
ABSTRACT
Enhancers control development and cellular function by spatiotemporal regulation of gene expression. Co-occurrence of acetylation of histone H3 at lysine 27 (H3K27ac) and mono methylation of histone H3 at lysine 4 (H3K4me1) has been widely used for identification of active enhancers. However, increasing evidence suggests that using this combination of marks alone for enhancer identification gives an incomplete picture of the active enhancer repertoire. We have shown that the H3 globular domain acetylations, H3K64ac and H3K122ac, and an H4 tail acetylation, H4K16ac, are enriched at active enhancers together with H3K27ac, and also at a large number of enhancers without detectable H3K27ac. We propose that acetylations at these lysine residues of histones H3 and H4 might function by directly affecting chromatin structure, nucleosome–nucleosome interactions, nucleosome stability, and transcription factor accessibility.
Introduction
Histone post-translational modification (PTM) status, DNA methylation, transcription factor (TF) binding, and DNA accessibility profiles are extensively used to understand the gene regulatory mechanisms in various cell and tissue types. Although much progress has been made in genome-wide profiling of chromatin modifications, occupancy of TFs, and coactivators, most of the data is still correlative, and it is difficult to delineate the importance of each of these dynamic features in regulating chromatin structure and/or gene expression (reviewed in ref.Citation1). Most histone tail PTMs function by recruiting chromatin proteins that have distinct domains that can ‘read’ specific histone modifications, which then affect various chromatin-mediated processes like transcription, DNA repair, and RNA processing.Citation2–6 An increasing number of such proteins have been identified using chromatin proteomic techniques (highlighted in ref.Citation7). Unlike histone H3 tail PTMs that depend on these effector proteins for their function, H4 tail modifications—H4K16ac and H4K20me—are known to directly affect chromatin structure by altering inter-nucleosomal interactions in vitro ().Citation8-10 PTMs can also occur at the histone globular domain that is also called histone fold domain. PTMs at the globular domain, especially at the lateral (outer) surface of the histone octamer, can directly affect nucleosomal stability and chromatin structure by altering the histone–DNA interactions ().Citation11-13
Figure 1. (A) H4K16ac role in increasing the accessibility of DNA to TFs basic amino acid residues at the histone H4 tail interaction with the acidic patches of H2A from neighboring nucleosomes, which is important for higher order chromatin folding. Mof-mediated acetylation of lysine 16 on the histone H4 (H4K16ac) tail disrupts the inter-nucleosomal interaction in vitro, which disrupts the higher order chromatin structure and might also increase the accessibility of TFs to DNA and increased eRNA transcription. (B) Working model for histone H3 globular acetylations at regulatory elements. Monomethylation of H3K4 residues (H3K4me1) away from transcription start sites (TSSs) is widely used to identify enhancers. Inactive enhancers are H3K4me1 positive, and they lack detectable level of histone acetylations. Poised enhancers are enriched for H3K27me3 along with H3K4me1 and H3K122ac. At active enhancers, EP300 acetylates nucleosomes at H3K27, H3K64, H3K122, and Mof acetylates H4K16. H3K122ac co-localizes with H2A.Z-containing nucleosomes, suggesting that H3K122ac might contribute to destabilization of nucleosomes at active and poised enhancers. Working model shows that EP300-mediated H3 globular domain acetylations lead to increased accessibility of nucleosomal DNA for TFs by reducing the affinity between histone and DNA. H3 globular acetylation facilitates the remodeling of nucleosomal DNA and contributes to destability of labile nucleosomes and eRNA transcription. Presence (+) or absence (−) of histone modifications, histone variants at enhancer classes is given below.
