Zhang X. Altered expression of piRNAs and their relation with clinicopathologic features of breast cancer. Clin Transl Oncol 2013; 15:563-568.
- Siddiqi S, Matushansky I. Piwis and piwi-interacting RNAs in the epigenetics of cancer. J Cell Biochem 2012; 113:373-380.
- Yan H, Wu QL, Sun CY, Ai LS, Deng J, Zhang L, Chen L, Chu ZB, Tang B, Wang K, et al. piRNA-823 contributes to tumorigenesis by regulating de novo DNA methylation and angiogenesis in multiple myeloma. Leukemia 2015; 29:196-206.
- Yang L, Bi L, Liu Q, Zhao M, Cao B, Li D, Xiu J. Hiwi promotes the proliferation of colorectal cancer cells via upregulating global DNA methylation. Dis Markers 2015; 2015:383056.
- Moyano M, Stefani G. piRNA involvement in genome stability and human cancer. J Hematol Oncol 2015; 8:38.
- Goh WS, Falciatori I, Tam OH, Burgess R, Meikar O, Kotaja N, Hammell M, Hannon GJ. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev 2015; 29:1032-1044.
- Zhang P, Kang JY, Gou LT, Wang J, Xue Y, Skogerboe G, Dai P, Huang DW, Chen R, Fu XD, et al. MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res 2015; 25:193-207.
- Yamtich J, Heo SJ, Dhahbi J, Martin DI, Boffelli D. piRNA-like small RNAs mark extended 3′UTRs present in germ and somatic cells. BMC Genomics 2015; 16:462.
- Ha H, Song J, Wang S, Kapusta A, Feschotte C, Chen KC, Xing J. A comprehensive analysis of piRNAs from adult human testis and their relationship with genes and mobile elements. BMC Genomics 2014; 15:545.
- Gebert D, Ketting RF, Zischler H, Rosenkranz D. piRNAs from pig testis provide evidence for a conserved role of the Piwi Pathway in post-transcriptional gene regulation in mammals. PLoS One 2015; 10:e0124860.
- Martinez VD, Vucic EA, Thu KL, Hubaux R, Enfield KS, Pikor LA, Becker-Santos DD, Brown CJ, Lam S, Lam WL. Unique somatic and malignant expression patterns implicate PIWI-interacting RNAs in cancer-type specific biology. Sci Rep 2015; 5:10423.
- Martinez VD, Enfield KS, Rowbotham DA, Lam WL. An atlas of gastric PIWI-interacting RNA transcriptomes and their utility for identifying signatures of gastric cancer recurrence. Gastric Cancer 2016; 19(2):660-5.
- Chu H, Hui G, Yuan L, Shi D, Wang Y, Du M, Zhong D, Ma L, Tong N, Qin C, et al. Identification of novel piRNAs in bladder cancer. Cancer Lett 2015; 356:561-567.
- Watanabe T, Cheng EC, Zhong M, Lin H. Retrotransposons and pseudogenes regulate mRNAs and lncRNAs via the piRNA pathway in the germline. Genome Res 2015; 25:368-380.
- Pantano L, Jodar M, Bak M, Ballescà JL, Tommerup N, Oliva R, Vavouri T. The small RNA content of human sperm reveals pseudogene-derived piRNAs complementary to protein-coding genes. RNA 2015; 21:1085-1095.
- Hirano T, Iwasaki YW, Lin ZY, Imamura M, Seki NM, Sasaki E, Saito K, Okano H, Siomi MC, Siomi H. Small RNA profiling and characterization of piRNA clusters in the adult testes of the common marmoset, a model primate. RNA 2014; 20:1223-1237.
- Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 2015; 16:71-84.
- Mani SR, Juliano CE. Untangling the web: the diverse functions of the PIWI/piRNA pathway. Mol Reprod Dev 2013; 80:632-664.