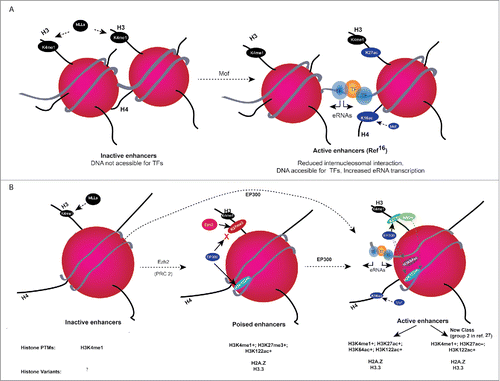
Use of histone modifications to differentiate enhancers from promoters
A large number of efforts have been made to map various histone modifications using chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) in a variety of cell types and organisms. Analysis of this data shows that the enrichment of most histone tail modifications correlate or anti-correlate with gene expression levels, and also that they are enriched at particular genomic features. For example, it is clear that H3K4me3 is present at promoters, whereas H3K4me1 is enriched at enhancers. This high H3K4me1 to H3K4me3 ratio is widely used to distinguish enhancers from promoters (reviewed in Ref.Citation14). Along with H3K4 methylation, active promoters and enhancers are enriched with histone acetylations, such as H3K27ac and H4K16ac.Citation15-19 Combinations of H3K4me1, H3K27ac, and H3K27me3 are widely used to classify active enhancers (H3K4me1 and H3K27ac positive) from inactive (H3K4me1 positive and H3K27ac negative) or poised enhancers (H3K4me1 and H3K27me3 positive).Citation15,17,20
Role of histone acetylations in regulation of gene expression
The functions of most histone tail modifications depend on effector proteins that recognize these modifications and bind to them. Exception to that is acetylation at H4K16 and methylation at H4K20, which have direct impacts on inter-nucleosomal interactions.Citation8,10 The H4 tail has been shown to interact with the acidic patch of H2A in the adjacent nucleosomes, and H4K16ac inhibits 30-nm chromatin fiber formation by destabilizing the inter-nucleosomal interactions ().
Similarly, PTMs located on the lateral surface of the histone octamer can directly alter contacts between histones and nucleosomal DNA and affect chromatin structure directly (reviewed in refCitation21). Acetylation of histone H3 at lysine 56 (H3K56ac) by EP300 is associated with DNA unwrapping from the nucleosome and has been implicated in chromatin assembly and genome stability.Citation22,23 Although this modification is abundant and better studied in yeast, H3K56ac is present at very low levels in mammals and shown to be enriched at regulatory regions in human stem cells.Citation24 Recently, two more lysine residues at the histone H3 globular domain, K122 and K64, have been shown to be acetylated by EP300.Citation11,12
Both H3K64ac and H3K122ac are located at the lateral surface of the histone octamer. H3K64ac, a modification at the lysine residue at the start of the first α-helix in the histone fold domain, destabilizes nucleosomes and facilitates nucleosome dynamics in vitro.Citation11 The H3K122 residue is located at the dyad axis where only one DNA fiber binds, and histone binding to DNA is at its highest strength (reviewed in ref.Citation25). Rob Schneider's group has elegantly shown that H3K122ac directly impacts histone–DNA binding in the dyad axis. Unlike acetylation on histone tails, H3K122ac is sufficient to stimulate transcription in vitro from chromatinized templatesCitation12 and promote nucleosome disassemblyCitation26 (). We have shown the presence of H3K122ac at promoters of expressed genes, and also at poised genes along with H3K27me3, H2A.Z, and H2A.Zac, which are known to be associated with poised genes.Citation27-29 Our H3K122ac ChIP-seq data together with data from in vitro transcription assays and induction studies done in Schizosaccharomyces pombe suggest the causal role of H3K122ac in rapid activation of poised genes upon differentiation signaling.Citation12,27
Although H3K64ac and H3K122ac are shown to be acetylated by EP300 acetyl transferase.Citation11,12 Recently, bromodomain-containing protein 4 (BRD4), a chromatin reader protein that recognizes and binds acetylated histones, also has been recently shown to possess acetyl transferase activity. BRD4 also suggested to acetylate several lysine residues of histone H3 and H4, including H3K122.Citation30 It has been proposed that BRD4 increases transcription by facilitating the eviction of nucleosomes by catalyzing the acetylation at H3K122. It remains to be investigated whether BRD4 can directly bind acetylated H3K122 or is recruited through histone tail acetylations. However, absence of histone tail acetylations at many H3K122ac sites suggests that there might be a histone acetylation independent mechanism of recruitment for BRD4 at these sites.
Complex pattern of histone modification at enhancers
Although H3K4me1, H3K27ac, and EP300-binding profile are widely used to predict enhancer activity,Citation15,17,20 it is becoming increasingly clear that this combination of histone marks and EP300 co-activator binding may not identify all active enhancers.Citation16,27,31,32 We have previously shown that acetylation of Lysine 16 of histone H4 (H4K16ac) marks active enhancers that are associated with pluripotency-associated genes in mouse embryonic stem cells. Interestingly, there are also some active enhancers, which are H3K27ac negative but are marked with H4K16ac.Citation16 KAT8/MOF which acetylates H4K16,Citation16 and the ATAC complex containing Gcn5/PCAF, and H3K9ac, which is preferentially catalyzed by Gcn5/PCAF have also been detected at enhancers, suggesting a wider role of different lysine acetyl transferase complexes in enhancer function.Citation16,31,33
Co-occurrence of H3 globular acetylations, H3K122ac and H3K64ac, with H3K4me1 at enhancer elements suggested their role in enhancer function. Moreover, we suggest H3K122ac can be used to identify active enhancer elements more robustly than the widely used H3K27ac mark, which misses a whole new class of enhancers.Citation27 Interestingly, H3K64ac is enriched at highly active enhancers and also at long stretches of highly active enhancers so-called super-enhancers (SEs).Citation27,34 SEs are known to transcribe higher levels of eRNAs compared to typical enhancers.Citation35 It is reasonable to assume that H3K64ac might contribute to transcription of eRNAs at highly active enhancer elements, including SEs. High level of H3K64ac but not H3K122ac at these SEs suggests that although these two globular domain modifications are present at regulatory elements, they might have different functional mechanisms in vivo. It will be interesting to study whether simultaneous acetylations at both H3K122 and H3K64 further increase eRNA transcription and contributes to enhancer activity.
Unique features of H3K122ac positive enhancers
Although high levels of H3K64ac and H3K122ac are detected at active enhancers as defined by the presence of H3K27ac, H3K122ac is also found at H3K27ac negative enhancers, a subset of which are H3K27me3 positive. H3K27me3 and H2A.Z are known to colocalize across polycomb-repressed genes and enhancers.Citation15,28,29 Co-occurrence of H3K122ac with H3K27me3 and H2A.Z at both poised promoters and at H3K27ac negative enhancers suggests that subset of H3K122ac positive enhancers are poised. The presence of H3K122ac, H2A.Z at poised promoters and enhancers hints at a causal role of H3K122ac and H2A.Z containing labile nucleosomes in increasing the accessibility of TFs for rapid induction of transcription upon differentiation signaling.Citation12,27
DNA methylations at enhancers
DNA methylation is associated with gene repression, and CpG islands located at promoters are generally unmethylated, in contrast to those at gene bodies of expressed genes that are methylated.Citation36 Intriguingly, recent studies also show a higher level of DNA methylation at active enhancers that transcribe eRNAs, although the significance of this is not known.Citation37,38 Like gene body methylation, enhancer methylation has been shown to be dependent on transcription and H3K36me3-mediated recruitment of DNMT3A/DNMT3B.Citation39 This new observation of eRNA-transcription-dependent DNA methylation at enhancers suggests that enhancers and genes share similar chromatin features. Increasing pieces of evidence show that like promoters, chromatin features at enhancers are more complex than previously thought (), and it is important to consider the combinatorial features of enhancers to decipher the enhancer activity in a given cell type.Citation16,27
Recent studies show correlation of active enhancers (H3K27ac positive) with DNA methylation at enhancers.Citation37 DNA methylation is known to inhibit the CTCF binding at many sites (reviewed inCitation40). We speculate that subclass of H3K122ac enhancers has a higher level of CTCF and other architectural proteins than H3K27ac enhancers; these multifunctional proteins might contribute to a unique mechanism of enhancer action. Further investigations are needed to improve our understanding of interplay among TFs, DNA methylation, CTCF and its interacting partners at H3K122ac enhancers.