- Bacolla A, Wang G, Vasquez KM. New perspectives on DNA and RNA triplexes as effectors of biological activity. PLoS Genet 2015; 11:e1005696.
- Schmitz KM, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 2010; 24:2264-2269.
- Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, Jaffrey SR. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014; 343:1002-1005.
- Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, Seidel R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci U S A 2014; 111:9798-9803.
- Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 2014; 32:569-576.
- Taylor BJ, Wu YL, Rada C. Active RNAP pre-initiation sites are highly mutated by cytidine deaminases in yeast, with AID targeting small RNA genes. Elife 2014; 3:e03553.
- Bach D, Peddi S, Mangeat B, Lakkaraju A, Strub K, Trono D. Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 2008; 5:54.
- Whitehurst AW. Cause and consequence of cancer/testis antigen activation in cancer. Annu Rev Pharmacol Toxicol 2014; 54:251-272.
- Tan Y, Liu L, Liao M, Zhang C, Hu S, Zou M, Gu M, Li X. Emerging roles for PIWI proteins in cancer. Acta Biochim Biophys Sin (Shanghai) 2015; 47:315-324.
- Ng KW, Anderson C, Marshall EA, Minatel BC, Enfield KS, Saprunoff HL, Lam WL, Martinez VD. Piwi-interacting RNAs in cancer: emerging functions and clinical utility. Mol Cancer 2016; 15:5.
- van Wolfswinkel JC. Piwi and potency: PIWI proteins in animal stem cells and regeneration. Integr Comp Biol 2014; 54:700-713.
- Akiyama Y, Komiyama M, Miyata H, Yagoto M, Ashizawa T, Iizuka A, Oshita C, Kume A, Nogami M, Ito I, et al. Novel cancer-testis antigen expression on glioma cell lines derived from high-grade glioma patients. Oncol Rep 2014; 31:1683-1690.
- Yoon H, Lee H, Kim HJ, You KT, Park YN, Kim H, Kim H. Tudor domain-containing protein 4 as a potential cancer/testis antigen in liver cancer. Tohoku J Exp Med 2011; 224:41-6.
- Scanlan MJ, Welt S, Gordon CM, Chen YT, Gure AO, Stockert E, Jungbluth AA, Ritter G, Jäger D, Jäger E, et al. Cancer-related serological recognition of human colon cancer: identification of potential diagnostic and immunotherapeutic targets. Cancer Res 2002; 62:4041-4047.
- Yokoe T, Tanaka F, Mimori K, Inoue H, Ohmachi T, Kusunoki M, Mori M. Efficient identification of a novel cancer/testis antigen for immunotherapy using three-step microarray analysis. Cancer Res 2008; 68:1074-1082.
- Pellon-Maison M, Montanaro MA, Lacunza E, Garcia-Fabiani MB, Soler-Gerino MC, Cattaneo ER, Quiroga IY, Abba MC, Coleman RA, Gonzalez-Baro MR. Glycerol-3-phosphate acyltranferase-2 behaves as a cancer testis gene and promotes growth and tumorigenicity of the breast cancer MDA-MB-231 cell line. PLoS One 2014; 9:e100896.
- Liu M, Chen J, Hu L, Shi X, Zhou Z, Hu Z, Sha J. HORMAD2/CT46.2, a novel cancer/testis gene, is ectopically expressed in lung cancer tissues. Mol Hum Reprod 2012; 18:599-604.
- Tsuchiya N, Ochiai M, Nakashima K, Ubagai T, Sugimura T, Nakagama H. SND1, a component of RNA-induced silencing complex, is up-regulated in human colon cancers and implicated in early stage colon carcinogenesis. Cancer Res 2007; 67:9568-9576.
- Xiao L, Wang Y, Zhou Y, Sun Y, Sun W, Wang L, Zhou C, Zhou J, Zhang J. Identification of a novel human cancer/testis gene MAEL that is regulated by DNA methylation. Mol Biol Rep 2010; 37:2355-2360.
- Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet 2013; 14:880-893.
- Conley AB, Miller WJ, Jordan IK. Human cis natural antisense transcripts initiated by transposable elements. Trends Genet 2008; 24:53-56.
- Pandey R, Mandal AK, Jha V, Mukerji M. Heat shock factor binding in Alu repeats expands its involvement in stress through an antisense mechanism. Genome Biol 2011; 12:R117.
- Balbin OA, Malik R, Dhanasekaran SM, Prensner JR, Cao X, Wu YM, Robinson D, Wang R, Chen G, Beer DG, et al. The landscape of antisense gene expression in human cancers. Genome Res 2015; 25:1068-1079.
- Carreira PE, Richardson SR, Faulkner GJ. L1 retrotransposons, cancer stem cells and oncogenesis. FEBS J 2014; 281:63-73.
- Mourier T, Nielsen LP, Hansen AJ, Willerslev E. Transposable elements in cancer as a by-product of stress-induced evolvability. Front Genet 2014; 5:156.
- Wilkins AS. The enemy within: an epigenetic role of retrotransposons in cancer initiation. Bioessays 2010; 32:856-865.
- Miousse IR, Chalbot MC, Lumen A, Ferguson A, Kavouras IG, Koturbash I. Response of transposable elements to environmental stressors. Mutat Res Rev Mutat Res 2015; 765:19-39.
- Poliseno L, Marranci A, Pandolfi PP. Pseudogenes in human cancer. Front Med (Lausanne) 2015; 2:68.
- Roberts TC, Morris KV. Not so pseudo anymore: pseudogenes as therapeutic targets. Pharmacogenomics 2013; 14:2023-2034.
- Johnsson P, Ackley A, Vidarsdottir L, Lui WO, Corcoran M, Grandér D, Morris KV. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat Struct Mol Biol 2013; 20:440-446.
- Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol 2012; 30:677-706.
- Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 2013; 13:759-771.
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015; 162:974-986.
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. DNA-Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015; 162:961-973.
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, Fend L, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014; 20:1301-1309.
- Huang J, Xie Y, Sun X, Zeh HJ, 3rd, Kang R, Lotze MT, Tang D. DAMPs, ageing, and cancer: The [DAMP Hypothesis]. Ageing Res Rev 2015; 24:3-16.
- Fucikova J, Moserova I, Urbanova L, Bezu L, Kepp O, Cremer I, Salek C, Strnad P, Kroemer G, Galluzzi L, et al. Prognostic and predictive value of DAMPs and DAMP-Associated processes in cancer. Front Immunol 2015; 6:402.
- Moris A, Murray S, Cardinaud S. AID and APOBECs span the gap between innate and adaptive immunity. Front Microbiol 2014; 5:534.
- Zanotti KJ, Gearhart PJ. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair (Amst) 2016; 38:110-116.
- Franchini DM, Petersen-Mahrt SK. AID and APOBEC deaminases: balancing DNA damage in epigenetics and immunity. Epigenomics 2014; 6:427-443.
- Ramiro AR, Barreto VM. Activation-induced cytidine deaminase and active cytidine demethylation. Trends Biochem Sci 2015; 40:172-181.
- Baixauli F, Lopez-Otin C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol 2014; 5:403.
- Zhang X, Yuan X, Shi H, Wu L, Qian H, Xu W. Exosomes in cancer: small particle, big player. J Hematol Oncol 2015; 8:83.
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borràs FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, et al. Biological properties of extracellular vesicles and their physiological functions. J Extra Cell Vesicles 2015; 4:27066.
- Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM. Exosome mediated communication within the tumor microenvironment. J Control Release 2015; 219:278-294.
- Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014; 159:499-513.
- Sousa D, Lima RT, Vasconcelos MH. Intercellular transfer of cancer drug resistance traits by extracellular vesicles. Trends Mol Med 2015; 21:595-608.