Enhancer RNAs (eRNA), their process of transcription in enhancer mechanism
Enhancer activity is correlated with bidirectional transcription from enhancer elements by RNA Polymerase II, producing a class of non-coding RNAs called eRNAsCitation41-43 which are degraded by exosome complexes.Citation44 There is a clear association of enhancer elements with bidirectional transcription as measured by capped analysis of gene expression (CAGE) tags,Citation43 and it has been suggested that such bi-directional unstable transcripts might be the best way to catalog active enhancers in a particular cell-type. However, recent computational analysis argues against the specificity of bidirectional transcription in identifying active enhancer elements, as it appears to arise due to promiscuous RNA Pol II activity at accessible chromatin in general and not just that specific to enhancers.Citation45 Importance of eRNAs in enhancer function is still controversial; however, there are few eRNAs that are shown to be functional in positively regulating target genes.Citation46,47 Some long noncoding RNAs (lncRNAs) such as Hottip are also shown to have enhancer-like function.Citation48,49
Causal role of histone H3 globular acetylations at enhancers
The functional significance of histone marks at enhancers is not known (reviewed in ref.Citation1). Most histone tail modifications depend on effector proteins, which bind to modified histone tails. Bromodomain-containing proteins—e.g. BRD2, 3, and 4—bind to acetylated histone tail residues at promoters as well as enhancers and have been shown to promote transcription of protein coding genes and eRNAs.Citation50 Acetylation at H3 globular domain residues, H3K64 and H3K122, located at the lateral (outer) surface of the histone octamer can directly destabilize nucleosomes and stimulate transcription.Citation21 Enrichment of these modifications at active promoters and enhancers suggests their causal role in stimulating transcription of genes and eRNAs. Our recent genome-wide studies in ESCs and cancer cells support the data from previous in vitro experiments and induction experiments done in S. pombe.Citation11,12 Together, we suggest a causal role of these H3 globular domain acetylations in stimulating transcription at both promoters and enhancers.Citation11,12,27,51 Intriguingly, we detect low level of CAGE tags in H3K122ac positive enhancers, which lack detectable level of histone tail acetylations. However, these enhancers indeed transcribe bidirectional eRNA, but are degraded by exosome complexes.Citation27,52 This data shows that many of the bidirectional eRNAs at new class of H3K122ac enhancers are either low abundant or rapidly degraded by exosome complexes; it is possible to miss identification of some of the H3K122ac positive active enhancers using CAGE based assays.
Importance of labile nucleosomes at enhancers
H3K122ac is detected at nucleosomes with histone variants H3.3 and H2A.Z at promoters and enhancers,Citation12,27 suggesting that the K122 residue might be acetylated on H3.3 molecules. Nucleosomes with these double histone variants and H3K122ac are known to be particularly unstable,Citation26,53 and such labile H3.3/H2A.Z nucleosomes are shown to occupy regulatory elements which are widely regarded as ‘nucleosome free.’Citation53,54 We speculate that acetylation of H3K122 contributes to the destabilization of H3.3/H2A.Z nucleosomes at promoter and enhancer elements and that this will aid the binding of TFs.
Future perspectives
Recent functional genomics data suggests that there are many more gene regulatory elements than previously thought (ENCODE consortium). Although a large number of disease-associated single nucleotide polymorphisms (SNPs) map to enhancer elements, a lack of information about gene-specific function of enhancers means that the causal role of most SNPs in disease is not known. Increasing evidence suggests that the accessible chromatin structure caused by labile nucleosomes and/or the act of transcription at enhancers might have a causal role in enhancer activity. Recent development in genome editing tools like TALE and CRISPR Cas9 and their versatility will no doubt be useful in deciphering the importance of DNA methylation, histone modifications, or the act of eRNA transcription at enhancers.Citation27,55-58
The role of histone modifications in enhancer function is generally not clear, and it has been challenging to find out the enhancer specific functions for histone modifications. Although H3K122ac is enriched at active enhancers along with H3K27ac, a subset of H3K122ac-marked novel enhancers is also enriched for H3K27me3, a marker of poised enhancers, suggesting a role for H3K122ac in maintaining accessible chromatin at both active and poised regulatory elements.Citation27 Investigations into the specific recruitment mechanisms of protein complexes that modify histone and non-histone proteins, together with the understanding of the importance of reader proteins that binds to these modifications will be useful in deciphering the importance of these PTMs in enhancer function.
The presence of H3 globular domain acetylations at the lateral surface of the histone octamer together with histone tail acetylations suggests that there are both effector dependent and independent functions for these modifications in enhancer function. Further investigations are needed to appreciate the functional importance of these complex and highly dynamic chromatin features at enhancers in maintaining the chromatin structure and regulation of gene expression.
In summary, it is important to address the cause/consequence relationship of dynamic features at enhancers like open chromatin, histone PTMs, histone variants, the process of bidirectional transcription, and DNA methylation in order to understand enhancer mechanisms. Future work will potentially unveil the complex interplay between the process of eRNA transcription, histone modifications, and protein complexes present at enhancer elements and their contribution to the distal regulation of gene expression.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
I thank Wendy Bickmore, Michael Moffat, Rob Schneider, and anonymous reviewers for constructive comments to improve this manuscript.
Funding
I also thank Medical Research Council, UK, University of Essex and Wellcome Trust seed award for funding my work.
References
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell 2013; 49:825-37; PMID:23473601; http://dx.doi.org/10.1016/j.molcel.2013.01.038
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381-95; PMID:21321607; http://dx.doi.org/10.1038/cr.2011.22
- Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet 2012; 8:e1002717; PMID:22615581; http://dx.doi.org/10.1371/journal.pgen.1002717
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science 2010; 327:996-1000; PMID:20133523; http://dx.doi.org/10.1126/science.1184208
- Pradeepa MM, Grimes GR, Taylor GC, Sutherland HG, Bickmore WA. Psip1/Ledgf p75 restrains Hox gene expression by recruiting both trithorax and polycomb group proteins. Nucleic Acids Res 2014; 42:9021-32; PMID:25056311; http://dx.doi.org/10.1093/nar/gku647
- Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sørensen CS, Petersen NH, Sorensen PH, Lukas C, Bartek J et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol 2012; 19:803-10; PMID:22773103; http://dx.doi.org/10.1038/nsmb.2314
- Baker M. Mass spectrometry for chromatin biology. Nat Methods 2012; 9:649-52; http://dx.doi.org/10.1038/nature01096
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006; 311:844-7; PMID:16469925; http://dx.doi.org/10.1126/science.1124000
- Allahverdi A, Yang R, Korolev N, Fan Y, Davey CA, Liu CF, Nordenskiöld L. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res 2011; 39:1680-91; PMID:21047799; http://dx.doi.org/10.1093/nar/gkq900
- Lu X, Simon MD, Chodaparambil JV, Hansen JC, Shokat KM, Luger K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat Struct Mol Biol 2008; 15:1122-4; PMID:18794842; http://dx.doi.org/10.1038/nsmb.1489
- Di Cerbo V, Mohn F, Ryan DP, Montellier E, Kacem S, Tropberger P, Kallis E, Holzner M, Hoerner L, Feldmann A et al. Acetylation of histone H3 at lysine 64 regulates nucleosome dynamics and facilitates transcription. Elife 2014; 3:e01632; PMID: 24668167; http://dx.doi.org/10.7554/eLife.01632
- Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, Li G, Mittler G, Liu ET, Bühler M et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 2013; 152:859-72; PMID:23415232; http://dx.doi.org/10.1016/j.cell.2013.01.032
- Mersfelder EL, Parthun MR. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res 2006; 34:2653-62; PMID:16714444; http://dx.doi.org/10.1093/nar/gkl338
- Kim TK, Shiekhattar R. Architectural and functional commonalities between enhancers and promoters. Cell 2015; 162:948-59; PMID:26317464; http://dx.doi.org/10.1016/j.cell.2015.08.008
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010; 107:21931-6; PMID: 21106759; http://dx.doi.org/10.1073/pnas.1016071107