- van der Grein SG, Nolte-'t Hoen EN. “Small Talk” in the innate immune system via RNA-containing extracellular vesicles. Front Immunol 2014; 5:542.
- Kim YJ, Maizel A, Chen X. Traffic into silence: endomembranes and post-transcriptional RNA silencing. EMBO J 2014; 33:968-980.
- Leung AK. The whereabouts of microRNA actions: Cytoplasm and beyond. Trends Cell Biol 2015; 25:601-610.
- Bento CF, Puri C, Moreau K, Rubinsztein DC. The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci 2013; 126:1059-1069.
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al. Autophagy in malignant transformation and cancer progression. EMBO J 2015; 34:856-880.
- Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013; 153:1461-1474.
- Lencer WI, DeLuca H, Grey MJ, Cho JA. Innate immunity at mucosal surfaces: the IRE1-RIDD-RIG-I pathway. Trends Immunol 2015; 36:401-409.
- Jheng JR, Ho JY, Horng JT. ER stress, autophagy, and RNA viruses. Front Microbiol 2014; 5:388.
- Squadrito ML, Baer C, Burdet F, Maderna C, Gilfillan GD, Lyle R, Ibberson M, De Palma M. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep 2014; 8:1432-1446.
- Ding SW, Lu R. Virus-derived siRNAs and piRNAs in immunity and pathogenesis. Curr Opin Virol 2011; 1:533-544.
- Dumesic PA, Madhani HD. Recognizing the enemy within: licensing RNA-guided genome defense. Trends Biochem Sci 2014; 39:25-34.
- Rechavi O. Guest list or black list: heritable small RNAs as immunogenic memories. Trends Cell Biol 2014; 24:212-220.
- Parrish NF, Fujino K, Shiromoto Y, Iwasaki YW, Ha H, Xing J, Makino A, Kuramochi-Miyagawa S, Nakano T, Siomi H, et al. piRNAs derived from ancient viral processed pseudogenes as transgenerational sequence-specific immune memory in mammals. RNA 2015; 21:1691-1703.
- Haase AD. A small RNA-based immune system defends germ cells against mobile genetic elements. Stem Cells Int 2016; 2016:7595791.
- Malone CD, Hannon GJ. Small RNAs as guardians of the genome. Cell 2009; 136:656-668.
- Yona AH, Frumkin I, Pilpel Y. A relay race on the evolutionary adaptation spectrum. Cell 2015; 163:549-559.
- Matsumoto N, Sato K, Nishimasu H, Namba Y, Miyakubi K, Dohmae N, Ishitani R, Siomi H, Siomi MC, Nureki O. Crystal structure and activity of the Endoribonuclease domain of the piRNA pathway factor maelstrom. Cell Rep 2015; 11:366-375.
- Reuter M, Chuma S, Tanaka T, Franz T, Stark A, Pillai RS. Loss of the Mili-interacting Tudor domain-containing protein-1 activates transposons and alters the Mili-associated small RNA profile. Nat Struct Mol Biol 2009; 16:639-646.
- Wasik KA, Tam OH, Knott SR, Falciatori I, Hammell M, Vagin VV, Hannon GJ. RNF17 blocks promiscuous activity of PIWI proteins in mouse testes. Genes Dev 2015; 29:1403-1415.
- Kasahara M, Clikeman JA, Bates DB, Kogoma T. RecA protein-dependent R-loop formation in vitro. Genes Dev 2000; 14:360-365.
- Khadka P, Croteau DL, Bohr VA. RECQL5 has unique strand annealing properties relative to the other human RecQ helicase proteins. DNA Repair (Amst) 2016; 37:53-66.
- Wahba L, Gore SK, Koshland D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. Elife 2013; 2:e00505.
- Rhodes D, Lipps HJ. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res 2015; 43:8627-8637.
- Wanrooij PH, Uhler JP, Shi Y, Westerlund F, Falkenberg M, Gustafsson CM. A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res 2012; 40:10334-10344.