- Taylor G, Eskeland R, Hekimoglu-Balkan B, Pradeepa M, Bickmore WA. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res 2013; 23:2053-65; PMID:23990607; http://dx.doi.org/10.1101/gr.155028.113
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011; 470:279-83; PMID:21160473; http://dx.doi.org/10.1038/nature09692
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108-12; PMID:19295514; http://dx.doi.org/10.1038/nature07829
- Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 2011; 21:1273-83; PMID:21632746; http://dx.doi.org/10.1101/gr.122382.111
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007; 39:311-8; http://dx.doi.org/10.1038/ng1966
- Tropberger P, Schneider R. Scratching the (lateral) surface of chromatin regulation by histone modifications. Nat Struct Mol Biol 2013; 20:657-61; PMID:23739170; http://dx.doi.org/10.1038/nsmb.2581
- Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell 2009; 36:153-63; PMID:19818718; http://dx.doi.org/10.1016/j.molcel.2009.07.027
- Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009; 459:113-7; PMID:19270680; http://dx.doi.org/10.1038/nature07861
- Xie W, Song C, Young NL, Sperling AS, Xu F, Sridharan R, Conway AE, Garcia BA, Plath K, Clark AT et al. Histone H3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell 2009; 33:417-27; PMID:19250903; http://dx.doi.org/10.1016/j.molcel.2009.02.004
- Lawrence M, Daujat S, Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet 2016; 32:42-56; PMID:26704082; http://dx.doi.org/10.1016/j.tig.2015.10.007
- Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ, Poirier MG. Histone fold modifications control nucleosome unwrapping and disassembly. Proc Natl Acad Sci U S A 2011; 108:12711-6; PMID:21768347; http://dx.doi.org/10.1073/pnas.1106264108
- Pradeepa MM, Grimes GR, Kumar Y, Olley G, Taylor GC, Schneider R, Bickmore WA. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet 2016; 48:681-6; PMID:27089178; http://dx.doi.org/10.1038/ng.3550
- Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, Young RA, Jaenisch R, Boyer LA. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 2008; 135:649-61; PMID:18992931; http://dx.doi.org/10.1016/j.cell.2008.09.056
- Illingworth RS, Botting CH, Grimes GR, Bickmore WA, Eskeland R. Prc1 and prc2 are not required for targeting of h2a.z to developmental genes in embryonic stem cells. PLoS One 2012; 7:1-13; http://dx.doi.org/10.1371/journal.pone.0034848
- Devaiah BN, Case-Borden C, Gegonne A, Hsu CH, Chen Q, Meerzaman D, Dey A, Ozato K, Singer DS. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat Struct Mol Biol 2016; 23:1-12; PMID:26733217; http://dx.doi.org/10.1038/nsmb.3228
- Karmodiya K, Krebs AR, Oulad-Abdelghani M, Kimura H, Tora L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 2012; 13:424; PMID:22920947; http://dx.doi.org/10.1186/1471-2164-13-424
- Chelmicki T, Dündar F, Turley MJ, Khanam T, Aktas T, Ramírez F, Gendrel AV, Wright PR, Videm P, Backofen R et al. MOF-associated complexes ensure stem cell identity and Xist repression. Elife 2014; 3:e02024; PMID:24842875; http://dx.doi.org/10.7554/eLife.02024
- Krebs AR, Karmodiya K, Lindahl-Allen M, Struhl K, Tora L. SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Mol Cell 2011; 44:410-23; PMID:22055187; http://dx.doi.org/10.1016/j.molcel.2011.08.037
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell 2013; 155:934-47; PMID:24119843; http://dx.doi.org/10.1016/j.cell.2013.09.053
- Hah N, Benner C, Chong LW, Yu RT, Downes M, Evans RM. Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proc Natl Acad Sci U S A 2015; 112:E297-302; PMID:25564661; http://dx.doi.org/10.1073/pnas.1424028112
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484-92; http://dx.doi.org/10.1038/nrg3230
- Charlet J, Duymich CE, Lay FD, Mundbjerg K, Dalsgaard Sørensen K, Liang G, Jones PA. Bivalent regions of cytosine methylation and H3K27 acetylation suggest an active role for DNA methylation at enhancers. Mol Cell 2016; 62:422-31; PMID:27153539; http://dx.doi.org/10.1016/j.molcel.2016.03.033
- Kaaij LJT, Mokry M, Zhou M, Musheev M, Geeven G, Melquiond AS, de Jesus Domingues AM, de Laat W, Niehrs C, Smith AD et al. Enhancers reside in a unique epigenetic environment during early zebrafish development. Genome Biol 2016; 17:146; PMID:27381023; http://dx.doi.org/10.1186/s13059-016-1013-1
- Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, Blanco E, Pons JI, Matallanas D, Von Kriegsheim A et al. Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell 2016; 19:1-11; PMID:27476967; http://dx.doi.org/10.1016/j.stem.2016.06.020
- Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 2014; 15:234-46; PMID:24614316; http://dx.doi.org/10.1038/nrg3663
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010; 465:182-7; PMID:20393465; http://dx.doi.org/10.1038/nature09033
- Arner E, Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W, Lilje B, Drabløs F, Lennartsson A, Rönnerblad M et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science (80-) 2015; 347:1010-5; http://dx.doi.org/10.1126/science.1259418
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T et al. An atlas of active enhancers across human cell types and tissues. Nature 2014; 507:455-61; PMID:24670763; http://dx.doi.org/10.1038/nature12787
- Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu ZP et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell 2015; 161:774-89; PMID:25957685; http://dx.doi.org/10.1016/j.cell.2015.04.034
- Young RS, Kumar Y, Bickmore WA, Taylor MS. Bidirectional transcription marks accessible chromatin and is not specific to enhancers. BiorXiv 2016; http://dx.doi.org/http://dx.doi.org/10.1101/048629
- Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Léveillé N, Kalluri R et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 2013; 49:524-35; PMID:23273978; http://dx.doi.org/10.1016/j.molcel.2012.11.021
- Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel G a, Shiekhattar R. Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature 2013; 494:497-501; PMID:23417068; http://dx.doi.org/10.1038/nature11884
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011; 472:120-4; PMID:21423168; http://dx.doi.org/10.1038/nature09819
- Pradeepa MM, Taylor GAG, Grimes GR, Wood A, Bickmore W. Psip1/p52 regulates distal Hoxa genes through activation of lncRNA Hottip. BiorXiv 2016; http://dx.doi.org/10.1101/061192
- Kanno T, Kanno Y, LeRoy G, Campos E, Sun H-W, Brooks SR, Vahedi G, Heightman TD, Garcia BA, Reinberg D et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 2014; 21:1047-57; PMID:25383670; http://dx.doi.org/10.1038/nsmb.2912
- Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 2005; 121:375-85; PMID:15882620; http://dx.doi.org/10.1016/j.cell.2005.03.011
- Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu ZP et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell 2015; 161:774-89; PMID:25957685; http://dx.doi.org/10.1016/j.cell.2015.04.034
- Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. H3.3/H2A.Z double variant-containing nucleosomes mark “nucleosome-free regions” of active promoters and other regulatory regions. Nat Genet 2009; 41:941-5; PMID:19633671; http://dx.doi.org/10.1038/ng.409
- Henikoff S. Labile H3.3+H2A.Z nucleosomes mark “nucleosome-free regions.” Nat Publ Gr 2009; 41:865-6; PMID: 19639024; http://dx.doi.org/10.1038/ng0809-865
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R. Editing DNA methylation in the mammalian genome. Cell 2016; 167:233-47.e17; PMID:27662091; http://dx.doi.org/10.1016/j.cell.2016.08.056
- Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, Bernstein BE. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol 2013; 31:1133-6; PMID:24013198; http://dx.doi.org/10.1038/nbt.2701
- King AD, Huang K, Rubbi L, Liu S, Wang C-Y, Wang Y, Pellegrini M, Fan G. Reversible regulation of promoter and enhancer histone landscape by DNA methylation in mouse embryonic stem cells. Cell Rep 2016; 17:289-302; PMID:27681438; http://dx.doi.org/10.1016/j.celrep.2016.08.083
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013; 152:1173-83; PMID:23452860; http://dx.doi.org/10.1016/j.cell.2013.02.022